Developments in Carbohydrate-Based Metzincin Inhibitors



Abstract
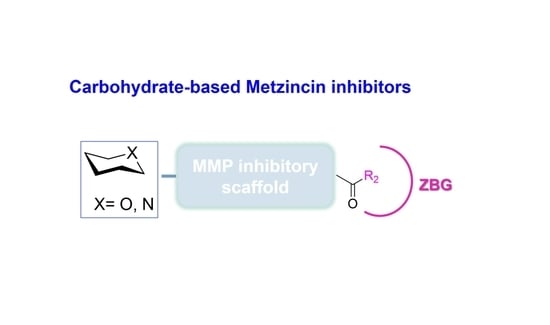
1. Introduction
1.1. Metzincins
1.2. Matrix Metalloproteinases
- Archetypal MMPs have a similar structure and are divided into:
- -
- Collagenases (MMP-1, MMP-8, MMP-13): specifically cleave the collagen triple helix. Moreover, their collagenolytic activity, due to a cooperation between the hemopexin and the catalytic domains, acts on a various number of ECM and non-ECM molecules.
- -
- Stromelysins (MMP-3, MMP-10): structurally similar to collagenases, show wide substrate specificity but are unable to degrade native collagen.
- -
- Others (MMP-12, MMP-19, MMP-20, and MMP-27): not classified in the previous categories. Metalloelastase (MMP-12) is mainly expressed in macrophages and digests elastin and other proteins.
- Gelatinases (MMP-2 or Gelatinase A and MMP-9 or Gelatinase B): owing to the presence of three fibronectin type II repeats inside the catalytic domain, they readily digest type IV collagen, gelatin, and a number of ECM molecules including laminin, fibronectin, and aggrecan core proteins.
- Matrilysins (MMP-7 and MMP-26): characterized by the lack of the hinge region and of hemopexin domain.
- Furin-activatable MMPs:
- -
- Secreted (MMP-11, MMP-21, and MMP-28): activated by furin-like proteases before secretion.
- -
- Membrane-type MMPs (MT-MMPs): classified in type I transmembrane proteins (MMP-14, MMP-15, MMP-16, and MMP-24) and type II transmembrane protein (MMP-23).
- -
- Glycosylphosphatidylinositol (GPI)-anchored proteins (MMP-17 and MMP-25).
MMP Catalytic Binding Site
1.3. ADAM Metalloproteinases
ADAM Ectodomain Shedding
1.4. Metzincin Inhibitors
2. Carbohydrate-Based Metzincin Inhibitors
2.1. Sugar-Based MMPIs
2.2. Azasugar-Based Inhibitors
2.2.1. Azasugar Biological Background
2.2.2. Azasugar-Based ADAM/MMP Inhibitors
3. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Bode, W.; Gomis-Rüth, F.-X.; Stöckler, W. Astacins, serralysins, snake venom and matrix metalloproteinases exhibit identical zinc-binding environments (HEXXHXXGXXH and Met-turn) and topologies and should be grouped into a common family, the ‘metzincins’. FEBS Lett. 1993, 331, 134–140. [Google Scholar] [CrossRef]
- Tallant, C.; García-Castellanos, R.; Baumann, U.; Gomis-Rüth, F.X. On the Relevance of the Met-turn Methionine in Metzincins. J. Biol. Chem. 2010, 285, 13951–13957. [Google Scholar] [CrossRef] [PubMed]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Nagase, H. Progress in matrix metalloproteinase research. Mol. Asp. Med. 2008, 29, 290–308. [Google Scholar] [CrossRef] [PubMed]
- Maskos, K. Crystal structures of MMPs in complex with physiological and pharmacological inhibitors. Biochimie 2005, 87, 249–263. [Google Scholar] [CrossRef]
- Ogata, H.; Decaneto, E.; Grossman, M.; Havenith, M.; Sagi, I.; Lubitz, W.; Knipp, M. Crystallization and preliminary X-ray crystallographic analysis of the catalytic domain of membrane type 1 matrix metalloproteinase. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2014, 70, 232–235. [Google Scholar] [CrossRef]
- Tallant, C.; Marrero, A.; Gomis-Rüth, F.X. Matrix metalloproteinases: Fold and function of their catalytic domains. Biochim. Biophys. Acta Bioenerg. 2010, 1803, 20–28. [Google Scholar] [CrossRef]
- Bertini, I.; Calderone, V.; Cosenza, M.; Fragai, M.; Lee, Y.-M.; Luchinat, C.; Mangani, S.; Terni, B.; Turano, P. Conformational variability of matrix metalloproteinases: Beyond a single 3D structure. Proc. Natl. Acad. Sci. USA 2005, 102, 5334–5339. [Google Scholar] [CrossRef]
- Page-McCaw, A.; Ewald, A.J.; Werb, Z. Matrix metalloproteinases and the regulation of tissue remodelling. Nat. Rev. Mol. Cell Biol. 2007, 8, 221–233. [Google Scholar] [CrossRef]
- Schechter, E.; Berger, A. On the size of the active site in proteases: Pronase. Biochem. Biophys. Res. Commun. 1972, 46, 1956–1960. [Google Scholar] [CrossRef]
- Gupta, S.P.; Patil, V.M. Specificity of Binding with Matrix Metalloproteinases. Exp. Suppl. 2012, 103, 35–56. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, A.; Beltrán-Debón, R.; Mulero, M.; Pujadas, G.; Garcia-Vallvé, S. Understanding the variability of the S1′ pocket to improve matrix metalloproteinase inhibitor selectivity profiles. Drug Discov. Today 2020, 25, 38–57. [Google Scholar] [CrossRef] [PubMed]
- Edwards, D.R.; Handsley, M.M.; Pennington, C.J. The ADAM metalloproteinases. Mol. Asp. Med. 2008, 29, 258–289. [Google Scholar] [CrossRef]
- Giebeler, N.; Zigrino, P. A Disintegrin and Metalloprotease (ADAM): Historical Overview of Their Functions. Toxins 2016, 8, 122. [Google Scholar] [CrossRef] [PubMed]
- Seals, D.F.; Courtneidge, S.A. The ADAMs family of metalloproteases: Multidomain proteins with multiple functions. Genes Dev. 2003, 17, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Shieh, H.-S.; Day, J.E.; Caspers, N.; Chrencik, J.E.; Williams, J.M.; Pegg, L.E.; Pauley, A.M.; Moon, A.F.; Krahn, J.M.; et al. Structure of human ADAM-8 catalytic domain complexed with batimastat. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 616–621. [Google Scholar] [CrossRef]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the α-Secretase ADAM10. Cell 2017, 171, 1638–1648. [Google Scholar] [CrossRef]
- Maskos, K.; Fernandez-Catalan, C.; Huber, R.; Bourenkov, G.P.; Bartunik, H.; Ellestad, G.A.; Reddy, P.; Wolfson, M.F.; Rauch, C.T.; Castner, B.J.; et al. Crystal structure of the catalytic domain of human tumor necrosis factor-α-converting enzyme. Proc. Natl. Acad. Sci. USA 1998, 95, 3408–3412. [Google Scholar] [CrossRef]
- Orth, P.; Reichert, P.; Wang, W.; Prosise, W.W.; Yarosh-Tomaine, T.; Hammond, G.; Ingram, R.N.; Xiao, L.; Mirza, U.A.; Zou, J.; et al. Crystal Structure of the Catalytic Domain of Human ADAM33. J. Mol. Biol. 2004, 335, 129–137. [Google Scholar] [CrossRef]
- Stone, A.L.; Kroeger, M.; Sang, Q.X.A. Structure–Function Analysis of the ADAM Family of Disintegrin-Like and Metalloproteinase-Containing Proteins (Review). J. Protein Chem. 1999, 18, 447–465. [Google Scholar] [CrossRef]
- Weber, S.; Saftig, P. Ectodomain shedding and ADAMs in development. Development 2012, 139, 3693–3709. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. The ADAMs: Signalling scissors in the tumour microenvironment. Nat. Rev. Cancer 2008, 8, 929–941. [Google Scholar] [CrossRef] [PubMed]
- Boucher, B.J. Matrix metalloproteinase protein inhibitors: Highlighting a new beginning for metalloproteinases in medicine. Met. Med. 2016, 3, 75–79. [Google Scholar] [CrossRef]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef]
- Nissinen, L.; Kähäri, V.-M. Matrix metalloproteinases in inflammation. Biochim. Biophys. Acta Gen. Subj. 2014, 1840, 2571–2580. [Google Scholar] [CrossRef]
- Hu, J.; Van den Steen, P.E.; Sang, Q.X.A.; Opdenakker, G. Matrix metalloproteinase inhibitors as therapy for inflammatory and vascular diseases. Nat. Rev. Drug Discov. 2007, 6, 480–498. [Google Scholar] [CrossRef]
- Muri, L.; Leppert, D.; Grandgirard, D.; Leib, S.L. MMPs and ADAMs in neurological infectious diseases and multiple sclerosis. Cell. Mol. Life Sci. 2019, 76, 3097–3116. [Google Scholar] [CrossRef]
- Mishra, H.K.; Ma, J.; Walcheck, B. Ectodomain Shedding by ADAM17: Its Role in Neutrophil Recruitment and the Impairment of This Process during Sepsis. Front. Cell. Infect. Microbiol. 2017, 7, 138. [Google Scholar] [CrossRef]
- Rivera, S.; García-González, L.; Khrestchatisky, M.; Baranger, K. Metalloproteinases and their tissue inhibitors in Alzheimer’s disease and other neurodegenerative disorders. Cell. Mol. Life Sci. 2019, 76, 3167–3191. [Google Scholar] [CrossRef]
- Marcello, E.; Borroni, B.; Pelucchi, S.; Gardoni, F.; Di Luca, M. ADAM10 as a therapeutic target for brain diseases: From developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. [Google Scholar] [CrossRef]
- Qian, M.; Shen, X.; Wang, H. The Distinct Role of ADAM17 in APP Proteolysis and Microglial Activation Related to Alzheimer’s Disease. Cell. Mol. Neurobiol. 2016, 36, 471–482. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Senn, N.; Riedl, R. Design and Structural Evolution of Matrix Metalloproteinase Inhibitors. Chem. Eur. J. 2019, 25, 7960–7980. [Google Scholar] [CrossRef] [PubMed]
- Fisher, J.F.; Mobashery, S. Recent advances in MMP inhibitor design. Cancer Metastasis Rev. 2006, 25, 115–136. [Google Scholar] [CrossRef] [PubMed]
- Verma, R.P. Hydroxamic Acids as Matrix Metalloproteinase Inhibitors. Exp. Suppl. 2012, 103, 137–176. [Google Scholar] [CrossRef]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J.H. Design and Therapeutic Application of Matrix Metalloproteinase Inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef]
- MacPherson, L.J.; Bayburt, E.K.; Capparelli, M.P.; Carroll, B.J.; Goldstein, R.; Justice, M.R.; Zhu, L.; Hu, S.-I.; Melton, R.A.; Fryer, L.; et al. Discovery of CGS 27023A, a Non-Peptidic, Potent, and Orally Active Stromelysin Inhibitor That Blocks Cartilage Degradation in Rabbits. J. Med. Chem. 1997, 40, 2525–2532. [Google Scholar] [CrossRef]
- Bertini, I.; Calderone, V.; Fragai, M.; Luchinat, C.; Mangani, S.; Terni, B. Crystal Structure of the Catalytic Domain of Human Matrix Metalloproteinase 10. J. Mol. Biol. 2004, 336, 707–716. [Google Scholar] [CrossRef]
- Rossello, A.; Nuti, E.; Orlandini, E.; Carelli, P.; Rapposelli, S.; Macchia, M.; Minutolo, F.; Carbonaro, L.; Albini, A.; Benelli, R.; et al. New N-arylsulfonyl-N-alkoxyaminoacetohydroxamic acids as selective inhibitors of gelatinase A (MMP-2). Bioorganic Med. Chem. 2004, 12, 2441–2450. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Renkiewicz, R.; Qiu, L.; Lesch, C.; Sun, X.; Devalaraja, R.; Cody, T.; Kaldjian, E.; Welgus, H.; Baragi, V. Broad-spectrum matrix metalloproteinase inhibitor marimastat-induced musculoskeletal side effects in rats. Arthritis Rheum. 2003, 48, 1742–1749. [Google Scholar] [CrossRef]
- Senn, N.; Ott, M.; Lanz, J.; Riedl, R. Targeted Polypharmacology: Discovery of a Highly Potent Non-Hydroxamate Dual Matrix Metalloproteinase (MMP)-10/-13 Inhibitor. J. Med. Chem. 2017, 60, 9585–9598. [Google Scholar] [CrossRef] [PubMed]
- Nara, H.; Sato, K.; Naito, T.; Mototani, H.; Oki, H.; Yamamoto, Y.; Kuno, H.; Santou, T.; Kanzaki, N.; Terauchi, J.; et al. Discovery of Novel, Highly Potent, and Selective Quinazoline-2-carboxamide-Based Matrix Metalloproteinase (MMP)-13 Inhibitors without a Zinc Binding Group Using a Structure-Based Design Approach. J. Med. Chem. 2014, 57, 8886–8902. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Fuerst, R.; Knapinska, A.M.; Taylor, A.B.; Smith, L.; Cao, X.; Hart, P.J.; Fields, G.B.; Roush, W.R. Structure-Based Design and Synthesis of Potent and Selective Matrix Metalloproteinase 13 Inhibitors. J. Med. Chem. 2017, 60, 5816–5825. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Sampson, N.S.; Li, J.; Kuscu, C.; Rizzo, R.C.; DeLeon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-Molecule Anticancer Compounds Selectively Target the Hemopexin Domain of Matrix Metalloproteinase-9. Cancer Res. 2011, 71, 4977–4988. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP Small-Molecule Inhibitors Based on Insights into Hemopexin Domain Function in Tumor Growth. Cancer Res. 2012, 72, 2339–2349. [Google Scholar] [CrossRef]
- Spicer, T.P.; Jiang, J.; Taylor, A.B.; Choi, J.Y.; Hart, P.J.; Roush, W.R.; Fields, G.B.; Hodder, P.S.; Minond, D. Characterization of Selective Exosite-Binding Inhibitors of Matrix Metalloproteinase 13 That Prevent Articular Cartilage Degradation In Vitro. J. Med. Chem. 2014, 57, 9598–9611. [Google Scholar] [CrossRef]
- Saftig, P.; Reiss, K. The “A Disintegrin and Metalloproteases” ADAM10 and ADAM17: Novel drug targets with therapeutic potential? Eur. J. Cell Biol. 2011, 90, 527–535. [Google Scholar] [CrossRef]
- Zunke, F.; Rose-John, S. The shedding protease ADAM17: Physiology and pathophysiology. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 2059–2070. [Google Scholar] [CrossRef]
- Fridman, J.S.; Caulder, E.; Hansbury, M.; Liu, X.; Yang, G.; Wang, Q.; Lo, Y.; Zhou, B.-B.S.; Pan, M.; Thomas, S.M.; et al. Selective Inhibition of ADAM Metalloproteases as a Novel Approach for Modulating ErbB Pathways in Cancer. Clin. Cancer Res. 2007, 13, 1892–1902. [Google Scholar] [CrossRef]
- Zhou, B.-B.S.; Peyton, M.; He, B.; Liu, C.; Girard, L.; Caudler, E.; Lo, Y.; Baribaud, F.; Mikami, I.; Reguart, N.; et al. Targeting ADAM-mediated ligand cleavage to inhibit HER3 and EGFR pathways in non-small cell lung cancer. Cancer Cell 2006, 10, 39–50. [Google Scholar] [CrossRef]
- Nuti, E.; Casalini, F.; Santamaria, S.; Fabbi, M.; Carbotti, G.; Ferrini, S.; Marinelli, L.; La Pietra, V.; Novellino, E.; Camodeca, C.; et al. Selective Arylsulfonamide Inhibitors of ADAM-17: Hit Optimization and Activity in Ovarian Cancer Cell Models. J. Med. Chem. 2013, 56, 8089–8103. [Google Scholar] [CrossRef] [PubMed]
- Minond, D.; Cudic, M.; Bionda, N.; Giulianotti, M.; Maida, L.; Houghten, R.A.; Fields, G.B. Discovery of Novel Inhibitors of a Disintegrin and Metalloprotease 17 (ADAM17) Using Glycosylated and Non-glycosylated Substrates. J. Biol. Chem. 2012, 287, 36473–36487. [Google Scholar] [CrossRef] [PubMed]
- Knapinska, A.M.; Dreymueller, D.; Ludwig, A.; Smith, L.; Golubkov, V.; Sohail, A.; Fridman, R.; Giulianotti, M.; LaVoi, T.M.; Houghten, R.A.; et al. SAR Studies of Exosite-Binding Substrate-Selective Inhibitors of A Disintegrin And Metalloprotease 17 (ADAM17) and Application as Selective in Vitro Probes. J. Med. Chem. 2015, 58, 5808–5824. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Tuccinardi, T.; Rossello, A. Matrix Metalloproteinase Inhibitors: New Challenges in the Era of Post Broad-Spectrum Inhibitors. Curr. Pharm. Des. 2007, 13, 2087–2100. [Google Scholar] [CrossRef] [PubMed]
- Camodeca, C.; Cuffaro, D.; Nuti, E.; Rossello, A. ADAM Metalloproteinases as Potential Drug Targets. Curr. Med. Chem. 2019, 26, 2661–2689. [Google Scholar] [CrossRef]
- Digilio, G.; Tuccinardi, T.; Casalini, F.; Cassino, C.; Dias, D.M.; Geraldes, C.F.G.C.; Catanzaro, V.; Maiocchi, A.; Rossello, A. Study of the binding interaction between fluorinated matrix metalloproteinase inhibitors and Human Serum Albumin. Eur. J. Med. Chem. 2014, 79, 13–23. [Google Scholar] [CrossRef]
- Gabius, H.-J. The Sugar Code. Fundamentals of Glycosciences; Wiley-VCH: Weinheim, Germany, 2009. [Google Scholar]
- Ernst, B.; Hart, G.W.; Sinaý, P. Carbohydrates in Chemistry and Biology; Wiley-VCH: Weinheim, Germany, 2000. [Google Scholar]
- Mullapudi, S.S.; Mitra, D.; Li, M.; Kang, E.-T.; Chiong, E.; Neoh, K.G. Potentiating anti-cancer chemotherapeutics and antimicrobials via sugar-mediated strategies. Mol. Syst. Des. Eng. 2020, 5, 772–791. [Google Scholar] [CrossRef]
- Micoli, F.; Costantino, P.; Adamo, R. Potential targets for next generation antimicrobial glycoconjugate vaccines. FEMS Microbiol. Rev. 2018, 42, 388–423. [Google Scholar] [CrossRef]
- Khatun, F.; Toth, I.; Stephenson, R.J. Immunology of carbohydrate-based vaccines. Adv. Drug Deliv. Rev. 2020. [Google Scholar] [CrossRef]
- Mettu, R.; Chen, C.-Y.; Wu, C.-Y. Synthetic carbohydrate-based vaccines: Challenges and opportunities. J. Biomed. Sci. 2020, 27, 1–22. [Google Scholar] [CrossRef]
- Gragnani, T.; Cuffaro, D.; Fallarini, S.; Lombardi, G.; D’Andrea, F.; Guazzelli, L. Selectively Charged and Zwitterionic Analogues of the Smallest Immunogenic Structure of Streptococcus Pneumoniae Type 14. Molecules 2019, 24, 3414. [Google Scholar] [CrossRef] [PubMed]
- Hossain, F.; Andreana, P.R. Developments in Carbohydrate-Based Cancer Therapeutics. Pharmaceuticals 2019, 12, 84. [Google Scholar] [CrossRef] [PubMed]
- D’Andrea, F.; Vagelli, G.; Granchi, C.; Guazzelli, L.; Tuccinardi, T.; Poli, G.; Iacopini, D.; Minutolo, F.; Di Bussolo, V. Synthesis and Biological Evaluation of New Glycoconjugated LDH Inhibitors as Anticancer Agents. Molecules 2019, 24, 3520. [Google Scholar] [CrossRef]
- Drinnan, N.B.; Vari, F. Aspects of the stability and bioavailability of carbohydrates and carbohydrate derivatives. Mini Revi. Med. Chem. 2003, 3, 633–649. [Google Scholar] [CrossRef] [PubMed]
- Macmillan, D.; Daines, A.M. Recent Developments in the Synthesis and Discovery of Oligosaccharides and Glycoconjugates for the Treatment of Disease. Curr. Med. Chem. 2003, 10, 2733–2773. [Google Scholar] [CrossRef] [PubMed]
- Makuch, S.; Woźniak, M.; Krawczyk, M.; Pastuch-Gawołek, G.; Szeja, W.; Agrawal, S. Glycoconjugation as a Promising Treatment Strategy for Psoriasis. J. Pharmacol. Exp. Ther. 2020, 373, 204–212. [Google Scholar] [CrossRef]
- Molejon, M.I.; Weiz, G.; Breccia, J.D.; Vaccaro, M.I. Glycoconjugation: An approach to cancer therapeutics. World J. Clin. Oncol. 2020, 11, 110–120. [Google Scholar] [CrossRef]
- Cuffaro, D.; Nuti, E.; Rossello, A. An overview of carbohydrate-based Carbonic Anhydrase inhibitors. J. Enzyme Inhib. Med. Chem. 2020. [Google Scholar] [CrossRef]
- Nativi, C.; Richichi, B.; Roelens, S. Carbohydrate-containing matrix metalloproteinase inhibitors. In Carbohydrates in Drug Design and Discovery; Jimenez-Barbero, J., Canada, F.J., Martin-Santamaria, S., Eds.; RSC Drug Discovery: Cambridge, UK, 2015. [Google Scholar]
- Fragai, M.; Nativi, C.; Richichi, B.; Venturi, C. Design In Silico, Synthesis and Binding Evaluation of a Carbohydrate-Based Scaffold for Structurally Novel Inhibitors of Matrix Metalloproteinases. ChemBioChem 2005, 6, 1345–1349. [Google Scholar] [CrossRef]
- Calderone, V.; Fragai, M.; Luchinat, C.; Nativi, C.; Richichi, B.; Roelens, S. A High-Affinity Carbohydrate-Containing Inhibitor of Matrix Metalloproteinases. ChemMedChem 2006, 1, 598–601. [Google Scholar] [CrossRef]
- Bartoloni, M.; Domínguez, B.E.; Dragoni, E.; Richichi, B.; Fragai, M.; André, S.; Gabius, H.-J.; Ardá, A.; Luchinat, C.; Jiménez-Barbero, J.; et al. Targeting Matrix Metalloproteinases: Design of a Bifunctional Inhibitor for Presentation by Tumour-Associated Galectins. Chem. A Eur. J. 2012, 19, 1896–1902. [Google Scholar] [CrossRef] [PubMed]
- Hugenberg, V.; Breyholz, H.-J.; Riemann, B.; Hermann, S.; Schober, O.; Schäfers, M.; Gangadharmath, U.; Mocharla, V.; Kolb, H.; Walsh, J.; et al. A New Class of Highly Potent Matrix Metalloproteinase Inhibitors Based on Triazole-Substituted Hydroxamates: (Radio)Synthesis and in Vitro and First in Vivo Evaluation. J. Med. Chem. 2012, 55, 4714–4727. [Google Scholar] [CrossRef] [PubMed]
- Nuti, E.; Cuffaro, D.; D’Andrea, F.; Rosalia, L.; Tepshi, L.; Fabbi, M.; Carbotti, G.; Ferrini, S.; Santamaria, S.; Camodeca, C.; et al. Sugar-Based Arylsulfonamide Carboxylates as Selective and Water-Soluble Matrix Metalloproteinase-12 Inhibitors. ChemMedChem 2016, 11, 1626–1637. [Google Scholar] [CrossRef] [PubMed]
- Cuffaro, D.; Camodeca, C.; D’Andrea, F.; Piragine, E.; Testai, L.; Calderone, V.; Orlandini, E.; Nuti, E.; Rossello, A. Matrix metalloproteinase-12 inhibitors: Synthesis, structure-activity relationships and intestinal absorption of novel sugar-based biphenylsulfonamide carboxylates. Bioorganic Med. Chem. 2018, 26, 5804–5815. [Google Scholar] [CrossRef]
- Santamaria, S.; Nuti, E.; Cercignani, G.; Marinelli, L.; La Pietra, V.; Novellino, E.; Rossello, A. N-O-Isopropyl sulfonamido-based hydroxamates: Kinetic characterisation of a series of MMP-12/MMP-13 dual target inhibitors. Biochem. Pharmacol. 2012, 84, 813–820. [Google Scholar] [CrossRef]
- Nuti, E.; Cuffaro, D.; Bernardini, E.; Camodeca, C.; Panelli, L.; Chaves, S.; Ciccone, L.; Tepshi, L.; Vera, L.; Orlandini, E.; et al. Development of Thioaryl-Based Matrix Metalloproteinase-12 Inhibitors with Alternative Zinc-Binding Groups: Synthesis, Potentiometric, NMR, and Crystallographic Studies. J. Med. Chem. 2018, 61, 4421–4435. [Google Scholar] [CrossRef]
- D’Andrea, F.; Nuti, E.; Becherini, S.; Cuffaro, D.; Husanu, E.; Camodeca, C.; De Vita, E.; Zocchi, M.R.; Poggi, A.; D’Arrigo, C.; et al. Design and Synthesis of Ionic Liquid-Based Matrix Metalloproteinase Inhibitors (MMPIs): A Simple Approach to Increase Hydrophilicity and to Develop MMPI-Coated Gold Nanoparticles. ChemMedChem 2019, 14, 686–698. [Google Scholar] [CrossRef]
- Nuti, E.; Panelli, L.; Casalini, F.; Avramova, S.I.; Orlandini, E.; Santamaria, S.; Nencetti, S.; Tuccinardi, T.; Martinelli, A.; Cercignani, G.; et al. Design, Synthesis, Biological Evaluation, and NMR Studies of a New Series of Arylsulfones As Selective and Potent Matrix Metalloproteinase-12 Inhibitors. J. Med. Chem. 2009, 52, 6347–6361. [Google Scholar] [CrossRef]
- Antoni, C.; Vera, L.; Devel, L.; Catalani, M.P.; Czarny, B.; Cassar-Lajeunesse, E.; Nuti, E.; Rossello, A.; Dive, V.; Stura, E.A. Crystallization of bi-functional ligand protein complexes. J. Struct. Biol. 2013, 182, 246–254. [Google Scholar] [CrossRef]
- Thorens, B.; Mueckler, M. Glucose transporters in the 21st Century. Am. J. Physiol. Endocrinol. Metab. 2010, 298, E141–E145. [Google Scholar] [CrossRef]
- Ponedel’kina, I.Y.; Gaskarova, A.R.; Khaybrakhmanova, E.A.; Lukina, E.S.; Odinokov, V.N. Hyaluronic acid based hydroxamate and conjugates with biologically active amines: In vitro effect on matrix metalloproteinase-2. Carbohydr. Polym. 2016, 144, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Compain, P.; Martin, O.R. Iminosugars: From Synthesis to Therapeutic Applications; Wiley-VCH: Weinheim, Germany, 2007. [Google Scholar]
- Iftikhar, M.; Wang, L.; Fang, Z. Synthesis of 1-Deoxynojirimycin: Exploration of Optimised Conditions for Reductive Amidation and Separation of Epimers. J. Chem. Res. 2017, 41, 460–464. [Google Scholar] [CrossRef]
- Wood, A.; Prichard, K.L.; Clarke, Z.; Houston, T.A.; Fleet, G.W.J.; Simone, M.I. Synthetic Pathways to 3,4,5-Trihydroxypiperidines from the Chiral Pool. Eur. J. Org. Chem. 2018, 2018, 6812–6829. [Google Scholar] [CrossRef]
- Cuffaro, D.; Landi, M.; D’Andrea, F.; Guazzelli, L. Preparation of 1,6-di-deoxy-d-galacto and 1,6-di-deoxy-l-altro nojirimycin derivatives by aminocyclization of a 1,5-dicarbonyl derivative. Carbohydr. Res. 2019, 482, 107744. [Google Scholar] [CrossRef]
- Dehoux-Baudoin, C.; Génisson, Y. C -Branched Imino Sugars: Synthesis and Biological Relevance. Eur. J. Org. Chem. 2019, 2019, 4765–4777. [Google Scholar] [CrossRef]
- Afarinkia, K.; Bahar, A. Recent advances in the chemistry of azapyranose sugars. Tetrahedron Asymmetry 2005, 16, 1239–1287. [Google Scholar] [CrossRef]
- Guazzelli, L.; Catelani, G.; D’Andrea, F.; Gragnani, T.; Griselli, A. Stereoselective Access to the β-D-N-Acetylhexosaminyl-(1→4)-1-deoxy-D-nojirimycin Disaccharide Series Avoiding the Glycosylation Reaction. Eur. J. Org. Chem. 2014, 2014, 6527–6537. [Google Scholar] [CrossRef]
- Guazzelli, L.; D’Andrea, F.; Sartini, S.; Giorgelli, F.; Confini, G.; Quattrini, L.; Piano, I.; Nencetti, S.; Orlandini, E.; Gargini, C.; et al. Synthesis and investigation of polyhydroxylated pyrrolidine derivatives as novel chemotypes showing dual activity as glucosidase and aldose reductase inhibitors. Bioorganic Chem. 2019, 92, 103298. [Google Scholar] [CrossRef]
- Bojarová, P.; Křen, V. Glycosidases: A key to tailored carbohydrates. Trends Biotechnol. 2009, 27, 199–209. [Google Scholar] [CrossRef]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef]
- Gao, K.; Zheng, C.; Wang, T.; Zhao, H.; Wang, J.; Wang, Z.; Zhai, X.; Jia, Z.; Chen, J.; Zhou, Y.; et al. 1-Deoxynojirimycin: Occurrence, Extraction, Chemistry, Oral Pharmacokinetics, Biological Activities and In Silico Target Fishing. Molecules 2016, 21, 1600. [Google Scholar] [CrossRef] [PubMed]
- Junge, B.; Matzke, M.; Stoltefuss, J. Chemistry and Structure—Activity Relationships of Glucosidase Inhibitors. In Handbook of Experimental Pharmacology; Springer: New York, NY, USA, 1996. [Google Scholar]
- Campbell, L.K.; Baker, D.E.; Campbell, R.K. Miglitol: Assessment of its role in the treatment of patients with diabetes mellitus. Ann. Pharmacother. 2000, 34, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.; Patrick, P.; Wishart, J.; Horo-witz, M.; Morley, J.E. The effects of miglitol on glucagon-like peptide-1 secretion and appetite sensations in obese type 2 diabetics. Diabetes Obes. Metab. 2002, 4, 329–335. [Google Scholar] [CrossRef] [PubMed]
- Giraldo, P.; Andrade-Campos, M.; Alfonso, P.; Irún, P.; Atutxa, K.; Acedo, A.; Barez, A.; Blanes, M.; Diaz-Morant, V.; Fernández-Galán, M.A.; et al. Twelve years of experience with miglustat in the treatment of type 1 Gaucher disease: The Spanish ZAGAL project. Blood Cells Mol. Dis. 2018, 68, 173–179. [Google Scholar] [CrossRef]
- Wang, R.-J.; Yang, C.-H.; Hu, M.-L. 1-Deoxynojirimycin Inhibits Metastasis of B16F10 Melanoma Cells by Attenuating the Activity and Expression of Matrix Metalloproteinases-2 and -9 and Altering Cell Surface Glycosylation. J. Agric. Food Chem. 2010, 58, 8988–8993. [Google Scholar] [CrossRef]
- Chan, K.-C.; Lin, M.-C.; Huang, C.-N.; Chang, W.-C.; Wang, C.-J. Mulberry 1-Deoxynojirimycin Pleiotropically Inhibits Glucose-Stimulated Vascular Smooth Muscle Cell Migration by Activation of AMPK/RhoB and Down-regulation of FAK. J. Agric. Food Chem. 2013, 61, 9867–9875. [Google Scholar] [CrossRef]
- Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, S.-I. Structure–activity relationships of azasugar-based MMP/ADAM inhibitors. Bioorganic Med. Chem. Lett. 2003, 13, 2737–2740. [Google Scholar] [CrossRef]
- Moriyama, H.; Tsukida, T.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, S. Design, synthesis and evaluation of novel azasugar-based MMP/ADAM inhibitors. Bioorganic Med. Chem. Lett. 2003, 13, 2741–2744. [Google Scholar] [CrossRef]
- Moriyama, H.; Tsukida, T.; Inoue, Y.; Yokota, K.; Yoshino, K.; Kondo, H.; Miura, A.N.; Nishimura, S.-I. Azasugar-Based MMP/ADAM Inhibitors as Antipsoriatic Agents. J. Med. Chem. 2004, 47, 1930–1938. [Google Scholar] [CrossRef]
- Chikaraishi, Y.; Shimazawa, M.; Yokota, K.; Yoshino, K.; Hara, H. CB-12181, a New Azasugar-Based Matrix Metalloproteinase/Tumor Necrosis Factor-α Converting Enzyme Inhibitor, Inhibits Vascular Endothelial Growth Factor-Induced Angiogenesis in Vitro and Retinal Neovascularization in Vivo. Curr. Neurovascular Res. 2009, 6, 140–147. [Google Scholar] [CrossRef]
- Sylte, I.; Dawadi, R.; Malla, N.; Von Hofsten, S.; Nguyen, T.-M.; Solli, A.I.; Berg, E.; Adekoya, O.A.; Svineng, G.; Winberg, J.-O. The selectivity of galardin and an azasugar-based hydroxamate compound for human matrix metalloproteases and bacterial metalloproteases. PLoS ONE 2018, 13, e0200237. [Google Scholar] [CrossRef] [PubMed]
- Tsukida, T.; Moriyama, H.; Inoue, Y.; Kondo, H.; Yoshino, K.; Nishimura, S. Synthesis and biological activity of selective azasugar-based TACE inhibitors. Bioorganic Med. Chem. Lett. 2004, 14, 1569–1572. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MMP-1 | MMP-2 | MMP-9 | MMP-12 | Ref |
|---|---|---|---|---|---|
| 1 | - a | - | - | 490,000 | [72] |
| 2 | - | - | - | 790,000 | [72] |
| 3 | 286 | - | - | 14.3 | [73] |
| 4 | - | - | 80 (Ki) | 17 (Ki) | [74] |
| 5 | - | 30 | 43 | - | [75] |
| 6 | - | 0.2 | 0.6 | 0.5 | [75] |
| 8 | - | - | 6800 | 63 | [76] |
| 9 | - | - | 9400 | 73 | [76] |
| 10 | - | - | 4400 | 72 | [76] |
| 11 | - | - | 3100 | 53 | [76] |
| 12 | - | - | 1220 | 36 | [76] |
| 13 | - | - | 1200 | 28 | [76] |
| 14 | 19,000 | 330 | 1200 | 18 | [76] |
| 15 | 40,000 | 320 | 5400 | 40 | [76] |
| 16 | 50,000 | 100 | 860 | 12 | [76] |
| 17 | - | - | 1470 | 42 | [76] |
| 18 | - | - | 880 | 19 | [77] |
| 19 | - | - | 1120 | 98 | [77] |
| 20 | - | - | 520 | 65 | [77] |
| 21 | - | - | 1200 | 18 | [77] |
| 22 | - | - | >50,000 | 1300 | [77] |
| 23 | - | - | 1235 | 83 | [77] |
| 24 | - | - | 890 | 41 | [77] |
| 25 | - | - | 2200 | 51 | [77] |
| Compound | ADAM17 | MMP-1 | MMP-3 | MMP-9 | Ref |
|---|---|---|---|---|---|
| 38 | 22 | 554 | 44 | 310 | [102] |
| 39 | 40 | 16 | 3.7 | 21 | [102] |
| 40 | 71 | 84 | 1.7 | 157 | [102] |
| 41 | 21 | 50 | 50 | 47 | [102] |
| 42 | 12 | 25 | 7.7 | 4.8 | [103] |
| 43 | 510 | >850 | 490 | 780 | [103] |
| 44 | 340 | 450 | 85 | 82 | [103] |
| 45 | 8.7 | >850 | 42 | 64 | [103] |
| 46 | 2.3 | 8.0 | 0.5 | 0.06 | [103] |
| 47 | 1.6 | 850 | 2.6 | 6.1 | [103] |
| 48 | 67 | 100 | 1.8 | 0.9 | [103] |
| 49 | 6.2 | 5.3 | 0.35 | 0.097 | [104] |
| 50 | 15 | 26 | 2 | 2 | [104] |
| 51 | 21 | 162 | 50 | 47 | [104] |
| 52 | 1.7 | >850 | 2.1 | 7.4 | [104] |
| 53 | 0.53 | 128 | 3.3 | 14 | [104] |
| 54 | 0.53 | 128 | 3.3 | 14.2 | [107] |
| 55 | 1.85 | 90 | 0.43 | 12 | [107] |
| 56 | 0.57 | >850 | 29.6 | 77 | [107] |
| 57 | 0.84 | 552 | 11.5 | 98 | [107] |
| 58 | 1.85 | >850 | 58 | 118 | [107] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuffaro, D.; Nuti, E.; D’Andrea, F.; Rossello, A. Developments in Carbohydrate-Based Metzincin Inhibitors. Pharmaceuticals 2020, 13, 376. https://doi.org/10.3390/ph13110376
Cuffaro D, Nuti E, D’Andrea F, Rossello A. Developments in Carbohydrate-Based Metzincin Inhibitors. Pharmaceuticals. 2020; 13(11):376. https://doi.org/10.3390/ph13110376
Chicago/Turabian StyleCuffaro, Doretta, Elisa Nuti, Felicia D’Andrea, and Armando Rossello. 2020. "Developments in Carbohydrate-Based Metzincin Inhibitors" Pharmaceuticals 13, no. 11: 376. https://doi.org/10.3390/ph13110376
APA StyleCuffaro, D., Nuti, E., D’Andrea, F., & Rossello, A. (2020). Developments in Carbohydrate-Based Metzincin Inhibitors. Pharmaceuticals, 13(11), 376. https://doi.org/10.3390/ph13110376