Determination of Antidepressants in Human Plasma by Modified Cloud-Point Extraction Coupled with Mass Spectrometry


Abstract
1. Introduction
2. Results and Discussion
2.1. Development of Cloud-Point Extraction
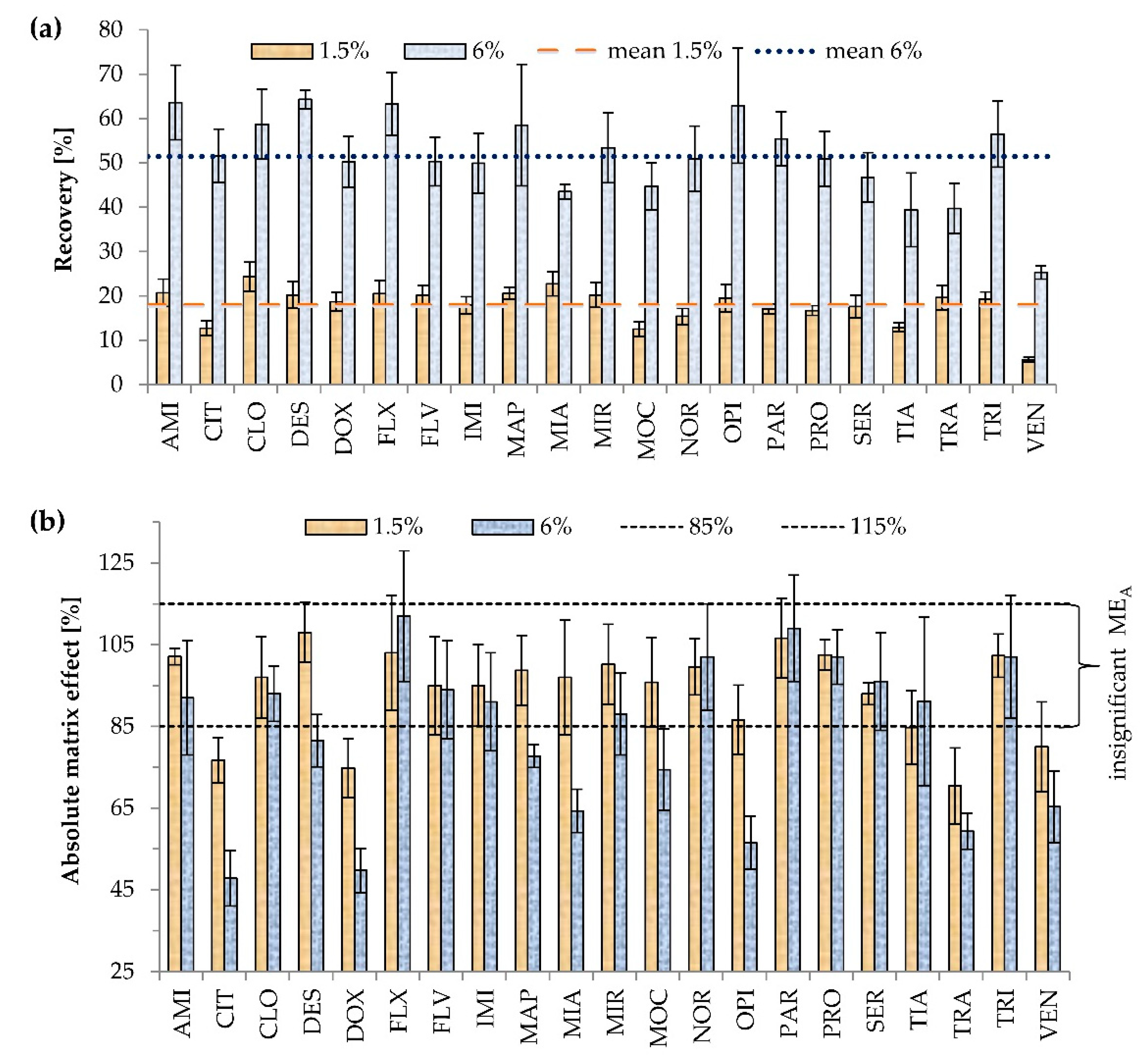
2.1.1. The Effects of Triton X-114
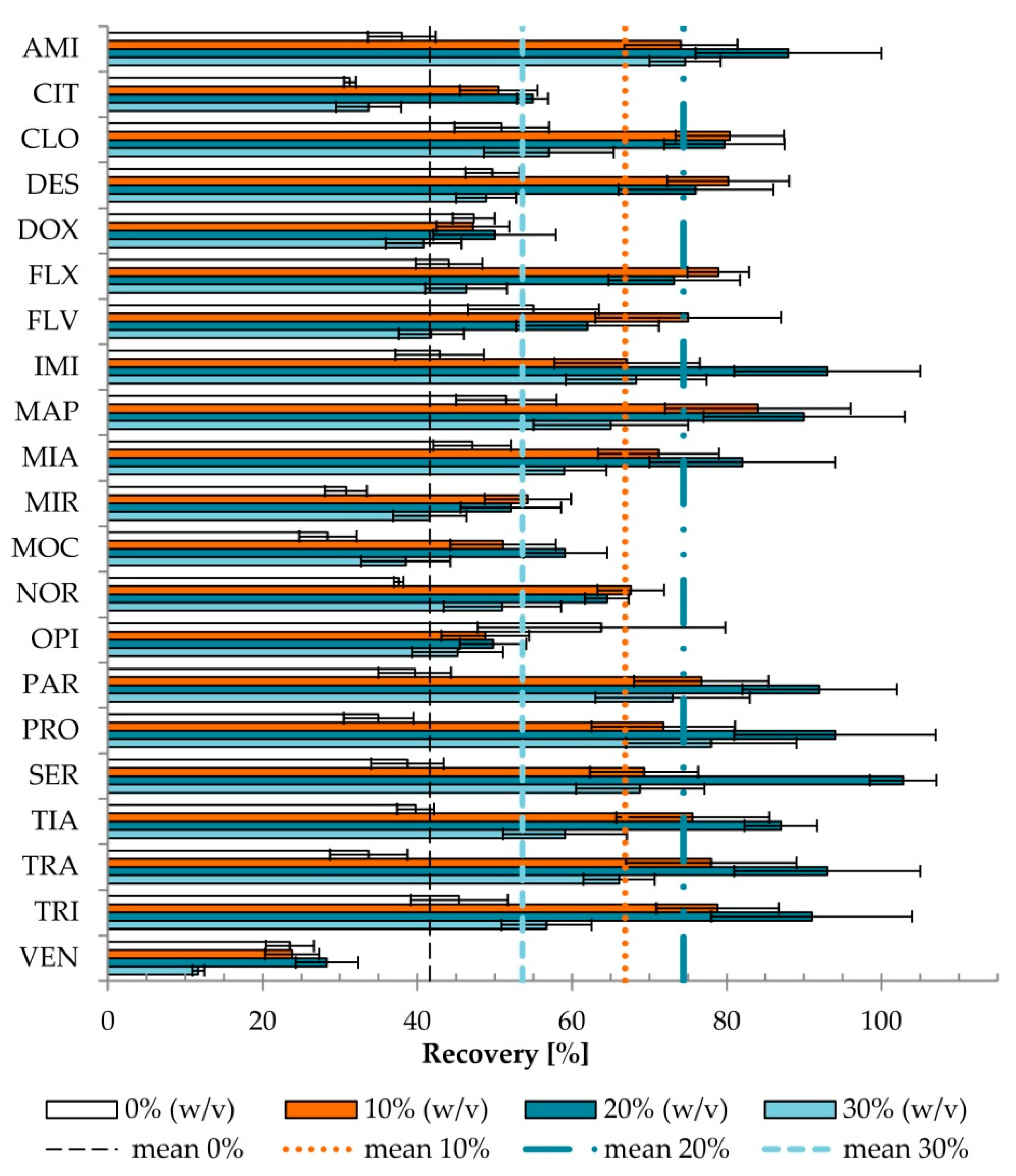
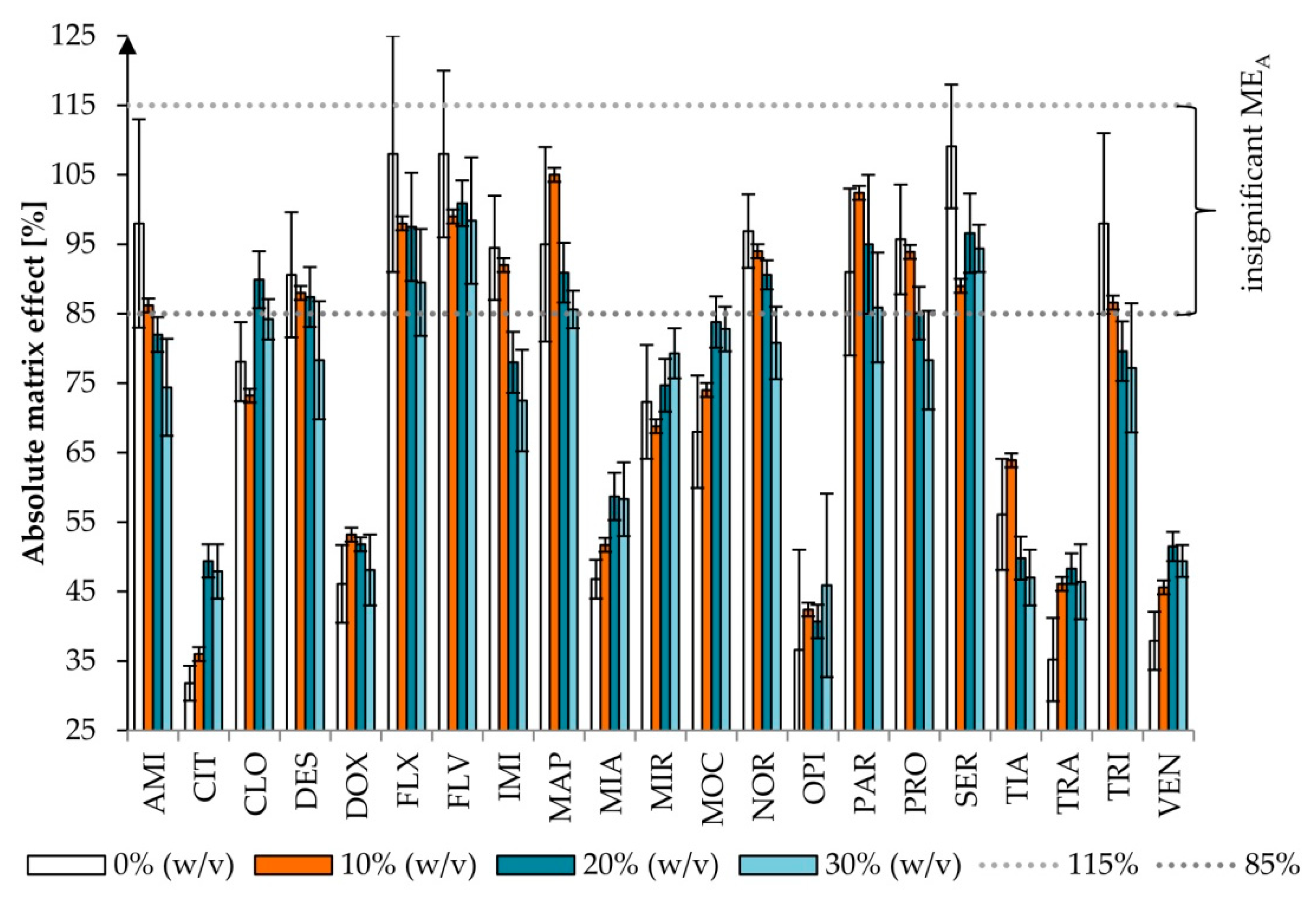
2.1.2. The Effects of Salt Addition
2.1.3. The Effects of pH
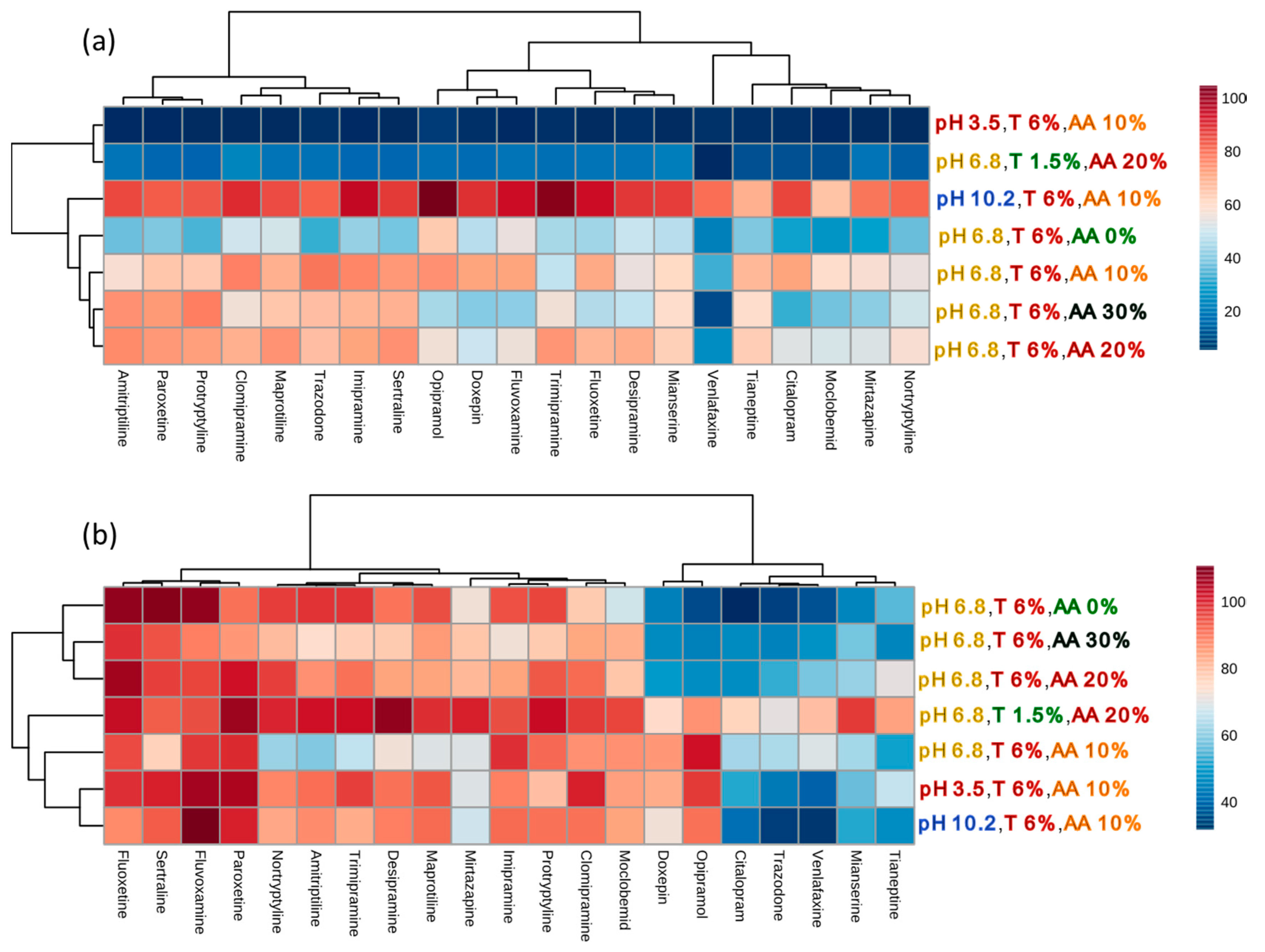
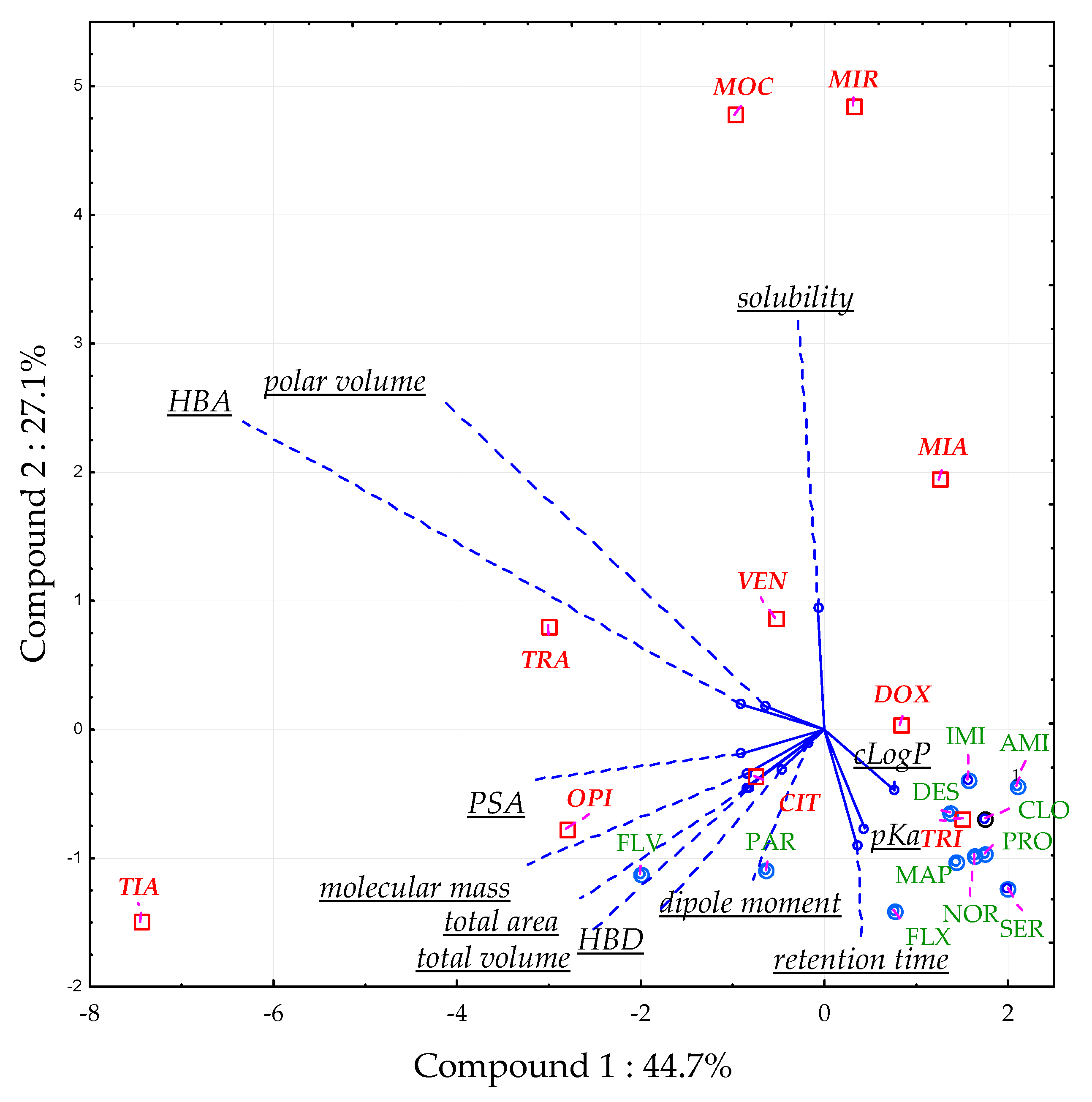
2.1.4. Principal Component Analysis (PCA)
2.2. Analytical Performance of the Proposed Method
2.2.1. Linearity
2.2.2. Limit of Quantification, Precision, and Accuracy
2.2.3. Stability
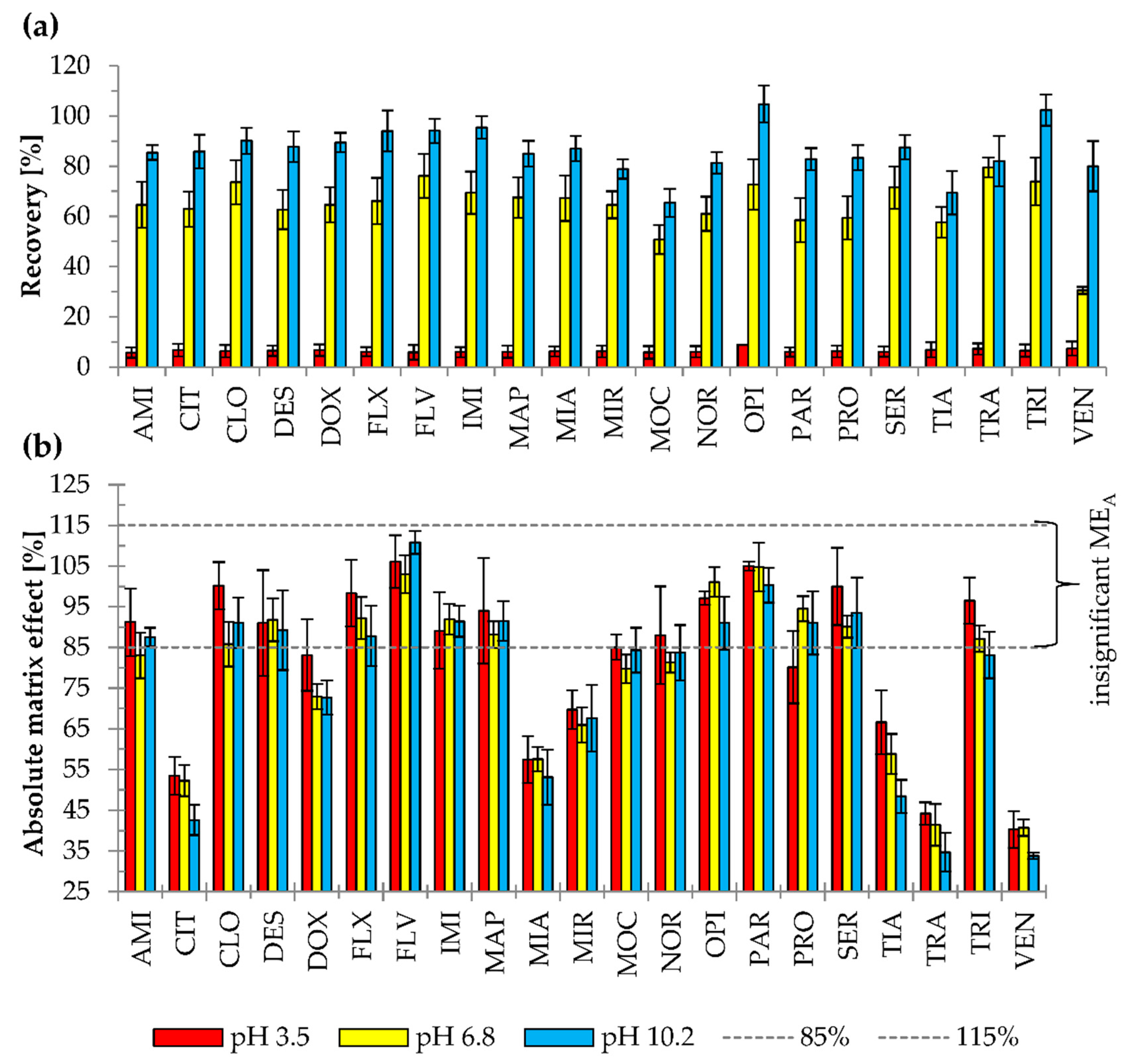
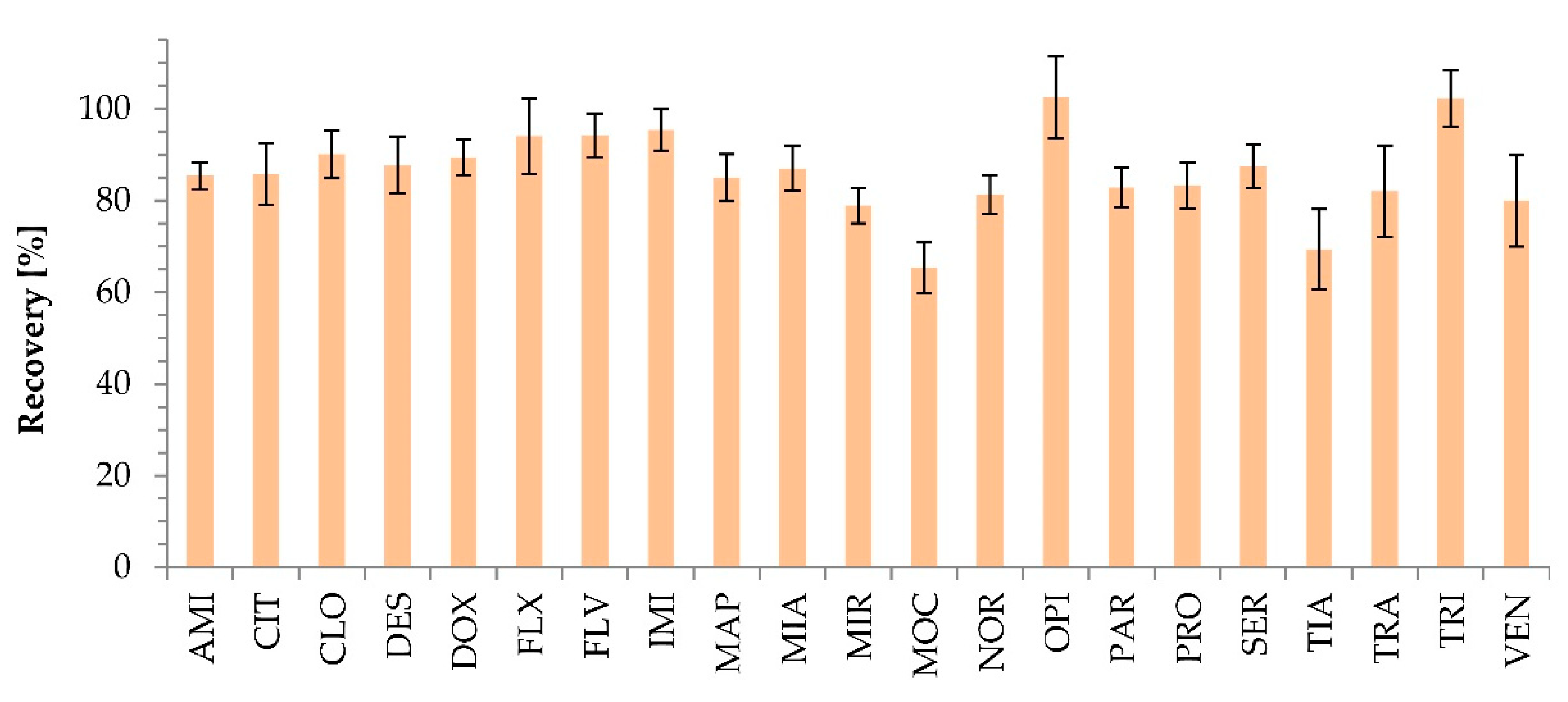
2.2.4. Matrix Effect and Recovery
3. Materials and Methods
3.1. Materials
3.2. Reference Standards and Chemicals
3.3. Chromatographic and Mass Spectrometric Conditions
3.4. Preparation of Solutions
3.5. Sample Preparation
3.5.1. CPE Procedure—Development
3.5.2. CPE Procedure—Validation
3.5.3. Preparation of Calibration Standards and Quality Samples for Validation Optimal CPE Conditions
3.6. Matrix Effect and Recovery Calculation
3.7. Statistical Analysis
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Parent ion [m/z] [M+H+] | Daughter ion [m/z] | Metabolite | Parent ion [m/z] [M+H+] | Daughter ion [m/z] | Source | Biofluid |
|---|---|---|---|---|---|---|---|
| Amitriptyline | 278 | 233 | Nortriptyline | 264 | 90 | [25] | human plasma |
| Citalopram | 325 | 109 | Desmethylcitalopram | 311 | 109 | [45,46] | human plasma whole blood |
| 262 | |||||||
| Citalopram N-oxide | 341 | 262 | human plasma whole blood | ||||
| 109 | |||||||
| Clomipramine | 315 | 86 | Norclomipramine | 301 | 72 | [47] | human plasma |
| Demetylclomipramine | 302 | 243 271 | [48] | whole blood | |||
| Hydroxydesipramine | 282 | 252 | |||||
| Doxepin | 280 | 107 | Nordoxepin | 266 | 107 | [49] | human plasma |
| Fluoxetine | 310 | 44 | Norfluoxetine | 296 | 134 | [47] | human plasma |
| Imipramine | 281 | 86 | Desipramine | 267 | 72 | This study | human plasma |
| Maprotiline | 278 | 250 | N-desmethylmaprotiline | 264 | 117 | [46] | human whole blood |
| 169 | |||||||
| Mirtazapine | 266 | 195 | Desmethyl mirtazapine | 252 | 195 | [50] | human bile |
| 8-Hydroxydesmethylmirtazapine | 282 | 211 | |||||
| Nortriptyline | 264 | 233 | 10- hydroxynortriptyline | 280.1 | 262.2 | [51] | human plasma |
| Paroxetine | 330 | 192 | 4-hydroxy-3-methoxy | 332 | 192 | [52] | human plasma |
| Sertraline | 306 | 158 | N-Desmethyl sertraline | 292.32 | 275.1 | [46] | human whole blood |
| Tianeptine | 437 | 292 | MC5 | 409 | 292 | [53] | rat plasma |
| 228 | |||||||
| Trazodone | 372 | 176 | m-Chlorophenylpiperazine | 197 | 118 | [49] | human plasma |
| Trimipramine | 295 | 100 | Desmethyltrimipramine | 281 | 86 | [54] | human hair |
| 2-Hydroxytrimipramine | 311 | 100 | |||||
| 2-Hydroxydesmethyltrimipramine | 297 | 86 | |||||
| Venlafaxine | 278 | 58 | O-Desmethyl venlafaxine | 264 | 58 | [46] | whole blood |
| 246 |
| Compound | R2 | Linear Equation y = ax + b | Standard Deviation a | Standard Deviation b |
|---|---|---|---|---|
| Amitriptyline | 0.998 | y = 0.001731x + 0.0100 | 0.000062 | 0.0016 |
| Citalopram | 0.993 | y = 0.001895x + 0.0016 | 0.000052 | 0.0011 |
| Clomipramine | 0.997 | y= 0.000728x + 0.002747 | 0.000017 | 0.000940 |
| Desipramine | 0.996 | y = 0.002608 ± 0.0008 | 0.000051 | 0.0015 |
| Doxepin | 0.994 | y= 0.001895x + 0.0119 | 0.000064 | 0.0075 |
| Fluoxetine | 0.997 | y = 0.002061x+ 0.0030 | 0.000075 | 0.0030 |
| Fluvoxamine | 0.994 | y = 0.000679x + 0.0079 | 0.000030 | 0.0032 |
| Imipramine | 0.997 | y = 0.00395x + 0.0054 | 0.00012 | 0.0057 |
| Maprotiline | 0.995 | y = 0.002716x + 0.0159 | 0.000050 | 0.0031 |
| Mianserin | 0.996 | y = 0.0002657x + (-0.000142) | 0.0000077 | 0.000265 |
| Mirtazapine | 0.997 | y = 0.003092x + (-0.0013) | 0.000081 | 0.0050 |
| Moclobemide | 0.990 | y = 0.0026x + (-0.0014) | 0.0011 | 0.0019 |
| Nortriptyline | 0.996 | y = 0.001011x + 0.000020 | 0.000027 | 0.000936 |
| Opipramol | 0.997 | y = 0.0260x + (-0.010) | 0.0044 | 0.017 |
| Paroxetine | 0.996 | y = 0.0001912x + 0.0050 | 0.0000063 | 0.0027 |
| Protriptyline | 0.997 | y = 0.002013x + 0.0050 | 0.000048 | 0.0040 |
| Sertraline | 0.998 | y = 0.000591x + 0.0035 | 0.000012 | 0.0015 |
| Tianeptine | 0.995 | y = 0.002512x + 0.0026 | 0.000084 | 0.0032 |
| Trazodone | 0.996 | y = 0.002249x + (-0.0026) | 0.000095 | 0.0025 |
| Trimipramine | 0.998 | y = 0.00573x + 0.0033 | 0.00023 | 0.0046 |
| Venlafaxine | 0.999 | y = 0.002268x + 0.0014 | 0.000086 | 0.0032 |
| Test Name | Autosampler Stability | Short-Term Stability | Freeze and Thaw Stability | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| (48h at 4°C) | (3h at Room Temperature) | (−20°C) | ||||||||||
| Drug Name | 16 | CV (%) | 625 ng/mL | CV (%) | 16 | CV (%) | 625 ng/mL | CV (%) | 16 | CV (%) | 625 ng/mL | CV (%) |
| ng/mL | ng/mL | ng/mL | ||||||||||
| Amitriptyline | 112.2 | 13 | 98.1 | 10 | 96 | 9 | 91.9 | 4 | 103.2 | 8 | 88.4 | 13 |
| Citalopram | 92.9 | 14 | 91.7 | 12 | 96.9 | 6 | 102.6 | 4 | 89.5 | 2 | 96.3 | 8 |
| Clomipramine | 102.8 | 1 | 96.3 | 4 | 101.2 | 4 | 98.3 | 5 | 102 | 9 | 97.2 | 4 |
| Desipramine | 108.7 | 6 | 99.1 | 13 | 92.7 | 6 | 93.1 | 3 | 101.5 | 5 | 92.3 | 5 |
| Doxepin | 110.3 | 9 | 90.6 | 11 | 94.1 | 3 | 91.1 | 2 | 91.3 | 7 | 86.5 | 12 |
| Fluoxetine | 104.9 | 14 | 98.8 | 9 | 97.7 | 5 | 94.2 | 1 | 110 | 7 | 90.9 | 8 |
| Fluvoxamine | 106.7 | 9 | 102.5 | 5 | 98.3 | 11 | 100 | 7 | 91.5 | 13 | 98.9 | 5 |
| Imipramine | 110.6 | 12 | 98.4 | 8 | 97.1 | 13 | 92.4 | 3 | 95.9 | 3 | 91.9 | 6 |
| Maprotiline | 104 | 9 | 106.2 | 6 | 113.3 | 2 | 98.4 | 7 | 110.5 | 3 | 96.4 | 7 |
| Mianserin | 106.9 | 13 | 91.1 | 6 | 95.2 | 3 | 99.4 | 4 | 91.5 | 2 | 91.3 | 6 |
| Mirtazapine | 107.2 | 10 | 92.4 | 9 | 92.7 | 11 | 94.6 | 6 | 99.7 | 13 | 88.3 | 12 |
| Moclobemide | 108 | 9 | 90.8 | 10 | 99.3 | 8 | 98.1 | 7 | 99.6 | 9 | 91.1 | 4 |
| Nortriptyline | 112.6 | 6 | 105.5 | 9 | 110.5 | 10 | 100.3 | 5 | 113.3 | 6 | 97.6 | 8 |
| Opipramol | 101.5 | 14 | 91.7 | 10 | 102.2 | 3 | 91.5 | 4 | 99.5 | 5 | 89.1 | 8 |
| Paroxetine | 109.3 | 6 | 94.2 | 6 | 108.5 | 8 | 101.6 | 9 | 107.1 | 9 | 91.3 | 13 |
| Protriptyline | 111.4 | 4 | 101.1 | 9 | 102.1 | 5 | 91.9 | 4 | 103.2 | 3 | 88 | 7 |
| Sertraline | 99.6 | 11 | 99.4 | 12 | 96.9 | 6 | 100.3 | 6 | 109.2 | 9 | 99.2 | 3 |
| Tianeptine | 96 | 10 | 90.4 | 12 | 93.5 | 7 | 102.7 | 6 | 93 | 6 | 100.8 | 9 |
| Trazodone | 97.2 | 5 | 96.6 | 12 | 99.7 | 7 | 104.7 | 5 | 92.1 | 8 | 100.3 | 8 |
| Trimipramine | 110 | 12 | 95.9 | 6 | 89.6 | 4 | 102.6 | 2 | 101.7 | 11 | 90.3 | 4 |
| Venlafaxine | 101.2 | 8 | 100.4 | 7 | 95.8 | 7 | 104.7 | 9 | 97.9 | 12 | 99.8 | 8 |
| QC Samples | Low QC (16 ng/mL) | High QC (625 ng/mL) | ||||
|---|---|---|---|---|---|---|
| Drug Name | MF Analyte | IS-Normalized MF | CV IS-Norm. MF [%] | MF Analyte | Is-Normalized MF | CV IS-Norm. MF [%] |
| Amitriptyline | 98.7 | 92.5 | 10.0 | 94.8 | 94.6 | 6.1 |
| Citalopram | 58.2 | 123.3 | 14.6 | 54.2 | 129.8 | 12.1 |
| Clomipramine | 108.3 | 106.2 | 9.3 | 95.1 | 99.1 | 2.7 |
| Desipramine | 95.6 | 90.2 | 14.6 | 95.4 | 95.5 | 8.9 |
| Doxepin | 79.2 | 73.3 | 13.7 | 73.9 | 73.7 | 6.5 |
| Fluoxetine | 106.3 | 99.1 | 9.3 | 96.6 | 96.5 | 10.0 |
| Fluvoxamine | 108.8 | 101.4 | 7.9 | 107.8 | 107.8 | 7.7 |
| Imipramine | 100.1 | 93.0 | 6.7 | 94.0 | 93.8 | 4.4 |
| Maprotiline | 102.1 | 95.7 | 9.4 | 97.7 | 97.5 | 2.6 |
| Mianserin | 64.8 | 63.1 | 15.0 | 58.7 | 61.0 | 8.9 |
| Mirtazapine | 72.6 | 68.1 | 14.8 | 70.4 | 70.4 | 3.3 |
| Moclobemide | 88.9 | 87.1 | 6.8 | 89.0 | 93.2 | 5.3 |
| Nortriptyline | 94.2 | 92.2 | 5.7 | 92.6 | 96.8 | 3.6 |
| Opipramol | 107.9 | 101.4 | 10.9 | 102.4 | 102.4 | 7.7 |
| Paroxetine | 115.6 | 111.8 | 6.7 | 107.1 | 107.1 | 7.6 |
| Protriptyline | 104.3 | 97.8 | 9.2 | 97.7 | 97.6 | 5.1 |
| Sertraline | 109.1 | 106.9 | 7.2 | 94.4 | 98.5 | 2.8 |
| Tianeptine | 73.8 | 142.1 | 6.3 | 64.3 | 153.9 | 13.9 |
| Trazodone | 53.2 | 108.0 | 12.4 | 48.0 | 108.1 | 11.9 |
| Trimipramine | 101.7 | 95.0 | 7.7 | 88.7 | 88.5 | 6.8 |
| Venlafaxine | 47.5 | 96.5 | 15.0 | 44.5 | 105.5 | 11.6 |
References
- Giebułtowicz, J.; Kojro, G.; Piotrowski, R.; Kułakowski, P.; Wroczyński, P. Cloud-point extraction is compatible with liquid chromatography coupled to electrospray ionization mass spectrometry for the determination of antazoline in human plasma. J. Pharm. Biomed. 2016, 128. [Google Scholar] [CrossRef]
- Madej, K.; Persona, K. Drug screening in human plasma by cloud-point extraction and HPLC. J. Open Chem. 2013, 11, 94. [Google Scholar] [CrossRef]
- Wentao, L.; Bi, K.; Liu, X.; Zhao, J.; Chen, X. Cloud-Point Extraction Combined with LC–MS for Analysis of Memantine in Rat Plasma. Chromatographia 2009, 69, 837–842. [Google Scholar] [CrossRef]
- Giebułtowicz, J.; Kojro, G.; Buś - Kwaśnik, K.; Rudzki, P.; Marszałek, R.; Leś, A.; Wroczyński, P. Cloud-point extraction is compatible with liquid chromatography coupled to electrospray ionization mass spectrometry for the determination of bisoprolol in human plasma. J. Chromatogr. A 2015, 1423. [Google Scholar] [CrossRef] [PubMed]
- Hunzicker, G.A.; Hein, G.J.; Hernández, S.R.; Altamirano, J.C. Cloud point extraction for analysis of antiretrovirals in human plasma by UFLC-ESI-MS/MS. Anal. Chem. Res. 2015, 6, 1–8. [Google Scholar] [CrossRef][Green Version]
- Rukhadze, M.D.; Tsagareli, S.K.; Sidamonidze, N.S.; Meyer, V.R. Cloud-point extraction for the determination of the free fraction of antiepileptic drugs in blood plasma and saliva. Anal. Biochem. 2000, 287, 279–283. [Google Scholar] [CrossRef]
- Tabrizi, A.B.; Naini, S.; Parnian, K.; Mohammadi, S.; Emami zad, F.; Anvarian, S.P.; Abdollahi, A. Determination of triamterene in human plasma and urine after its cloud point extraction. Quím. Nova 2014, 37, 1182–1187. [Google Scholar] [CrossRef]
- Qin, X.Y.; Meng, J.; Li, X.Y.; Zhou, J.; Sun, X.L.; Wen, A.D. Determination of venlafaxine in human plasma by high-performance liquid chromatography using cloud-point extraction and spectrofluorimetric detection. J. Chromatogr. B. 2008, 872, 38–42. [Google Scholar] [CrossRef]
- Zhao, Q.; Ding, J.; Jin, H.; Ding, L.; Ren, N.-Q. A Green Method Using a Micellar System for Determination of Andrographolide and Dehydroandrographolide in Human Plasma. J. Chromatogr. Sci. 2012, 51. [Google Scholar] [CrossRef]
- Filik, H.; Şener, İ.; Cekiç, S.; Kilic, E.; Apak, R. Spectrophotometric determination of paracetamol in urine with tetrahydroxycalix[4]arene as a coupling reagent and preconcentration with Triton X-114 using cloud point extraction. Chem. Pharm. Bull. 2006, 54, 891–896. [Google Scholar] [CrossRef]
- Wang, C.C.; Luconi, M.O.; Masi, A.N.; Fernández, L. Determination of terazosin by cloud point extraction-fluorimetric combined methodology. Talanta 2007, 72, 1779–1785. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Wang, J.; Yang, Y.; Lu, Y. The Determination of Nitrate and Nitrite in Human Urine and Blood by High-Performance Liquid Chromatography and Cloud-Point Extraction. J. Chromatogr. Sci. 2015, 53, 1169–1177. [Google Scholar] [CrossRef] [PubMed]
- World Health Organization. Depression. Available online: https://www.who.int/news-room/fact-sheets/detail/depression (accessed on 8 August 2020).
- Methling, M.; Krumbiegel, F.; Hartwig, S.; Parr, M.K.; Tsokos, M. Toxicological findings in suicides — frequency of antidepressant and antipsychotic substances. Forensic Sci. Med. Pathol. 2019, 15, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Pompili, M.; Serafini, G.; Innamorati, M.; Ambrosi, E.; Giordano, G.; Girardi, P.; Tatarelli, R.; Lester, D. Antidepressants and Suicide Risk: A Comprehensive Overview. Pharmaceuticals 2010, 3, 2861–2883. [Google Scholar] [CrossRef] [PubMed]
- Hawton, K.; Bergen, H.; Simkin, S.; Cooper, J.; Waters, K.; Gunnell, D.; Kapur, N. Toxicity of antidepressants: Rates of suicide relative to prescribing and non-fatal overdose. Br. J. Psychiatry 2010, 196, 354–358. [Google Scholar] [CrossRef] [PubMed]
- Jia, E.; Bartlett, M. Recent Advances in Liquid Chromatographic Methods for the Determination of Selective Serotonin Reuptake Inhibitors (SSRI) and Serotonin Norepinephrine Reuptake Inhibitors (SNRI). Biomed. Chromatogr. 2019, 34. [Google Scholar] [CrossRef]
- Vaghar-Lahijani, G.; Aberoomand-Azar, P.; Saber-Tehrani, M.; Soleimani, M. Application of ionic liquid-based ultrasonic-assisted microextraction coupled with HPLC for determination of citalopram and nortriptyline in human plasma. J. Liq. Chromatogr. Relat. 2017, 40, 1–7. [Google Scholar] [CrossRef]
- Duverneuil, C.; de la Grandmaison, G.L.; de Mazancourt, P.; Alvarez, J.-C. A high-performance liquid chromatography method with photodiode-array UV detection for therapeutic drug monitoring of the nontricyclic antidepressant drugs. Ther. Drug Monit. 2003, 25, 565–573. [Google Scholar] [CrossRef]
- Del Mar Ramírez Fernández, M.; Wille Sm Fau-Samyn, N.; Samyn, N. Quantitative method validation for the analysis of 27 antidepressants and metabolites in plasma with ultraperformance liquid chromatography-tandem mass spectrometry. Ther. Drug Monit. 2012, 34, 11–24. [Google Scholar] [CrossRef]
- De Castro, A.; Fernandez, M.d.M.R.; Laloup, M.; Samyn, N.; De Boeck, G.; Wood, M.; Maes, V.; López-Rivadulla, M. High-throughput on-line solid-phase extraction–liquid chromatography–tandem mass spectrometry method for the simultaneous analysis of 14 antidepressants and their metabolites in plasma. J. Chromatogr. A 2007, 1160, 3–12. [Google Scholar] [CrossRef]
- Ansermot, N.; Brawand-Amey, M.; Eap, C.B. Simultaneous quantification of selective serotonin reuptake inhibitors and metabolites in human plasma by liquid chromatography—Electrospray mass spectrometry for therapeutic drug monitoring. J. Chromatogr. B 2012, 885–886, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Petrides, A.K.; Moskowitz, J.; Johnson-Davis, K.L.; Jannetto, P.J.; Langman, L.J.; Clarke, W.; Marzinke, M.A. The development and validation of a turbulent flow-liquid chromatography-tandem mass spectrometric method for the simultaneous quantification of citalopram, sertraline, bupropion and hydroxybupropion in serum. Clin. Biochem. 2014, 47, 73–79. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.G.; Tavares, I.M.; Barbosa, A.F.; Bettini, J.; Figueiredo, E.C. Analysis of tricyclic antidepressants in human plasma using online-restricted access molecularly imprinted solid phase extraction followed by direct mass spectrometry identification/quantification. Talanta 2017, 163, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Berm, E.J.J.; Paardekooper, J.; Brummel-Mulder, E.; Hak, E.; Wilffert, B.; Maring, J.G. A simple dried blood spot method for therapeutic drug monitoring of the tricyclic antidepressants amitriptyline, nortriptyline, imipramine, clomipramine, and their active metabolites using LC-MS/MS. Talanta 2015, 134, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Nielsen, M.K.; Johansen, S.S. Simultaneous determination of 25 common pharmaceuticals in whole blood using ultra-performance liquid chromatography-tandem mass spectrometry. J. Anal. Toxicol. 2012, 36, 497–506. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Santos-Neto, A.J.; Bergquist, J.; Lanças, F.M.; Sjöberg, P.J.R. Simultaneous analysis of five antidepressant drugs using direct injection of biofluids in a capillary restricted-access media-liquid chromatography-tandem mass spectrometry system. J. Chromatogr. A 2008, 1189, 514–522. [Google Scholar] [CrossRef] [PubMed]
- De Souza, I.D.; Domingues, D.S.; Queiroz, M.E.C. Hybrid silica monolith for microextraction by packed sorbent to determine drugs from plasma samples by liquid chromatography–tandem mass spectrometry. Talanta 2015, 140, 166–175. [Google Scholar] [CrossRef]
- Remane, D.; Meyer, M.R.; Wissenbach, D.K.; Maurer, H.H. Full validation and application of an ultra high performance liquid chromatographic-tandem mass spectrometric procedure for target screening and quantification of 34 antidepressants in human blood plasma as part of a comprehensive multi-analyte approach. Anal. Bioanal. Chem. 2011, 400, 2093. [Google Scholar] [CrossRef]
- Roemmelt, A.T.; Steuer, A.E.; Kraemer, T. Liquid Chromatography, In Combination with a Quadrupole Time-of-Flight Instrument, with Sequential Window Acquisition of All Theoretical Fragment-Ion Spectra Acquisition: Validated Quantification of 39 Antidepressants in Whole Blood As Part of a Simultaneous Screening and Quantification Procedure. Anal. Chem. 2015, 87, 9294–9301. [Google Scholar] [CrossRef]
- Kojro, G.; Rudzki, P.J.; Pisklak, D.M.; Giebułtowicz, J. Matrix effect screening for cloud-point extraction combined with liquid chromatography coupled to mass spectrometry: Bioanalysis of pharmaceuticals. J. Chromatogr. A 2019, 1591, 44–54. [Google Scholar] [CrossRef]
- Kojro, G.; Wroczyński, P. Cloud Point Extraction in the Determination of Drugs in Biological Matrices. J. Chromatogr. Sci. 2020, 58, 151–162. [Google Scholar] [CrossRef] [PubMed]
- Matuszewski, B.K.; Constanzer, M.L.; Chavez-Eng, C.M. Strategies for the Assessment of Matrix Effect in Quantitative Bioanalytical Methods Based on HPLC−MS/MS. Anal. Chem. 2003, 75, 3019–3030. [Google Scholar] [CrossRef] [PubMed]
- More, P.R.; Arya, S.S. A novel, green cloud point extraction and separation of phenols and flavonoids from pomegranate peel: An optimization study using RCCD. J. Environ. Chem. Eng. 2019, 7, 103306. [Google Scholar] [CrossRef]
- Koshy, L.; Saiyad, A.H.; Rakshit, A.K. The effects of various foreign substances on the cloud point of Triton X 100 and Triton X 114. Colloid Polym. Sci. 1996, 274, 582–587. [Google Scholar] [CrossRef]
- Quina, F.H.; Hinze, W.L. Surfactant-Mediated Cloud Point Extractions: An Environmentally Benign Alternative Separation Approach. Ind. Eng. Chem. Res 1999, 38, 4150–4168. [Google Scholar] [CrossRef]
- Madej, K. Microwave-assisted and cloud-point extraction in determination of drugs and other bioactive compounds. Trends Anal. Chem. 2009, 28, 436–446. [Google Scholar] [CrossRef]
- Sayem Alam, M.; Mandal, A.B. The clouding phenomena of mixed surfactant (non-ionic Triton X-114+cationic gemini 16-5-16) solutions: Influence of inorganic and organic additives on the cloud point. J. Mol. Liq. 2015, 212, 237–244. [Google Scholar] [CrossRef]
- Pourreza, N.; Elhami, S. Spectrophtometric determination of malachite green in fish farming water samples after cloud point extraction using nonionic surfactant Triton X-100. Anal. Chim. Acta 2007, 596, 62–65. [Google Scholar] [CrossRef]
- Kori, S. Cloud point extraction coupled with back extraction: A green methodology in analytical chemistry. Forensic Sci. Res. 2019, 1–15. [Google Scholar] [CrossRef]
- Zhang, H.; Choi, H.-K. Analysis of meloxicam by high-performance liquid chromatography with cloud-point extraction. Anal. Bioanal. Chem. 2008, 392, 947–953. [Google Scholar] [CrossRef]
- Han, F.; Yin, R.; Shi, X.-l.; Jia, Q.; Liu, H.-z.; Yao, H.-m.; Xu, L.; Li, S.-m. Cloud point extraction-HPLC method for determination and pharmacokinetic study of flurbiprofen in rat plasma after oral and transdermal administration. J. Chromatogr. B 2008, 868, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Guideline on bioanalytical method validation. Committee for Medicinal Products for Human Use (CHMP), EMEA/CHMP/EWP/192217/2009, EuropeanMedicines Agency. 2011. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-bioanalytical-method-validation_en.pdf (accessed on 5 October 2020).
- Guidance for Industry: Bioanalytical Method Validation. Food and Drug Administration. Center for Drug Evaluation and Research and Center for Veterinary Medicine. 2001. Available online: http://academy.gmp-compliance.org/guidemgr/files/4252FNL.PDF (accessed on 5 October 2020).
- Jiang, T.; Rong, Z.; Peng, L.; Chen, B.; Xie, Y.; Chen, C.; Sun, J.; Xu, Y.; Lu, Y.; Chen, H. Simultaneous determination of citalopram and its metabolite in human plasma by LC–MS/MS applied to pharmacokinetic study. J. Chromatogr. B 2010, 878, 615–619. [Google Scholar] [CrossRef] [PubMed]
- Zhu, R.; Dong, X.; Zhang, D.; Liu, X.; Ye, Y.; Jiang, Y. Simultaneous Quantification of 38 Psychotropic Drugs and Relevant Metabolites in Blood using LC–MS-MS. J. Anal. Toxicol. 2020. [Google Scholar] [CrossRef] [PubMed]
- De Castro, A.; Concheiro, M.; Quintela, O.; Cruz, A.; López-Rivadulla, M. LC–MS/MS method for the determination of nine antidepressants and some of their main metabolites in oral fluid and plasma: Study of correlation between venlafaxine concentrations in both matrices. J. Pharm. Biomed. 2008, 48, 183–193. [Google Scholar] [CrossRef]
- Titier, K.; Castaing, N.; Le-Déodic, M.; Le-bars, D.; Moore, N.; Molimard, M. Quantification of Tricyclic Antidepressants and Monoamine Oxidase Inhibitors by High-Performance Liquid Chromatography-Tandem Mass Spectrometry in Whole Blood. J. Anal. Toxicol. 2007, 31, 200–207. [Google Scholar] [CrossRef][Green Version]
- Patel, B.N.; Sharma, N.; Sanyal, M.; Shrivastav, P.S. High throughput and sensitive determination of trazodone and its primary metabolite, m-chlorophenylpiperazine, in human plasma by liquid chromatography–tandem mass spectrometry. J. Chromatogr. B 2008, 871, 44–54. [Google Scholar] [CrossRef]
- Wenzel, S.; Aderjan, R.; Mattern, R.; Pedal, I.; Skopp, G. Tissue distribution of mirtazapine and desmethylmirtazapine in a case of mirtazapine poisoning. Forensic Sci. Int. 2006, 156, 229–236. [Google Scholar] [CrossRef]
- Hotha, K.K.; Ravindranath, L.K.; Veera, K.N.J.; Kumar, K.K.; Reddy, Y.R.; Jagadeesh, B.; Rao, D.V.N.; Bharathi, D.V.; Mullangi, R. Development and validation of a highly sensitive LC-MS/MS method for simultaneous quantitation of nortriptyline and 10-hydroxynortriptyline in human plasma: Application to a human pharmacokinetic study. Biomed. Chromatogr. 2010, 24, 1113–1119. [Google Scholar] [CrossRef]
- Segura, M.; Ortuño, J.; Farré, M.; Pacifici, R.; Pichini, S.; Joglar, J.; Segura, J.; Torre, R.d.l. Quantitative determination of paroxetine and its 4-hydroxy-3-methoxy metabolite in plasma by high-performance liquid chromatography/electrospray ion trap mass spectrometry: Application to pharmacokinetic studies. Rapid Commun. Mass Spectrom. 2003, 17, 1455–1461. [Google Scholar] [CrossRef][Green Version]
- Szafarz, M.; Wencel, A.; Pociecha, K.; Fedak, F.A.; Wlaź, P.; Wyska, E. Pharmacokinetic study of tianeptine and its active metabolite MC5 in rats following different routes of administration using a novel liquid chromatography tandem mass spectrometry analytical method. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2018, 391, 185–196. [Google Scholar] [CrossRef]
- Doherty, B.; Rodriguez, V.; Leslie, J.C.; McClean, S.; Smyth, W.F. An electrospray ionisation tandem mass spectrometric investigation of selected psychoactive pharmaceuticals and its application in drug and metabolite profiling by liquid chromatography/electrospray ionisation tandem mass spectrometry. Rapid Commun. Mass Spectrom. 2007, 21, 2031–2038. [Google Scholar] [CrossRef] [PubMed]







| LLOQ 10 ng/mL | Low 16 ng/mL | Medium QC 375 ng/mL | High QC 625 ng/mL | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Precision | Accuracy | Precision | Accuracy | Precision | Accuracy | Precision | Accuracy | ||
| Compound | [%] | [%] | [%] | [%] | [%] | [%] | [%] | [%] | |
| Amitriptyline | intra-run | 1.5 | 92 | 5.5 | 107 | 5.9 | 86 | 5.4 | 98 |
| between-run | 8.2 | 104 | 6.4 | 103 | 2.3 | 88 | 1.5 | 99 | |
| Citalopram | intra- run | 6.1 | 93 | 4.8 | 90 | 7.7 | 89 | 6.1 | 109 |
| between-run | 2.6 | 93 | 3.4 | 90 | 5.3 | 86 | 10 | 99 | |
| Clomipramine | intra- run | 6.7 | 88 | 3.7 | 98 | 10 | 104 | 9.1 | 105 |
| between-run | 9 | 100 | 5.4 | 98 | 3.3 | 101 | 3 | 108 | |
| Desipramine | intra- run | 5.1 | 106 | 5.6 | 101 | 11 | 92 | 5.6 | 95 |
| between-run | 14 | 102 | 6.9 | 99 | 3 | 89 | 4.1 | 99 | |
| Doxepin | intra-run | 3.4 | 104 | 6.8 | 99 | 7.6 | 110 | 8.4 | 113 |
| between-run | 6.2 | 110 | 3 | 96 | 2.5 | 103 | 5 | 109 | |
| Fluoxetine | intra-run | 6.7 | 108 | 5.4 | 109 | 8.1 | 91 | 0.8 | 97 |
| between-run | 0.8 | 109 | 0.6 | 108 | 0.3 | 91 | 0.4 | 98 | |
| Fluvoxamine | intra-run | 5.9 | 108 | 9.6 | 102 | 7.8 | 108 | 5.6 | 109 |
| between-run | 11 | 85 | 4.3 | 95 | 1.3 | 100 | 9 | 112 | |
| Imipramine | intra-run | 4.3 | 94 | 3.3 | 99 | 7.7 | 85 | 6.7 | 104 |
| between-run | 9.3 | 89 | 13 | 92 | 4.3 | 89 | 0.3 | 112 | |
| Maprotiline | intra-run | 2.7 | 96 | 6.1 | 89 | 12 | 92 | 7.1 | 104 |
| between-run | 7.9 | 97 | 5.2 | 88 | 1.2 | 93 | 0.7 | 105 | |
| Mianserin | intra-run | 3.8 | 113 | 3.9 | 101 | 8.8 | 114 | 11 | 93 |
| between-run | 2.9 | 112 | 3.5 | 96 | 14 | 97 | 11 | 105 | |
| Mirtazapine | intra-run | 6.2 | 97 | 13 | 101 | 11 | 95 | 5 | 100 |
| between-run | 12 | 102 | 6.2 | 104 | 2.8 | 93 | 7.2 | 107 | |
| Moclobemide | intra-run | 2.4 | 102 | 4.3 | 85 | 11 | 90 | 11 | 98 |
| between-run | 11 | 110 | 7.9 | 89 | 4.4 | 86 | 1.6 | 96 | |
| Nortriptyline | intra-run | 8.7 | 93 | 6.9 | 102 | 8.1 | 102 | 4.1 | 110 |
| between-run | 9.6 | 95 | 14 | 99 | 6.7 | 109 | 0.2 | 110 | |
| Opipramol | intra-run | 5.6 | 93 | 5.2 | 90 | 3.2 | 92 | 9.2 | 105 |
| between-run | 10 | 101 | 3.6 | 87 | 3.5 | 89 | 4.7 | 110 | |
| Paroxetine | intra-run | 7.5 | 105 | 7.4 | 101 | 12 | 100 | 11 | 91 |
| between-run | 4.5 | 106 | 2.9 | 102 | 0.8 | 99 | 6.7 | 97 | |
| Protriptyline | intra-run | 3.7 | 94 | 4.8 | 97 | 11 | 90 | 7.3 | 94 |
| between-run | 9.5 | 104 | 8.5 | 95 | 2.1 | 88 | 6.8 | 100 | |
| Sertraline | intra-run | 4.4 | 109 | 2 | 104 | 3.2 | 107 | 5.3 | 105 |
| between-run | 6.1 | 109 | 2.5 | 101 | 7 | 100 | 1.5 | 107 | |
| Tianeptine | intra-run | 2 | 103 | 3.2 | 93 | 11 | 93 | 9 | 111 |
| between-run | 3.2 | 106 | 2.9 | 97 | 1.8 | 95 | 5.8 | 105 | |
| Trazodone | intra-run | 2.4 | 111 | 4.1 | 88 | 6.1 | 89 | 5.6 | 105 |
| between-run | 9 | 103 | 3.3 | 92 | 1.6 | 91 | 2.7 | 102 | |
| Trimipramine | intra-run | 6.1 | 85 | 3.7 | 98 | 6 | 88 | 5.3 | 100 |
| between-run | 11 | 100 | 3.7 | 101 | 0.2 | 87 | 1.9 | 102 | |
| Venlafaxine | intra-run | 5.3 | 106 | 2.4 | 100 | 9.7 | 92 | 3.3 | 103 |
| between-run | 3.4 | 102 | 3 | 104 | 6 | 87 | 4.8 | 98 | |
| Study | This study | del Fernández et al. 2012 [20] | Madej and Persona 2013 [2] | Qin et al. 2008 [8] | Ansermot et al. 2012 [22] | de Castro et al. 2007 [21] | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Extraction | CPE | LLE | CPE | CPE | SPE | on-Line SPE | |||||||
| Analytical Method | LC/MS | LC/MS | LC/DAD | LC/FL | LC/MS | LC/MS | |||||||
| QC | Low | High | |||||||||||
| Amitryptyline | 85.4 | 70 | 67 | ↑ | 70 | ↑ | - | - | 99.9 | ↓ | |||
| Citalopram | 85.8 | 85 | 81 | ↑ | - | - | 83 | ↑ | 99.9 | ↓ | |||
| Clomipramine | 90.1 | 64 | 66 | ↑ | - | - | - | 99.7 | ↓ | ||||
| Desipramine | 87.7 | 72 | 74 | ↑ | - | - | - | 99.8 | ↓ | ||||
| Doxepine | 89.4 | 70 | 60 | ↑ | - | - | - | ||||||
| Fluoxetine | 94.0 | 74 | 72 | ↑ | - | - | 82 | ↑ | 99.9 | ↓ | |||
| Fluvoxamine | 94.1 | 70 | 71 | ↑ | - | - | - | 99.7 | ↓ | ||||
| Imipramine | 95.4 | 76 | 78 | ↑ | - | - | - | 99.9 | ↓ | ||||
| Maprotyline | 85.0 | 77 | 74 | ↑ | - | - | - | ||||||
| Mianserine | 87.0 | 86 | 83 | ↑ | - | - | - | ||||||
| Mirtazapine | 78.9 | 83 | 84 | ↓ | - | - | - | ||||||
| Moclobemide | 65.4 | 71 | 73 | ↓ | - | - | - | ||||||
| Nortryptyline | 81.3 | 71 | 73 | ↑ | - | - | 85 | ↓ | 99.6 | ↓ | |||
| Opipramol | 102.5 | - | - | 25 | ↑ | - | - | ||||||
| Paroxetine | 82.8 | 69 | 73 | ↑ | - | - | 74 | ↑ | 99.8 | ↓ | |||
| Protryptiline | 83.3 | - | - | - | - | - | |||||||
| Sertraline | 87.5 | 63 | 70 | ↑ | - | - | 80 | ↑ | 99.9 | ↓ | |||
| Tianeptine | 69.4 | - | - | - | - | - | |||||||
| Trazodone | 82.0 | 87 | 88 | ↓ | - | - | - | 99.9 | ↓ | ||||
| Trimipramine | 102.3 | - | - | - | - | - | |||||||
| Venlafaxine | 80.0 | 83 | 81 | ↓ | - | 89 | ↓ | - | 99.8 | ↓ | |||
| Mean | 86.2 | 75 | 75 | 48 | 89 | 81 | 99.8 | ||||||
| Range | 65.4–102.5 | 63–87 | 60–88 | 25–70 | 74–85 | 99.6–99.9 | |||||||
| Drug Name | Parent Ion [m/z] [M+H+] | Daughter Ion [m/z] | DP [V] | CE [V] | CXP [V] |
|---|---|---|---|---|---|
| Amitriptyline | 278 | 233 | 91 | 25 | 20 |
| Citalopram | 325 | 109 | 86 | 39 | 8 |
| Clomipramine | 315 | 86 | 81 | 29 | 6 |
| Desipramine | 267 | 72 | 81 | 31 | 12 |
| Doxepin | 280 | 107 | 71 | 33 | 6 |
| Fluoxetine | 310 | 44 | 56 | 37 | 6 |
| Fluoxetine-d5 | 315 | 44 | 71 | 41 | 6 |
| Fluvoxamine | 319 | 71 | 71 | 33 | 4 |
| Imipramine | 281 | 86 | 76 | 25 | 6 |
| Maprotiline | 278 | 250 | 86 | 27 | 16 |
| Mianserin | 265 | 208 | 96 | 31 | 12 |
| Mirtazapine | 266 | 195 | 46 | 37 | 10 |
| Moclobemide | 269 | 182 | 76 | 27 | 10 |
| Nortriptyline | 264 | 233 | 71 | 23 | 14 |
| Opipramol | 364 | 171 | 91 | 31 | 14 |
| Paroxetine | 330 | 192 | 51 | 31 | 14 |
| Protriptyline | 264 | 191 | 96 | 41 | 14 |
| Sertraline | 306 | 159 | 51 | 35 | 12 |
| Sertraline-d3 | 309 | 275 | 56 | 17 | 16 |
| Tianeptine | 437 | 292 | 61 | 25 | 8 |
| Trazodone | 372 | 176 | 81 | 37 | 10 |
| Trimipramine | 295 | 100 | 71 | 25 | 6 |
| Venlafaxine | 278 | 58 | 56 | 47 | 10 |
| Venlafaxine-d6 | 284 | 64 | 66 | 49 | 10 |
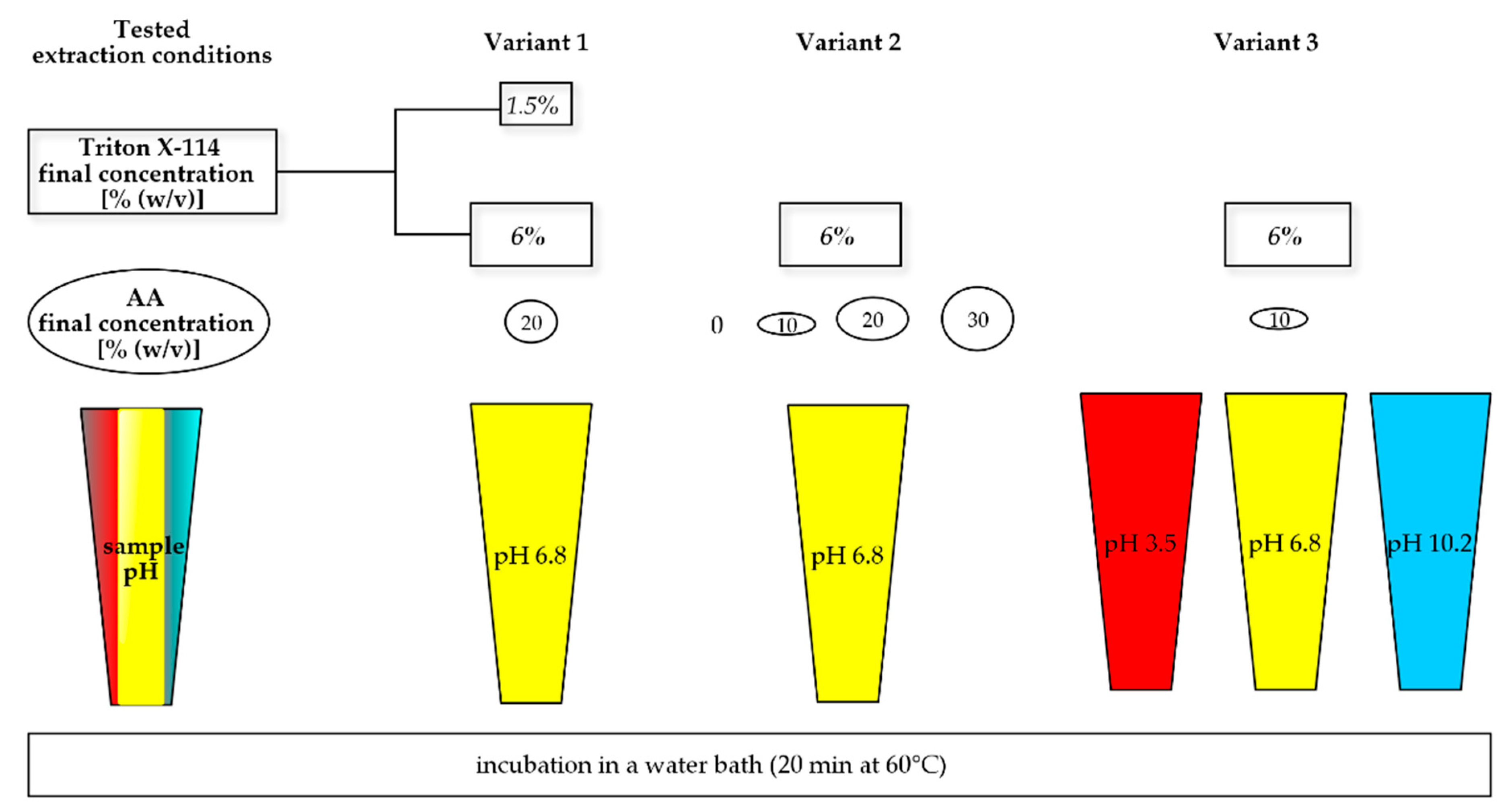
| Part 1. Development—Changes in Each Part | |||
|---|---|---|---|
| Variant | Triton X-114 in Sample [% (w/v)] | Sample pH | AA [% (w/v)] |
| Concentration of surfactant (a) | (a1) 1.5 (a2) 6 | 6.8 1 | 20 |
| Concentration of salt (b) | 6 | 6.8 1 | (b1) 0 (b2) 10 (b3) 20 (b4) 30 |
| Sample pH (c) | 6 | (c1) 3.5 2(c2) 6.8 1 (c3) 10.2 3 | 10 |
| Part 2. Validation method on sample C | |||
| Optimal method | 6 | 10.2 3 | 10 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gniazdowska, E.; Korytowska, N.; Kłudka, G.; Giebułtowicz, J. Determination of Antidepressants in Human Plasma by Modified Cloud-Point Extraction Coupled with Mass Spectrometry. Pharmaceuticals 2020, 13, 458. https://doi.org/10.3390/ph13120458
Gniazdowska E, Korytowska N, Kłudka G, Giebułtowicz J. Determination of Antidepressants in Human Plasma by Modified Cloud-Point Extraction Coupled with Mass Spectrometry. Pharmaceuticals. 2020; 13(12):458. https://doi.org/10.3390/ph13120458
Chicago/Turabian StyleGniazdowska, Elżbieta, Natalia Korytowska, Grzegorz Kłudka, and Joanna Giebułtowicz. 2020. "Determination of Antidepressants in Human Plasma by Modified Cloud-Point Extraction Coupled with Mass Spectrometry" Pharmaceuticals 13, no. 12: 458. https://doi.org/10.3390/ph13120458
APA StyleGniazdowska, E., Korytowska, N., Kłudka, G., & Giebułtowicz, J. (2020). Determination of Antidepressants in Human Plasma by Modified Cloud-Point Extraction Coupled with Mass Spectrometry. Pharmaceuticals, 13(12), 458. https://doi.org/10.3390/ph13120458