4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases

 , , ,
, , , 
 and
and 
Abstract
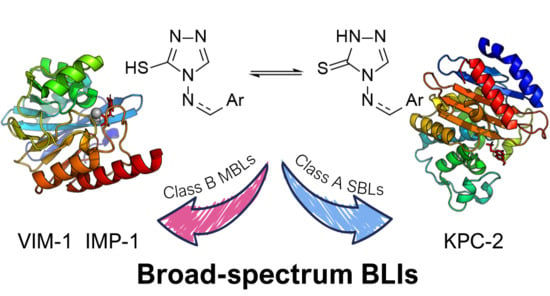
1. Introduction
2. Results and Discussion
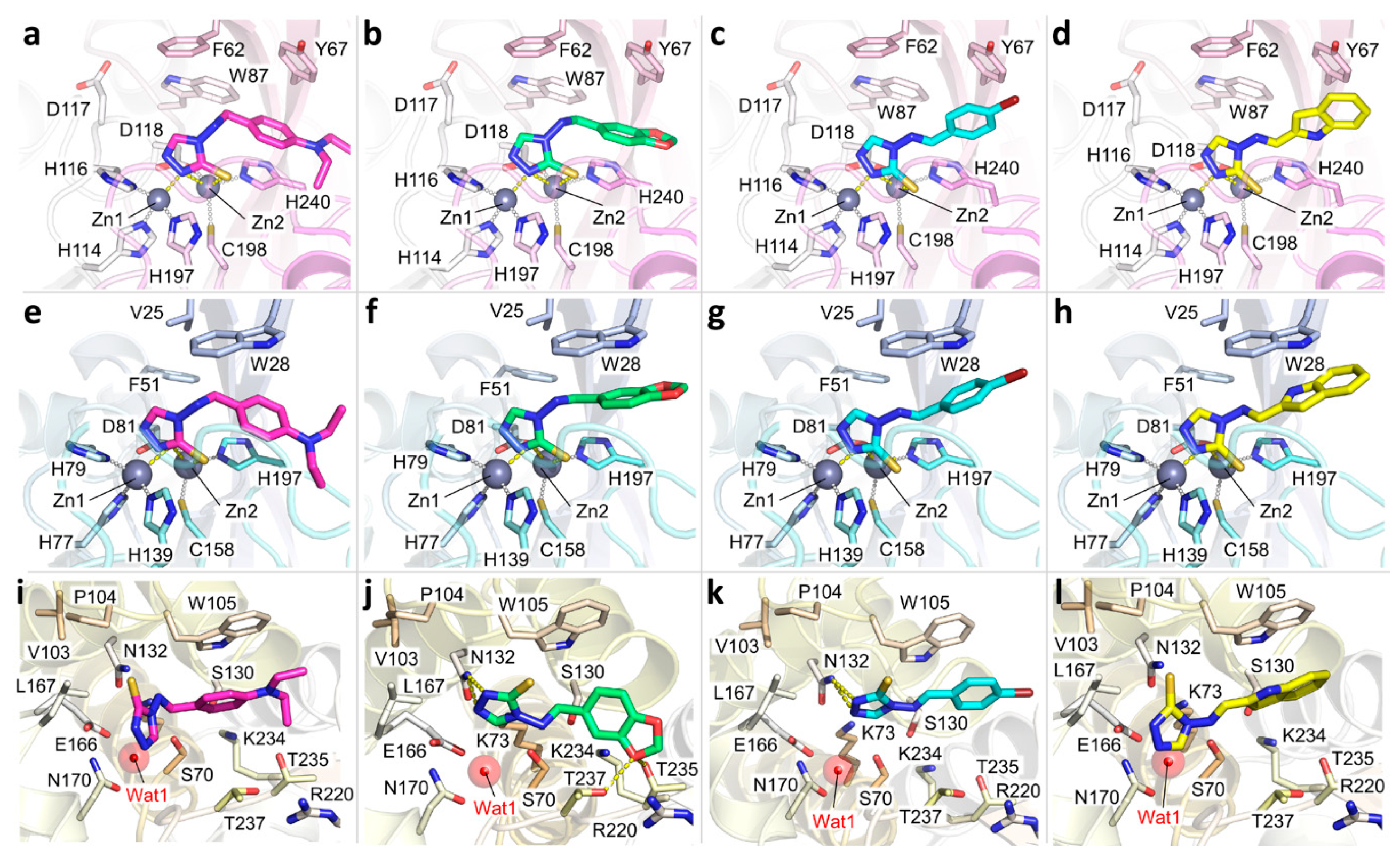
2.1. Compounds Design and Synthesis
2.2. Compounds In-Vitro Validation vs. SBLs and MBLs
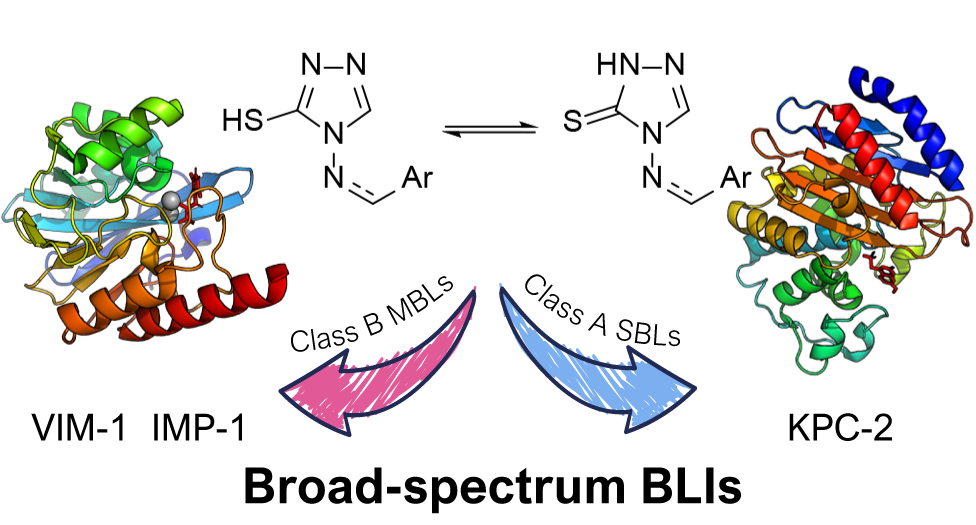
2.3. Molecular Docking and Molecular Dynamics
2.3.1. MBLs
2.3.2. KPC-2
2.4. Determination of Minimum Inhibitory Concentration (MIC) against Clinical Strains
3. Conclusions
4. Materials and Methods
4.1. Chemistry
4.1.1. General Procedure for the Synthesis of Amines 1a–g
4.1.2. General Procedure for the Synthesis of Imines 2a–g
4.1.3. Spectroscopic Data
4.2. Static Docking
4.3. MD Docking
4.4. Proteins Production and Purification
4.4.1. KPC-2
4.4.2. VIM-1
4.4.3. IMP-1
4.5. In Vitro Enzyme Inhibition Assays Against KPC-2, VIM-1 and IMP-1
4.6. MICs Assays
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- World Health Organization. Antimicrobial Resistance: Global Report on Surveillance; WHO Librrary Catalog Data; WHO: Geneva, Switzerland, 2016. [Google Scholar]
- Culyba, M.J.; Mo, C.Y.; Kohli, R.M. Targets for Combating the Evolution of Acquired Antibiotic Resistance. Biochemistry 2015, 54, 3573–3582. [Google Scholar] [CrossRef] [PubMed]
- Bellio, P.; Di, L.; Mancini, A.; Piovano, M.; Nicoletti, M.; Brisdelli, F.; Tondi, D.; Cendron, L.; Franceschini, N.; Amicosante, G.; et al. Phytomedicine Original article SOS response in bacteria: Inhibitory activity of lichen secondary metabolites against Escherichia coli RecA protein. Phytomedicine 2017, 29, 11–18. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, F.; Baldelli, V.; Halliday, N.; Pantalone, P.; Polticelli, F.; Fiscarelli, E.; Williams, P.; Visca, P.; Leoni, L.; Rampioni, G. Identification of FDA-Approved Drugs as Antivirulence Agents Targeting the pqs Quorum-Sensing System of Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 2018, 62, e01296-18. [Google Scholar] [CrossRef] [PubMed]
- Mobarki, N.; Almerabi, B.; Hattan, A. Antibiotic Resistance Crisis. Int. J. Med. Dev. Ctries. 2019, 40, 561–564. [Google Scholar] [CrossRef]
- Mojica, M.F.; Bonomo, R.A.; Fast, W. B1-Metallo-β-Lactamases: Where do we stand? Curr. Drugs Targets 2016, 17, 1029–1050. [Google Scholar] [CrossRef]
- Walsh, T.R. The emergence and implications of metallo-β-lactamases in Gram-negative bacteria. Clin. Microbiol. Infect. Suppl. 2005, 11, 2–9. [Google Scholar] [CrossRef]
- Hall, B.G.; Barlow, M. Revised Ambler classification of β-lactamases. J. Antimicrob. Chemother. 2005, 55, 1050–1051. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global spread of carbapenemase producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef]
- Mehta, S.C.; Rice, K.; Palzkill, T. Natural Variants of the KPC-2 Carbapenemase have Evolved Increased Catalytic Efficiency for Ceftazidime Hydrolysis at the Cost of Enzyme Stability. PLoS Pathog. 2015, 11, 1–20. [Google Scholar] [CrossRef]
- Linciano, P.; Cendron, L.; Gianquinto, E.; Spyrakis, F.; Tondi, D. Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect. Dis. 2019, 5, 9–34. [Google Scholar] [CrossRef]
- Drawz, S.M.; Bonomo, R.A. Three decades of β-lactamase inhibitors. Clin. Microbiol. Rev. 2010, 23, 160–201. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, M.; Iyobe, S.; Inoue, M.; Mitsuhashi, S. Transferable imipenem resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother. 1991, 35, 147–151. [Google Scholar] [CrossRef] [PubMed]
- Poirel, L.; Naas, T.; Nicolas, D.; Collet, L.; Bellais, S.; Cavallo, J.D.; Nordmann, P. Characterization of VIM-2, a carbapenem-hydrolyzing metallo-beta-lactamase and its plasmid- and integron-borne gene from a Pseudomonas aeruginosa clinical isolate in France. Antimicrob. Agents Chemother. 2000, 44, 891–897. [Google Scholar] [CrossRef] [PubMed]
- Lauretti, L.; Riccio, M.L.; Mazzariol, A.; Cornaglia, G.; Amicosante, G.; Fontana, R.; Rossolini, G.M. Cloning and characterization of blaVIM, a new integron-borne metallo-beta-lactamase gene from a Pseudomonas aeruginosa clinical isolate. Antimicrob. Agents Chemother. 1999, 43, 1584–1590. [Google Scholar] [CrossRef] [PubMed]
- Edelstein, M.V.; Skleenova, E.N.; Shevchenko, O.V.; D’souza, J.W.; Tapalski, D.V.; Azizov, I.S.; Sukhorukova, M.V.; Pavlukov, R.A.; Kozlov, R.S.; Toleman, M.A.; et al. Spread of extensively resistant VIM-2-positive ST235 Pseudomonas aeruginosa in Belarus, Kazakhstan, and Russia: A longitudinal epidemiological and clinical study. Lancet Infect. Dis. 2013, 13, 867–876. [Google Scholar] [CrossRef]
- Von Wintersdorff, C.J.H.; Penders, J.; Van Niekerk, J.M.; Mills, N.D.; Majumder, S.; Van Alphen, L.B.; Savelkoul, P.H.M.; Wolffs, P.F.G. Dissemination of antimicrobial resistance in microbial ecosystems through horizontal gene transfer. Front. Microbiol. 2016, 7, 1–10. [Google Scholar] [CrossRef]
- Cahill, S.T.; Cain, R.; Wang, D.Y.; Lohans, C.T.; Wareham, D.W.; Oswin, H.P.; Mohammed, J.; Spencer, J.; Fishwick, C.W.G.; McDonough, M.A.; et al. Cyclic Boronates Inhibit All Classes of β-Lactamases. Antimicrob. Agents Chemother. 2017, 61, e02260-16. [Google Scholar] [CrossRef]
- Krajnc, A.; Brem, J.; Hinchliffe, P.; Calvopiña, K.; Panduwawala, T.D.; Lang, P.A.; Kamps, J.J.A.G.; Tyrrell, J.M.; Widlake, E.; Saward, B.G.; et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-β-Lactamases. J. Med. Chem. 2019, 62, 8544–8556. [Google Scholar] [CrossRef]
- Liu, B.; Trout, R.E.L.; Chu, G.-H.; McGarry, D.; Jackson, R.W.; Hamrick, J.C.; Daigle, D.M.; Cusick, S.M.; Pozzi, C.; De Luca, F.; et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-β-lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem. 2019. [Google Scholar] [CrossRef]
- Karaiskos, I.; Lagou, S.; Pontikis, K.; Rapti, V.; Poulakou, G. The “Old” and the “New” antibiotics for MDR Gram-negative pathogens: For whom, when, and how. Front. Public Health 2019, 7, 1–25. [Google Scholar] [CrossRef]
- Ke, W.; Bethel, C.R.; Thomson, J.M.; Bonomo, R.A.; Van Den Akker, F. Crystal structure of KPC-2: Insights into carbapenemase activity in class A β-lactamases. Biochemistry 2007, 46, 5732–5740. [Google Scholar] [CrossRef] [PubMed]
- Pemberton, O.A.; Zhang, X.; Chen, Y. Molecular Basis of Substrate Recognition and Product Release by the Klebsiella pneumoniae Carbapenemase (KPC-2). J. Med. Chem. 2017, 60, 3525–3530. [Google Scholar] [CrossRef] [PubMed]
- Torelli, N.J.; Akhtar, A.; Defrees, K.; Jaishankar, P.; Pemberton, O.A.; Zhang, X.; Johnson, C.; Renslo, A.R.; Chen, Y. Active-Site Druggability of Carbapenemases and Broad-Spectrum Inhibitor Discovery. ACS Infect. Dis. 2019, 5, 1013–1021. [Google Scholar] [CrossRef] [PubMed]
- Docquier, J.D.; Mangani, S. An update on β-lactamase inhibitor discovery and development. Drug Resist. Updates 2018, 36, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Cendron, L.; Quotadamo, A.; Maso, L.; Bellio, P.; Montanari, M.; Celenza, G.; Venturelli, A.; Costi, M.P.; Tondi, D. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med. Chem. Lett. 2019, 10, 650–655. [Google Scholar] [CrossRef]
- Leiris, S.; Coelho, A.; Castandet, J.; Bayet, M.; Lozano, C.; Bougnon, J.; Bousquet, J.; Everett, M.; Lemonnier, M.; Sprynski, N.; et al. SAR Studies Leading to the Identification of a Novel Series of Metallo-β-lactamase Inhibitors for the Treatment of Carbapenem-Resistant Enterobacteriaceae Infections That Display Efficacy in an Animal Infection Model. ACS Infect. Dis. 2019, 5, 131–140. [Google Scholar] [CrossRef]
- Santucci, M.; Spyrakis, F.; Cross, S.; Quotadamo, A.; Farina, D.; Tondi, D.; De Luca, F.; Docquier, J.-D.; Prieto, A.I.; Ibacache, C.; et al. Computational and biological profile of boronic acids for the detection of bacterial serine- and metallo-β-lactamases. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Linciano, P.; Vicario, M.; Kekez, I.; Bellio, P.; Celenza, G.; Martín-Blecua, I.; Blázquez, J.; Cendron, L.; Tondi, D. Phenylboronic Acids Probing Molecular Recognition against Class A and Class C β-lactamases. Antibiotics 2019, 8, 171. [Google Scholar] [CrossRef]
- Spyrakis, F.; Celenza, G.; Marcoccia, F.; Santucci, M.; Cross, S.; Bellio, P.; Cendron, L.; Perilli, M.; Tondi, D. Structure-based virtual screening for the discovery of novel inhibitors of New Delhi metallo-β-lactamase-1. ACS Med. Chem. Lett. 2018, 9, 45–50. [Google Scholar] [CrossRef]
- Sevaille, L.; Gavara, L.; Bebrone, C.; De Luca, F.; Nauton, L.; Achard, M.; Mercuri, P.; Tanfoni, S.; Borgianni, L.; Guyon, C.; et al. 1,2,4-Triazole-3-thione Compounds as Inhibitors of Dizinc Metallo-β-lactamases. ChemMedChem 2017, 12, 972–985. [Google Scholar] [CrossRef]
- Vella, P.; Hussein, W.M.; Leung, E.W.W.; Clayton, D.; Ollis, D.L.; Mitić, N.; Schenk, G.; McGeary, R.P. The identification of new metallo-β-lactamase inhibitor leads from fragment-based screening. Bioorg. Med. Chem. Lett. 2011, 21, 3282–3285. [Google Scholar] [CrossRef]
- Christopeit, T.; Carlsen, T.J.O.; Helland, R.; Leiros, H.K.S. Discovery of Novel Inhibitor Scaffolds against the Metallo-β-lactamase VIM-2 by Surface Plasmon Resonance (SPR) Based Fragment Screening. J. Med. Chem. 2015, 58, 8671–8682. [Google Scholar] [CrossRef] [PubMed]
- Kwapien, K.; Damergi, M.; Nader, S.; El Khoury, L.; Hobaika, Z.; Maroun, R.G.; Piquemal, J.-P.; Gavara, L.; Berthomieu, D.; Hernandez, J.-F.; et al. Calibration of 1,2,4-Triazole-3-Thione, an Original Zn-Binding Group of Metallo-β-Lactamase Inhibitors. Validation of a Polarizable MM/MD Potential by Quantum Chemistry. J. Phys. Chem. B 2017, 121, 6295–6312. [Google Scholar] [CrossRef]
- Olsen, L.; Jost, S.; Adolph, H.-W.; Pettersson, I.; Hemmingsen, L.; Jørgensen, F.S. New leads of metallo-β-lactamase inhibitors from structure-based pharmacophore design. Bioorg. Med. Chem. 2006, 14, 2627–2635. [Google Scholar] [CrossRef] [PubMed]
- Song, W.-H.; Liu, M.-M.; Zhong, D.-W.; Zhu, Y.; Bosscher, M.; Zhou, L.; Ye, D.-Y.; Yuan, Z.-H. Tetrazole and triazole as bioisosteres of carboxylic acid: Discovery of diketo tetrazoles and diketo triazoles as anti-HCV agents. Bioorg. Med. Chem. Lett. 2013, 23, 4528–4531. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Linciano, P.; Celenza, G.; Bellio, P.; Papaioannou, S.; Blazquez, J.; Cendron, L.; Brenk, R.; Tondi, D. In silico identification and experimental validation of hits active against KPC-2 β-lactamase. PLoS ONE 2018, 13, 1–22. [Google Scholar] [CrossRef]
- Tondi, D.; Venturelli, A.; Bonnet, R.; Pozzi, C.; Shoichet, B.K.; Costi, M.P. Targeting class A and C serine β-lactamases with a broad-spectrum boronic acid derivative. J. Med. Chem. 2014, 57, 5449–5458. [Google Scholar] [CrossRef]
- Tondi, D.; Cross, S.; Venturelli, A.; Costi, M.P.; Cruciani, G.; Spyrakis, F. Decoding the Structural Basis For Carbapenem Hydrolysis By Class A beta-lactamases: Fishing For A Pharmacophore. Curr. Drug Targets 2016, 17, 983–1005. [Google Scholar] [CrossRef]
- Quotadamo, A.; Linciano, P.; Davoli, P.; Tondi, D.; Costi, M.P.; Venturelli, A. An Improved Synthesis of CENTA, a Chromogenic Substrate for β-Lactamases. Synlett 2016, 27, 2447–2450. [Google Scholar]
- Mroczek, T.; Plech, T.; Wujec, M. Novel Concept of Discrimination of 1,2,4-Triazole-3-thione and 3-Thiol Tautomers. J. Chromatogr. Sci. 2017, 55, 117–129. [Google Scholar] [CrossRef]
- Raper, E.S. Complexes of heterocyclic thionates. Part 1. Complexes of monodentate and chelating ligands. Coord. Chem. Rev. 1996, 153, 199–255. [Google Scholar] [CrossRef]
- Raper, E.S. Copper complexes of heterocyclic thioamides and related ligands. Coord. Chem. Rev. 1994, 129, 91–156. [Google Scholar] [CrossRef]
- Plech, T.; Luszczki, J.J.; Wujec, M.; Flieger, J.; Pizoń, M. Synthesis, characterization and preliminary anticonvulsant evaluation of some 4-alkyl-1,2,4-triazoles. Eur. J. Med. Chem. 2013, 60, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Yung-Chi, C.; Prusoff, W.H. Relationship between the inhibition constant (KI) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar] [CrossRef]
- Tamilselvi, A.; Mugesh, G. Metallo-β-lactamase-Catalyzed Hydrolysis of Cephalosporins: Some Mechanistic Insights into the Effect of Heterocyclic Thiones on Enzyme Activity. Inorg. Chem. 2011, 50, 749–756. [Google Scholar] [CrossRef]
- Nauton, L.; Kahn, R.; Garau, G.; Hernandez, J.F.; Dideberg, O. Structural insights into the design of inhibitors for the L1 metallo-beta-lactamase from Stenotrophomonas maltophilia. J. Mol. Biol. 2008, 375, 257–269. [Google Scholar] [CrossRef]
- Brem, J.; Cain, R.; Cahill, S.; McDonough, M.A.; Clifton, I.J.; Jiménez-Castellanos, J.C.; Avison, M.B.; Spencer, J.; Fishwick, C.W.G.; Schofield, C.J. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat. Commun. 2016, 7, 12406. [Google Scholar] [CrossRef]
- Al-Awadi, N.; Ibrahim, Y.; Kaul, K.; Dib, H. Gas-phase elimination reactions of 4-arylideneimino-1,2,4-triazol-3(2H)-ones and their 3(2H)-thione analogues. J. Phys. Org. Chem. 2001, 14, 521–525. [Google Scholar] [CrossRef]
- Singh, K.; Singh, D.P.; Singh Barwa, M.; Tyagi, P.; Mirza, Y. Some bivalent metal complexes of Schiff bases containing N and S donor atoms. J. Enzyme Inhib. Med. Chem. 2006, 21, 749–755. [Google Scholar] [CrossRef]
- Mashayekhi, V.; Haj Mohammad Ebrahim Tehrani, K.; Amidi, S.; Kobarfard, F. Synthesis of Novel Indole Hydrazone Derivatives and Evaluation of Their Antiplatelet Aggregation Activity. Chem. Pharm. Bull. 2013, 61, 144–150. [Google Scholar] [CrossRef]
- Smičius, R.; Burbuliene, M.M.; Jakubkienė, V.; Udrwėnaitė, E.; Vainilavičius, P. Convenient way to 5-substituted 4-amino-2,3-dihydro-4H-1,2,4-triazole-3-thiones. J. Heterocycl. Chem. 2007, 44, 279–284. [Google Scholar] [CrossRef]
- Jangale, A.D.; Kumavat, P.P.; Wagh, Y.B.; Tayade, Y.A.; Mahulikar, P.P.; Dalal, D.S. Green Process Development for the Synthesis of Aliphatic Symmetrical N,N′-Disubstituted Thiourea Derivatives in Aqueous Medium. Synth. Commun. 2015, 45, 376–385. [Google Scholar] [CrossRef]
- Milletti, F.; Storchi, L.; Sforna, G.; Cruciani, G. New and Original pKa Prediction Method Using Grid Molecular Interaction Fields. J. Chem. Inf. Model. 2007, 47, 2172–2181. [Google Scholar] [CrossRef] [PubMed]
- Klingler, F.M.; Wichelhaus, T.A.; Frank, D.; Cuesta-Bernal, J.; El-Delik, J.; Müller, H.F.; Sjuts, H.; Göttig, S.; Koenigs, A.; Pos, K.M.; et al. Approved drugs containing thiols as inhibitors of metallo-β-lactamases: Strategy to combat multidrug-resistant bacteria. J. Med. Chem. 2015, 58, 3626–3630. [Google Scholar] [CrossRef] [PubMed]
- Büttner, D.; Kramer, J.S.; Klingler, F.M.; Wittmann, S.K.; Hartmann, M.R.; Kurz, C.G.; Kohnhäuser, D.; Weizel, L.; Brüggerhoff, A.; Frank, D.; et al. Challenges in the Development of a Thiol-Based Broad-Spectrum Inhibitor for Metallo-β-Lactamases. ACS Infect. Dis. 2018, 4, 360–372. [Google Scholar] [CrossRef]
- Badarau, A.; Llinas, A.; Laws, A.P.; Damblon, C.; Page, M.I. Inhibitors of metallo-beta-lactamase generated from beta-lactam antibiotics. Biochemistry 2005, 44, 8578–8589. [Google Scholar] [CrossRef]
- Spyrakis, F.; Santucci, M.; Maso, L.; Cross, S.; Gianquinto, E.; Sannio, F.; Verdirosa, F.; De Luca, F.; Docquier, J.-D.; Cendron, L.; et al. Development of β-lactamase broad-spectrum inhibitors: Virtual screening and in vitro validation to address antimicrobial resistance. Sci. Rep. under review.
- Salimraj, R.; Hinchliffe, P.; Kosmopoulou, M.; Tyrrell, J.M.; Brem, J.; van Berkel, S.S.; Verma, A.; Owens, R.J.; McDonough, M.A.; Walsh, T.R.; et al. Crystal structures of VIM-1 complexes explain active site heterogeneity in VIM-class metallo-β-lactamases. FEBS J. 2019, 286, 169–183. [Google Scholar] [CrossRef]
- Toney, J.H.; Hammond, G.G.; Fitzgerald, P.M.D.; Sharma, N.; Balkovec, J.M.; Rouen, G.P.; Olson, S.H.; Hammond, M.L.; Greenlee, M.L.; Gao, Y.D. Succinic Acids as Potent Inhibitors of Plasmid-borne IMP-1 Metallo-β-lactamase. J. Biol. Chem. 2001, 276, 31913–31918. [Google Scholar] [CrossRef]
- Decherchi, S.; Bottegoni, G.; Spitaleri, A.; Rocchia, W.; Cavalli, A. BiKi Life Sciences: A New Suite for Molecular Dynamics and Related Methods in Drug Discovery. J. Chem. Inf. Model. 2018, 58, 219–224. [Google Scholar] [CrossRef]
- Celenza, G.; Vicario, M.; Bellio, P.; Linciano, P.; Perilli, M.; Oliver, A.; Blazquez, J.; Cendron, L.; Tondi, D. Phenylboronic Acid Derivatives as Validated Leads Active in Clinical Strains Overexpressing KPC-2: A Step against Bacterial Resistance. ChemMedChem 2018. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.Y.; Shoichet, B.K. A detergent-based assay for the detection of promiscuous inhibitors. Nat. Protoc. 2006, 1, 550–553. [Google Scholar] [CrossRef] [PubMed]
- Weinsten, M.P.; Patel, J.B.; Bobenchik, A.M.; Campeau, S.; Culle, S.K.; Galas, M.F.; Gold, H.; Humphries, R.M.; Kirn, T.J., Jr.; Lewis, K.S.; et al. Performance Standards for Antimicrobial Susceptibility Testing, 29th ed.; Clinical and Laboratory Standards Institute: Irving, TX, USA, 2019; ISBN 9781684400324. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
| Code | Structure | VIM-1 Ki (µM) a | IMP-1 Ki (µM) a | KPC-2 Ki (µM) a |
|---|---|---|---|---|
| 1d |  | 224 ± 11 | 173 ± 6 | 263 ± 13 |
| 1f |  | 120 ± 4 | 125 ± 5 | 138 ± 3 |
| 2b |  | 58 ± 2 | 285 ± 13 | 158 ± 7 |
| 2g |  | 95 ± 3 | 104 ± 4 | 139 ± 4 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Linciano, P.; Gianquinto, E.; Montanari, M.; Maso, L.; Bellio, P.; Cebrián-Sastre, E.; Celenza, G.; Blázquez, J.; Cendron, L.; Spyrakis, F.; et al. 4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases. Pharmaceuticals 2020, 13, 52. https://doi.org/10.3390/ph13030052
Linciano P, Gianquinto E, Montanari M, Maso L, Bellio P, Cebrián-Sastre E, Celenza G, Blázquez J, Cendron L, Spyrakis F, et al. 4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases. Pharmaceuticals. 2020; 13(3):52. https://doi.org/10.3390/ph13030052
Chicago/Turabian StyleLinciano, Pasquale, Eleonora Gianquinto, Martina Montanari, Lorenzo Maso, Pierangelo Bellio, Esmeralda Cebrián-Sastre, Giuseppe Celenza, Jesús Blázquez, Laura Cendron, Francesca Spyrakis, and et al. 2020. "4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases" Pharmaceuticals 13, no. 3: 52. https://doi.org/10.3390/ph13030052
APA StyleLinciano, P., Gianquinto, E., Montanari, M., Maso, L., Bellio, P., Cebrián-Sastre, E., Celenza, G., Blázquez, J., Cendron, L., Spyrakis, F., & Tondi, D. (2020). 4-Amino-1,2,4-triazole-3-thione as a Promising Scaffold for the Inhibition of Serine and Metallo-β-Lactamases. Pharmaceuticals, 13(3), 52. https://doi.org/10.3390/ph13030052