The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors


Abstract
1. Introduction
2. Regulation of MMP Activity
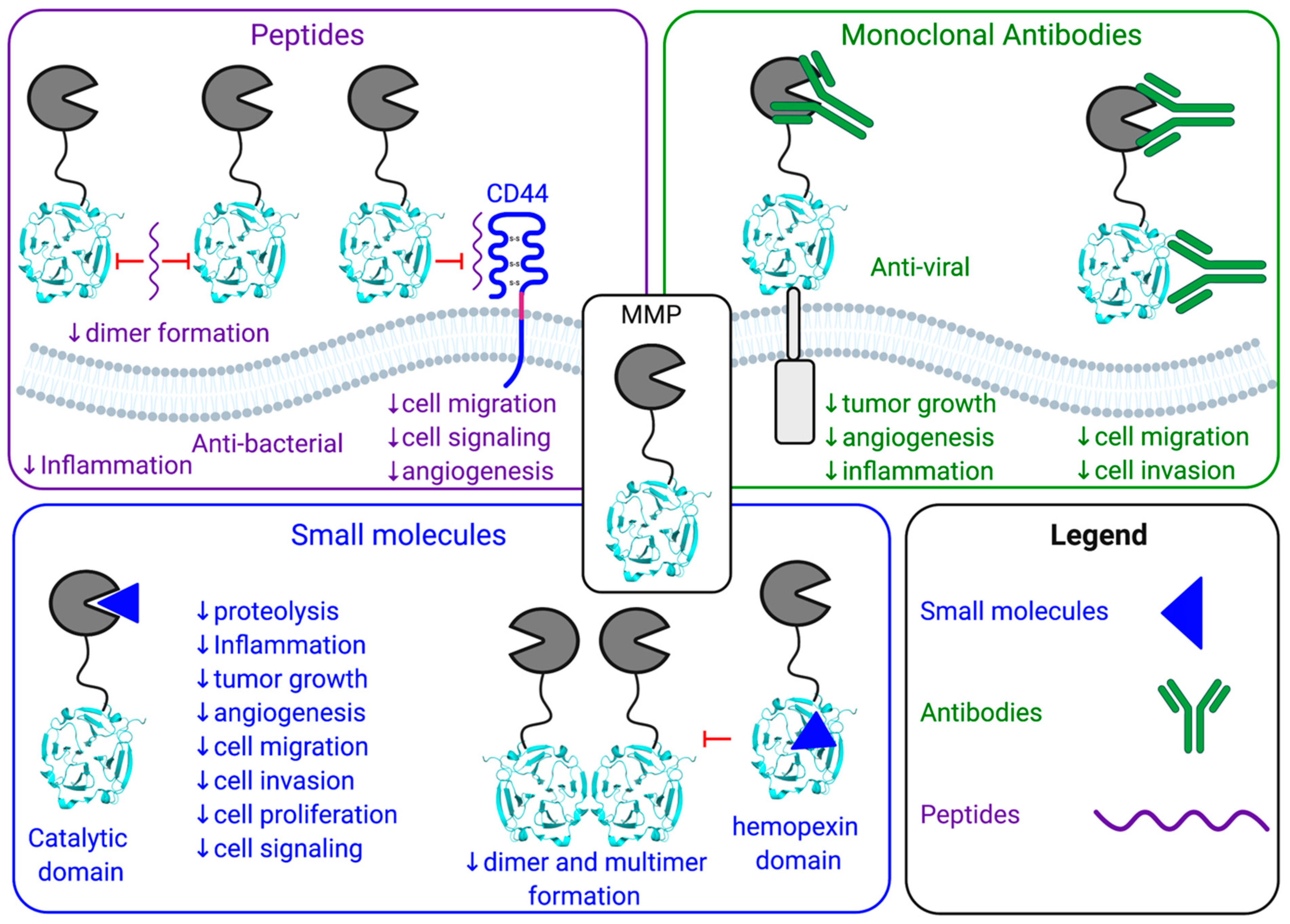
3. Non-Proteolytic Functions of MMPs
4. Strategies for the Development of Protease Inhibitors
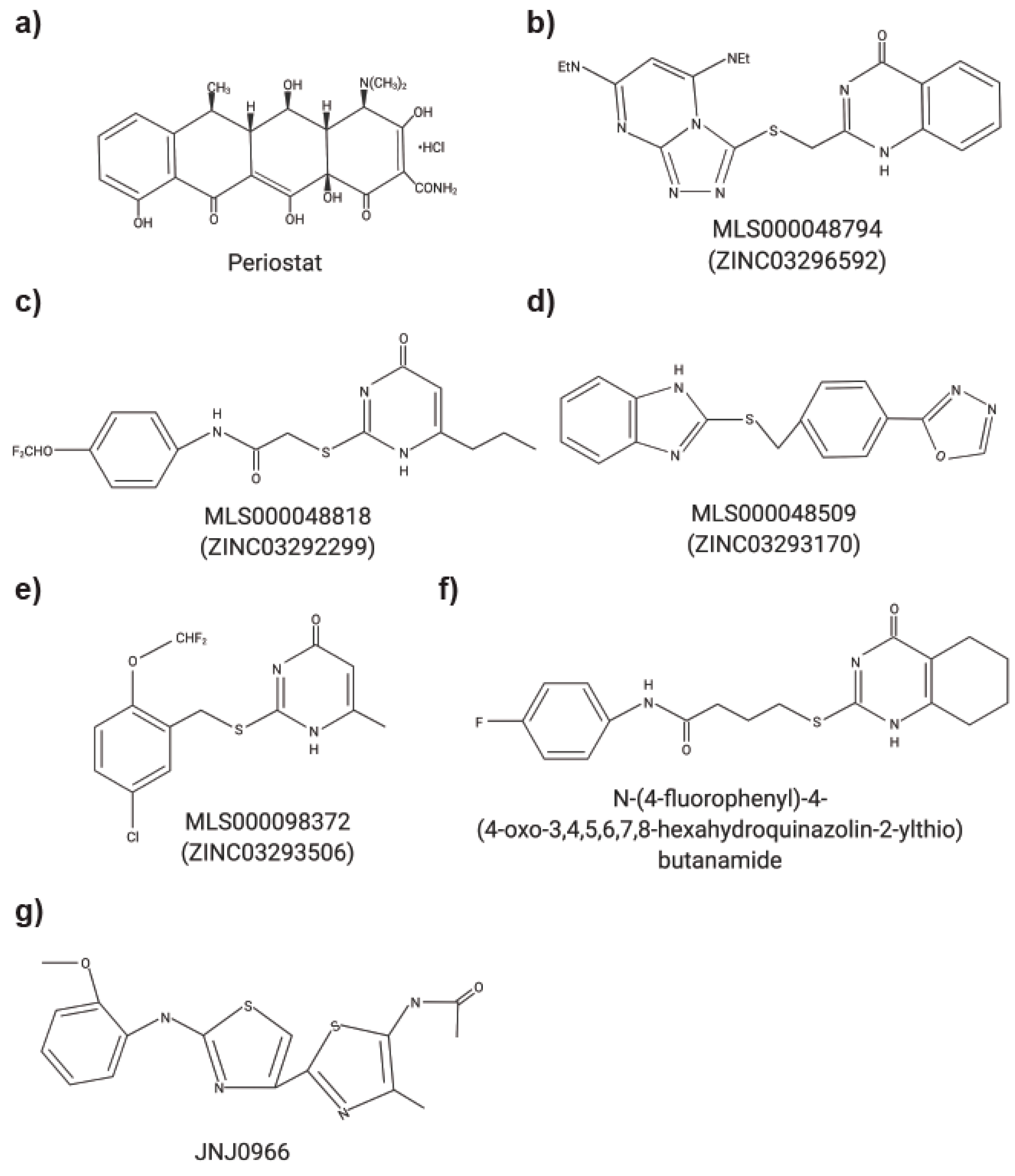
5. Small Molecule MMP Inhibitors
6. MMP Inhibition Using Selective Antibody
7. MMP Substrates Extend Beyond Matrix Proteins
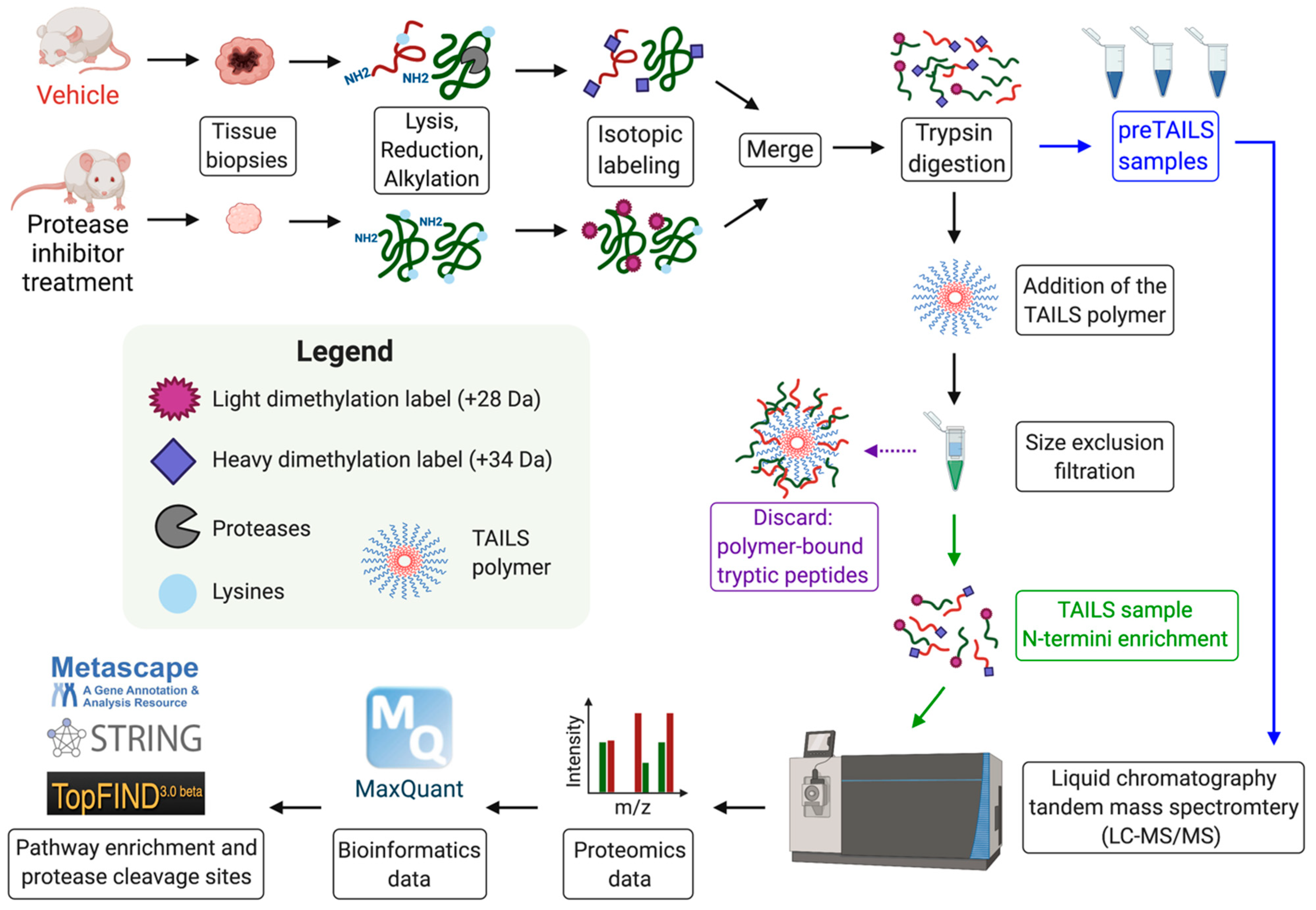
8. Identification of Novel MMP Substrates Using N-Terminomics/TAILS
9. Conclusions and Perspective: Next Generation of MMP Inhibitors
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Young, D.; Das, N.; Anowai, A.; Dufor, A. Matrix Metalloproteases as Influencers of the Cells’ Social Media. Int. J. Mol. Sci. 2019, 20, 3847. [Google Scholar] [CrossRef]
- Dufour, A.; Overall, C.M. Subtracting Matrix Out of the Equation: New Key Roles of Matrix Metalloproteinases in Innate Immunity and Disease. In Matrix Metalloproteinase Biology, 1st ed.; Sagi, I., Gaffney, J., Eds.; John Wiley & Sons, Inc.: Franklin Township, NJ, USA, 2015; pp. 131–152. [Google Scholar] [CrossRef]
- Dufour, A.; Overall, C.M. Missing the target: Matrix metalloproteinase antitargets in inflammation and cancer. Trends Pharmacol. Sci. 2013, 34, 233–242. [Google Scholar] [CrossRef]
- Mainoli, B.; Hirota, S.; Edgington-Mitchell, L.E. Proteomics and Imaging in Crohn’s Disease: TAILS of Unlikely Allies. Trends Pharmacol. Sci. 2020, 41, 74–84. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Validating matrix metalloproteinases as drug targets and anti-targets for cancer therapy. Nat. Rev. Cancer 2006, 6, 227–239. [Google Scholar] [CrossRef]
- Hunninghake, G.M.; Cho, M.H.; Tesfaigzi, Y.; Soto-Quiros, M.E.; Avila, L.; Lasky-Su, J.; Stidley, C.; Melén, E.; Söderhäll, C.; Hallberg, J.; et al. MMP12, lung function, and COPD in high-risk populations. N. Engl. J. Med. 2009, 361, 2599–2608. [Google Scholar] [CrossRef]
- Mallia-Milanes, B.; Dufour, A.; Philp, C.; Solis, N.; Klein, T.; Fischer, M.; Bolton, C.E.; Shapiro, S.; Overall, C.M.; Johnson, S.R. TAILS proteomics reveals dynamic changes in airway proteolysis controlling protease activity and innate immunity during COPD exacerbations. Am. J. Physiol. Cell. Mol. Physiol. 2018, 315, L1003–L1014. [Google Scholar] [CrossRef]
- Murphy, G.; Nagase, H. Reappraising metalloproteinases in rheumatoid arthritis and osteoarthritis: Destruction or repair? Nat. Clin. Pract. Rheumatol. 2008, 4, 128–135. [Google Scholar] [CrossRef]
- Checchi, V.; Maravic, T.; Bellini, P.; Generali, L.; Consolo, U.; Breschi, L.; Mazzoni, A. The role of matrix metalloproteinases in periodontal disease. Int. J. Environ. Res. Public Health 2020, 17, 4923. [Google Scholar] [CrossRef]
- Franco, C.; Patricia, H.-R.; Timo, S.; Claudia, B.; Marcela, H. Matrix metalloproteinases as regulators of periodontal inflammation. Int. J. Mol. Sci. 2017, 18, 440. [Google Scholar] [CrossRef]
- Chopra, S.; Overall, C.M.; Dufour, A. Matrix metalloproteinases in the CNS: Interferons get nervous. Cell. Mol. Life Sci. 2019, 76, 3083–3095. [Google Scholar] [CrossRef]
- Moore, C.S.; Crocker, S.J. An alternate perspective on the roles of TIMPs and MMPs in pathology. Am. J. Pathol. 2012, 180, 12–16. [Google Scholar] [CrossRef] [PubMed]
- Crocker, S.J.; Pagenstecher, A.; Campbell, I.L. The TIMPs tango with MMPs and more in the central nervous system. J. Neurosci. Res. 2004, 75, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Fischer, T.; Riedl, R. Challenges with matrix metalloproteinase inhibition and future drug discovery avenues. Expert Opin. Drug Discov. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- de Bruyn, M.; Vandooren, J.; Ugarte-Berzal, E.; Arijs, I.; Vermeire, S.; Opdenakker, G. The molecular biology of matrix metalloproteinases and tissue inhibitors of metalloproteinases in inflammatory bowel diseases. Crit. Rev. Biochem. Mol. Biol. 2016, 51, 295–358. [Google Scholar] [CrossRef]
- Dufour, A.; Zucker, S.; Sampson, N.S.; Kuscu, C.; Cao, J. Role of matrix metalloproteinase-9 dimers in cell migration design of inhibitory peptides. J. Biol. Chem. 2010, 285, 35944–35956. [Google Scholar] [CrossRef]
- Alford, V.M.; Kamat, A.; Ren, X.; Kumar, K.; Gan, Q.; Awwa, M.; Tong, M.; Seeliger, M.A.; Cao, J.; Ojima, I.; et al. Targeting the hemopexin-like domain of latent matrix metalloproteinase-9 (proMMP-9) with a small molecule inhibitor prevents the formation of focal adhesion junctions. ACS Chem. Biol. 2017, 12, 2788–2803. [Google Scholar] [CrossRef]
- Houghton, A.M.; Hartzell, W.O.; Robbins, C.S.; Gomis-Rüth, F.X.; Shapiro, S.D. Macrophage elastase kills bacteria within murine macrophages. Nature 2009, 460, 637–641. [Google Scholar] [CrossRef]
- Manka, S.W.; Carafoli, R.; Visse, R.; Bihan, D.; Raynal, N.; Farndale, R.W.; Murphy, G.; Enghild, J.J.; Hohenester, E.; Nagase, H. Structural insights into triple-helical collagen cleavage by matrix metalloproteinase 1. Proc. Natl. Acad. Sci. USA 2012, 109, 12461–12466. [Google Scholar] [CrossRef]
- Vandenbroucke, R.E.; Libert, C. Is there new hope for therapeutic matrix metalloproteinase inhibition? Nat. Rev. Drug Discov. 2014, 13, 904–927. [Google Scholar] [CrossRef]
- Cha, H.; Kopetzki, E.; Huber, R.; Lanzendörfer, M.; Brandstetter, H. Structural basis of the adaptive molecular recognition by MMP9. J. Mol. Biol. 2002, 320, 1065–1079. [Google Scholar] [CrossRef]
- Hariono, M.; Nuwarda, R.F.; Yusuf, M.; Rollando, R.; Jenie, R.I.; Al-Najjar, B.; Julianus, J.; Putra, K.C.; Nugroho, E.S.; Wisnumurti, Y.K.; et al. Arylamide as Potential Selective Inhibitor for Matrix Metalloproteinase 9 (MMP9): Design, Synthesis, Biological Evaluation, and Molecular Modeling. J. Chem. Inf. Model. 2019, 60, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Feinberg, T.Y.; Zheng, H.; Lui, R.; Wicha, M.S.; Yu, S.M.; Weiss, S.J. Divergent matrix-remodeling strategies distinguish developmental from neoplastic mammary epithelial cell invasion programs. Dev. Cell 2018, 47, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Remacle, A.G.; Golubkov, V.S.; Shiryaev, S.A.; Dahl, R.; Stebbins, J.L.; Chernov, A.V.; Cheltsov, A.V.; Pellecchia, M.; Strongin, A.Y. Novel MT1-MMP small-molecule inhibitors based on insights into hemopexin domain function in tumor growth. Cancer Res. 2012, 72, 2339–2349. [Google Scholar] [CrossRef] [PubMed]
- Zarrabi, K.; Dufour, A.; Li, J.; Kuscu, C.; Pulkoski-Gross, A.; Zhi, J.; Hu, Y.; Sampson, N.S.; Zucker, S.; Cao, J. Inhibition of matrix metalloproteinase 14 (MMP-14)-mediated cancer cell migration. J. Biol. Chem. 2011, 286, 33167–33177. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Li, S.; Zhang, J. Harnessing allostery: A novel approach to drug discovery. Med. Res. Rev. 2014, 34, 1242–1285. [Google Scholar] [CrossRef]
- Levin, M.; Udi, Y.; Solomonov, I.; Siga, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. Biochim. Biophys. Acta (BBA) Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef]
- Overall, C.M.; Kleifeld, O. Towards third generation matrix metalloproteinase inhibitors for cancer therapy. Br. J. Cancer 2006, 94, 941–946. [Google Scholar] [CrossRef]
- Scannevin, R.H.; Alexander, R.; Haarlander, T.M.; Burke, S.L.; Singer, M.; Huo, C.; Zhang, Y.M.; Maguire, D.; Spurlino, J.; Deckman, I.; et al. Discovery of a highly selective chemical inhibitor of matrix metalloproteinase-9 (MMP-9) that allosterically inhibits zymogen activation. J. Biol. Chem. 2017, 292, 17963–17974. [Google Scholar] [CrossRef]
- Fields, G.B. The rebirth of matrix metalloproteinase inhibitors: Moving beyond the dogma. Cells 2019, 8, 984. [Google Scholar] [CrossRef]
- Van Wart, H.E.; Birkedal-Hansen, H. The cysteine switch: A principle of regulation of metalloproteinase activity with potential applicability to the entire matrix metalloproteinase gene family. Proc. Natl. Acad. Sci. USA 1990, 87, 5578–5582. [Google Scholar] [CrossRef]
- Wetmore, D.R.; Hardman, K.D. Roles of the propeptide and metal ions in the folding and stability of the catalytic domain of stromelysin (matrix metalloproteinase 3). Biochemistry 1996, 35, 6549–6558. [Google Scholar] [CrossRef] [PubMed]
- Pirard, B. Insight into the structural determinants for selective inhibition of matrix metalloproteinases. Drug Discov. Today 2007, 12, 640–646. [Google Scholar] [CrossRef] [PubMed]
- Coussens, L.M.; Fingleton, B.; Matrisian, L.M. Matrix metalloproteinase inhibitors and cancer—Trials and tribulations. Science 2002, 295, 2387–2392. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef]
- Hande, K.R.; Collier, M.; Paradiso, L.; Stuart-Smith, J.; Dixon, M.; Clendeninn, N.; Yeun, G.; Alberti, D.; Binger, K.; Wilding, G. Phase I and pharmacokinetic study of prinomastat, a matrix metalloprotease inhibitor. Clin. Cancer Res. 2004, 10, 909–915. [Google Scholar] [CrossRef]
- Leighl, N.B.; Paz-Ares, L.; Douillard, J.-Y.; Peschel, C.; Arnold, A.; Depierre, A.; Santoro, A.; Betticher, D.C.; Gatzemeier, U.; Jassem, J.; et al. Randomized phase III study of matrix metalloproteinase inhibitor BMS-275291 in combination with paclitaxel and carboplatin in advanced non-small-cell lung cancer: National Cancer Institute of Canada-Clinical Trials Group Study BR. 18. J. Clin. Oncol. 2005, 23, 2831–2839. [Google Scholar] [CrossRef]
- Douillard, J.-Y.; Peschel, C.; Shepherd, F.; Paz-Ares, L.; Arnold, A.; Davis, M.; Tonato, M.; Smylie, M.; Tu, D.; Voi, M.; et al. Randomized phase II feasibility study of combining the matrix metalloproteinase inhibitor BMS-275291 with paclitaxel plus carboplatin in advanced non-small cell lung cancer. Lung Cancer 2004, 46, 361–368. [Google Scholar] [CrossRef]
- Lu, C.; Lee, J.; Komaki, R.; Herbst, R.S.; Feng, L.; Evans, W.K.; Choy, H.; Desjardins, P.; Esparaz, B.T.; Truong, M.T.; et al. Chemoradiotherapy with or without Ae-941 in Stage III Non–Small cell lung cancer: A randomized Phase III trial. JNCI J. Natl. Cancer Inst. C 2020, 102, 859–865. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Schulz, J.; Nemunaitis, J.; Brown, P.D.; Baillet, M.; Buckels, J.A. A double-blind placebo-controlled, randomised study comparing gemcitabine and marimastat with gemcitabine and placebo as first line therapy in patients with advanced pancreatic cancer. Br. J. Cancer 2002, 87, 161–167. [Google Scholar] [CrossRef]
- Preshaw, P.M.; Hefti, A.F.; Novak, M.J.; Michalowicz, B.S.; Pihlstrom, B.L.; Schoor, R.; Trummel, C.L.; Dean, J.; Van Dyke, T.E.; Walker, C.B.; et al. Subantimicrobial dose doxycycline enhances the efficacy of scaling and root planing in chronic periodontitis: A multicenter trial. J. Periodontol. 2004, 75, 1068–1076. [Google Scholar] [CrossRef]
- Peterson, J.T. Matrix metalloproteinase inhibitor development and the remodeling of drug discovery. Heart Fail. Rev. 2004, 9, 63–79. [Google Scholar] [CrossRef] [PubMed]
- Golub, L.M.; McNamara, T.F.; Ryan, M.E.; Kohut, B.; Blieden, T.; Payonk, G.; Sipos, T.; Baron, H.J. Adjunctive treatment with subantimicrobial doses of doxycycline: Effects on gingival fluid collagenase activity and attachment loss in adult periodontitis. J. Clin. Periodontol. 2001, 28, 146–156. [Google Scholar] [CrossRef] [PubMed]
- Baker, P.J.; Evans, R.T.; Coburn, R.A.; Genco, R.J. Tetracycline and its derivatives strongly bind to and are released from the tooth surface in active form. J. Periodontol. 1983, 54, 580–585. [Google Scholar] [CrossRef] [PubMed]
- Payne, J.B.; Stoner, J.A.; Nummikoski, P.V.; Reinhardt, R.A.; Goren, A.D.; Wolff, M.S.; Lee, H.M.; Lynch, J.C.; Valente, R.; Golub, L.M. Subantimicrobial dose doxycycline effects on alveolar bone loss in post-menopausal women. J. Clin. Periodontol. 2007, 34, 776–787. [Google Scholar] [CrossRef] [PubMed]
- Minagar, A.; Alexander, J.S.; Schwendimann, R.N.; Kelley, R.E.; Gonzalez-Toledo, E.; Jimenez, J.J.; Mauro, L.; Jy, W.; Smith, S.J. Combination therapy with interferon beta-1a and doxycycline in multiple sclerosis: An open-label trial. Arch. Neurol. 2008, 65, 199–204. [Google Scholar] [CrossRef]
- Frankwich, K.; Tibble, C.; Torres-Gonzalez, M.; Bonner, M.; Lefkowitz, R.; Tyndall, M.; Schmid-Schönbein, G.W.; Villarreal, F.; Heller, M.; Herbst, K. Proof of Concept: Matrix metalloproteinase inhibitor decreases inflammation and improves muscle insulin sensitivity in people with type 2 diabetes. J. Inflamm. 2012, 9, 35. [Google Scholar] [CrossRef]
- Talmi-Frank, D.; Altboum, Z.; Solomonov, I.; Udi, Y.; Jaitin, D.A.; Klepfish, M.; David, E.; Zhuravlev, A.; Keren-Shaul, H.; Winter, D.R.; et al. Extracellular matrix proteolysis by MT1-MMP contributes to influenza-related tissue damage and mortality. Cell Host Microbe 2016, 20, 458–470. [Google Scholar] [CrossRef]
- Marchant, D.J.; Bellac, C.L.; Moraes, T.J.; Wadsworth, S.J.; Dufour, A.; Butler, G.S.; Bilawchuk, L.M.; Hendry, R.G.; Robertson, A.G.; Cheung, C.T.; et al. A new transcriptional role for matrix metalloproteinase-12 in antiviral immunity. Nat. Med. 2014, 20, 493–502. [Google Scholar] [CrossRef]
- Dufour, A.; Sampson, N.S.; Li, J.; Kuscu, C.; Rizzo, R.C.; Deleon, J.L.; Zhi, J.; Jaber, N.; Liu, E.; Zucker, S.; et al. Small-molecule anticancer compounds selectively target the hemopexin domain of matrix metalloproteinase-9. Cancer Res. 2011, 71, 4877–4888. [Google Scholar] [CrossRef]
- Lipsky, P.E.; van der Heijde, D.M.; Clair, E.W.S.; Furst, D.E.; Breedveld, F.C.; Kalden, J.R.; Smolen, J.S.; Weisman, M.; Emery, P.; Feldmann, M.; et al. Anti-Tumor Necrosis Factor Trial in Rheumatoid Arthritis with Concomitant Therapy Study Group. Infliximab and methotrexate in the treatment of rheumatoid arthritis. N. Engl. J. Med. 2000, 343, 1594–1602. [Google Scholar] [CrossRef]
- Starnes, H.; Pearce, M.K.; Tewari, A.; Yim, J.H.; Zou, J.C.; Abrams, J.S. Anti-IL-6 monoclonal antibodies protect against lethal Escherichia coli infection and lethal tumor necrosis factor-alpha challenge in mice. J. Immunol. 1990, 145, 4185–4191. [Google Scholar] [PubMed]
- Hudziak, R.M.; Lewis, G.D.; Winget, M.; Fendly, B.M.; Shepard, H.M.; Ullrich, A. p185HER2 monoclonal antibody has antiproliferative effects in vitro and sensitizes human breast tumor cells to tumor necrosis factor. Mol. Cell. Biol. 1989, 9, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Milgrom, H.; Fick, R.B.; Su, J.Q.; Reimann, J.D.; Bush, R.K.; Watrous, M.L.; Metzger, W.J. Treatment of allergic asthma with monoclonal anti-IgE antibody. N. Engl. J. Med. 1999, 341, 1966–1973. [Google Scholar] [CrossRef] [PubMed]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143–147. [Google Scholar] [CrossRef]
- Martens, E.; Leyssen, A.; Aelst, I.V.; Fiten, P.; Piccard, H.; Hu, J.; Descamps, F.J.; van den Steen, P.E.; Proost, P.; van Damme, J.; et al. A monoclonal antibody inhibits gelatinase B/MMP-9 by selective binding to part of the catalytic domain and not to the fibronectin or zinc binding domains. Biochim. Biophys. Acta (BBA) Gen. Subj. 2007, 1770, 178–186. [Google Scholar] [CrossRef]
- Paemen, L.; Martens, E.; Masure, S.; Opdenakker, G. Monoclonal antibodies specific for natural human neutrophil gelatinase B used for affinity purification, quantitation by two-site ELISA and inhibition of enzymatic activity. Eur. J. Biochem. 1995, 234, 759–765. [Google Scholar] [CrossRef]
- Devy, L.; Huang, L.; Naa, L.; Yanamandra, N.; Pieters, H.; Frans, N.; Chang, E.; Tao, Q.; Vanhove, M.; Lejeune, A.; et al. Selective inhibition of matrix metalloproteinase-14 blocks tumor growth, invasion, and angiogenesis. Cancer Res. 2009, 69, 1517–1526. [Google Scholar] [CrossRef]
- Udi, Y.; Grossman, M.; Solomonov, I.; Dym, O.; Rozenberg, H.; Moreno, V.; Cuniasse, P.; Dive, V.; Arroyo, A.G.; Sagi, I. Inhibition mechanism of membrane metalloprotease by an exosite-swiveling conformational antibody. Structure 2015, 23, 104–115. [Google Scholar] [CrossRef]
- Dean, R.A.; Cox, J.H.; Bellac, C.L.; Doucet, A.; Starr, A.E.; Overal, C.M. Macrophage-specific metalloelastase (MMP-12) truncates and inactivates ELR + CXC chemokines and generates CCL2, -7, -8, and -13 antagonists: Potential role of the macrophage in terminating polymorphonuclear leukocyte influx. Blood 2008, 112, 3455–3464. [Google Scholar] [CrossRef]
- McQuibban, G.A.; Gong, J.-H.; Tam, E.M.; McCulloch, C.A.; Clark-Lewis, I.; Overall, C.M. Inflammation dampened by gelatinase a cleavage of monocyte chemoattractant protein-3. Science 2000, 289, 1202–1206. [Google Scholar] [CrossRef]
- Starr, A.E.; Dufour, A.; Maier, J.; Overall, C.M. Biochemical analysis of matrix metalloproteinase activation of chemokines CCL15 and CCL23 and increased glycosaminoglycan binding of CCL16. J. Biol. Chem. 2012, 287, 5848–5860. [Google Scholar] [CrossRef] [PubMed]
- Dufour, A.; Bellac, C.L.; Eckhard, U.; Solis, N.; Klein, T.; Kappelhoff, R.; Fortelny, N.; Jobin, P.; Rozmus, J.; Mark, J.; et al. C-terminal truncation of IFN-γ inhibits proinflammatory macrophage responses and is deficient in autoimmune disease. Nat. Commun. 2018, 9, 2416. [Google Scholar] [CrossRef] [PubMed]
- McQuibban, G.A.; Gong, J.-H.; Wong, J.P.; Wallace, J.L.; Clark-Lewis, I.; Overall, C.M. Matrix metalloproteinase processing of monocyte chemoattractant proteins generates CC chemokine receptor antagonists with anti-inflammatory properties in vivo. Blood J. Am. Soc. Hematol. 2002, 100, 1160–1167. [Google Scholar] [CrossRef]
- Corry, D.B.; Kiss, A.; Song, L.-Z.; Song, L.; Xu, J.; Lee, S.; Werb, Z.; Kheradmand, F. Overlapping and independent contributions of MMP2 and MMP9 to lung allergic inflammatory cell egression through decreased CC chemokines. FASEB J. 2004, 18, 995–997. [Google Scholar] [CrossRef]
- Kleifeld, O.; Doucet, A.; auf dem Keller, U.; Prudova, A.; Schilling, O.; Kainthan, R.K.; Starr, A.E.; Foster, L.J.; Kizhakkedathu, J.N.; Overall, C.M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010, 28, 281–288. [Google Scholar] [CrossRef]
- Prudova, A.; Gocheva, V.; auf dem Keller, U.; Eckhard, U.; Olson, O.C.; Akkari, L.; Butler, G.S.; Fortelny, N.; Lange, P.F.; Mark, J.C.; et al. TAILS N-terminomics and proteomics show protein degradation dominates over proteolytic processing by cathepsins in pancreatic tumors. Cell Rep. 2016, 16, 1762–1773. [Google Scholar] [CrossRef]
- Kleifeld, O.; Doucet, A.; Prudova, A.; Keller, U.a.d.; Gioia, M.; Kizhakkedathu, J.N.; Overall, C.M. Identifying and quantifying proteolytic events and the natural N terminome by terminal amine isotopic labeling of substrates. Nat. Protoc. 2011, 6, 1578–1611. [Google Scholar] [CrossRef]
- Wold, F. In vivo chemical modification of proteins (post-translational modification). Annu. Rev. Biochem. 1981, 50, 783–814. [Google Scholar] [CrossRef]
- Biancur, D.E.; Paulo, J.A.; Matachowska, B.; del Rey, M.Q.; Sousa, C.M.; Wang, X.; Sohn, A.S.W.; Chu, G.C.; Gygi, S.P.; Harper, J.W.; et al. Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Bellac, C.L.; Dufour, A.; Krisinger, J.M.; Loonchanta, A.; Starr, A.E.; Keller, U.A.d.; Lange, P.F.; Goebeler, V.; Kappelhoff, R.; Butler, G.S.; et al. Macrophage matrix metalloproteinase-12 dampens inflammation and neutrophil influx in arthritis. Cell Rep. 2014, 9, 618–663. [Google Scholar] [CrossRef]
- Starr, A.E.; Bellac, C.L.; Dufour, A.; Goebeler, V.; Overall, C.M. Biochemical characterization and N-terminomics analysis of leukolysin, the membrane-type 6 matrix metalloprotease (MMP25) chemokine and vimentin cleavages enhance cell migration and macrophage phagocytic activities. J. Biol. Chem. 2012, 287, 13382–13395. [Google Scholar] [CrossRef] [PubMed]
- Schlage, P.; Egli, F.E.; Nanni, P.; Wang, L.W.; Kizhakkedathu, J.N.; Apte, S.S.; Keller, U.a.d. Time-resolved analysis of the matrix metalloproteinase 10 substrate degradome. Mol. Cell. Proteom. 2014, 13, 580–593. [Google Scholar] [CrossRef] [PubMed]
- auf dem Keller, U.; Bellac, C.L.; Li, Y.; Lou, Y.; Lange, P.F.; Ting, R.; Harwig, C.; Kappelhoff, R.; Dedhar, S.; Adam, M.J.; et al. Novel matrix metalloproteinase inhibitor [18F] marimastat-aryltrifluoroborate as a probe for in vivo positron emission tomography imaging in cancer. Cancer Res. 2010, 70, 7562–7569. [Google Scholar] [CrossRef] [PubMed]
- Prudova, A.; auf dem Keller, U.; Butler, G.S.; Overall, C.M. Multiplex N-terminome analysis of MMP-2 and MMP-9 substrate degradomes by iTRAQ-TAILS quantitative proteomics. Mol. Cell. Proteom. 2010, 9, 894–911. [Google Scholar] [CrossRef]
- Butler, G.S.; Overall, C.M. Proteomic identification of multitasking proteins in unexpected locations complicates drug targeting. Nat. Rev. Drug Discov. 2009, 8, 935–948. [Google Scholar] [CrossRef]
- Hanifeh, M.; Rajamaki, M.M.; Makitalo, L.; Syrjä, P.; Sankari, S.; Kilpinen, S.; Spillmann, T. Identification of matrix metalloproteinase-2 and-9 activities within the intestinal mucosa of dogs with chronic enteropathies. Acta Vet. Scand. 2018, 60, 16. [Google Scholar] [CrossRef]
- Moesgaard, S.G.; Aupperle, H.; Rajamaki, M.M.; Falk, T.; Rasmussen, C.E.; Zois, N.E.; Olsen, L.H. Matrix metalloproteinases (MMPs), tissue inhibitors of metalloproteinases (TIMPs) and transforming growth factor-β (TGF-β) in advanced canine myxomatous mitral valve disease. Res. Vet. Sci. 2014, 97, 560–567. [Google Scholar] [CrossRef]
- Qiu, Z.; Hu, J.; Van den Steen, P.E.; Opdenakker, G. Targeting matrix metalloproteinases in acute inflammatory shock syndromes. Comb. Chem. High Throughput Screen. 2012, 15, 555–570. [Google Scholar] [CrossRef]
- Xia, L.; Zhu, Z.; Zhang, L.; Xu, Y.; Chen, G.; Luo, J. EZH2 enhances expression of CCL5 to promote recruitment of macrophages and invasion in lung cancer. Biotechnol. Appl. Biochem. 2019. [Google Scholar] [CrossRef]
- Wojtowicz-Praga, S.; Low, J.; Marshall, J.; Ness, E.; Dickson, R.; Barter, J.; Sale, M.; McCann, P.; Moore, J.; Cole, A.; et al. Phase I trial of a novel matrix metalloproteinase inhibitor batimastat (BB-94) in patients with advanced cancer. Invest. New Drugs 1996, 14, 193–202. [Google Scholar] [CrossRef]
- Nemunaitis, J.; Poole, C.; Primrose, J.; Rosemurgy, A.; Malfetano, J.; Brown, P.; Berrington, A.; Cornish, A.; Lynch, K.; Rasmussen, H.; et al. Combined analysis of studies of the effects of the matrix metalloproteinase inhibitor marimastat on serum tumor markers in advanced cancer: Selection of a biologically active and tolerable dose for longer-term studies. Clin. Cancer Res. 1998, 4, 1101–1109. [Google Scholar] [PubMed]
- Bramhall, S.R.; Rosemurgy, A.; Brwon, P.D.; Bowry, C.; Buckels, J.A.; Marimastat Pancreatic Cancer Study Group. Marimastat as first-line therapy for patients with unresectable pancreatic cancer: A randomized trial. J. Clin. Oncol. 2001, 19, 3447–3455. [Google Scholar] [CrossRef]
- Bramhall, S.R.; Hallissey, M.T.; Whiting, J.; Scholefield, J.; Tierney, G.; Stuart, R.C.; Hawkins, R.E.; McCulloch, P.; Maughan, T.; Brown, P.D.; et al. Marimastat as maintenance therapy for patients with advanced gastric cancer: A randomised trial. Br. J. Cancer 2002, 86, 1864–1870. [Google Scholar] [CrossRef] [PubMed]
- Sparano, J.A.; Bernardo, P.; Stephenson, P.; Gradishar, W.J.; Ingle, J.N.; Zucker, S.; Davidson, N.E. Randomized phase III trial of marimastat versus placebo in patients with metastatic breast cancer who have responding or stable disease after first-line chemotherapy: Eastern Cooperative Oncology Group trial E2196. J. Clin. Oncol. 2004, 22, 4683–4690. [Google Scholar] [CrossRef]
- Levitt, N.C.; Eskens, F.A.L.M.; O’Byrne, K.J.; Propper, D.J.; Denis, L.J.; Owen, S.J.; Choi, L.; Foekens, J.A.; Wilner, S.; Wood, J.M.; et al. Phase I and pharmacological study of the oral matrix metalloproteinase inhibitor, MMI270 (CGS27023A), in patients with advanced solid cancer. Clin. Cancer Res. 2001, 7, 1912–1922. [Google Scholar] [PubMed]
- Bissett, D.; O’Byrne, K.J.; von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III study of matrix metalloproteinase inhibitor prinomastat in non-small-cell lung cancer. J. Clin. Oncol. 2004, 23, 842–849. [Google Scholar] [CrossRef]
- Heath, E.I.; Burtness, B.A.; Kleinberg, L.; Salem, R.R.; Yang, S.C.; Heitmiller, R.F.; Canto, M.I.; Knisely, J.P.S.; Topazian, M.; Montgomery, E.; et al. Phase II, parallel-design study of preoperative combined modality therapy and the matrix metalloprotease (mmp) inhibitor prinomastat in patients with esophageal adenocarcinoma. Invest. New Drugs 2006, 24, 135–140. [Google Scholar] [CrossRef]
- Erlichman, C.; Adjei, A.A.; Alberts, S.R.; Sloan, J.A.; Goldberg, R.M.; Pitot, H.C.; Rubin, J.; Atherton, P.J.; Klee, G.G.; Humphrey, R. Phase I study of the matrix metal loproteinase inhibitor, BAY 12–9566. Ann. Oncol. 2001, 12, 389–395. [Google Scholar] [CrossRef]
- Moore, M.J.; Hamm, J.; Dancey, J.; Eisenberg, P.D.; Dagenais, M.; Fields, A.; Hagan, K.; Greenberg, B.; Colwell, B.; Zee, B.; et al. Comparison of gemcitabine versus the matrix metalloproteinase inhibitor BAY 12-9566 in patients with advanced or metastatic adenocarcinoma of the pancreas: A phase III trial of the National Cancer Institute of Canada Clinical Trials Group. J. Clin. Oncol. 2003, 21, 3296–3302. [Google Scholar] [CrossRef]
- Hirte, H.; Vergote, I.B.; Jeffrey, J.R.; Grimshaw, R.N.; Coppieters, S.; Schwartz, B.; Tu, D.; Sadura, A.; Brundage, M.; Seymour, L. A phase III randomized trial of BAY 12-9566 (tanomastat) as maintenance therapy in patients with advanced ovarian cancer responsive to primary surgery and paclitaxel/platinum containing chemotherapy: A National Cancer Institute of Canada Clinical Trials Group Study. Gynecol. Oncol. 2006, 102, 300–308. [Google Scholar] [CrossRef]
- Rudek, M.A.; Figg, W.D.; Dyer, V.; Dahut, W.; Turner, M.L.; Steinberg, S.M.; Liewehr, D.J.; Kohler, D.R.; Pluda, J.M.; Reed, E. Phase I clinical trial of oral COL-3, a matrix metalloproteinase inhibitor, in patients with refractory metastatic cancer. J. Clin. Oncol. 2001, 19, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Cianfrocca, M.; Cooley, T.P.; Lee, J.Y.; Rudek, M.A.; Scadden, D.T.; Ratner, L.; Pluda, J.M.; Figg, W.D.; Krown, S.E.; Dezube, B.J. Matrix metalloproteinase inhibitor COL-3 in the treatment of AIDS-related Kaposi’s sarcoma: A phase I AIDS malignancy consortium study. J. Clin. Oncol. 2001, 20, 153–159. [Google Scholar]
- Chu, Q.S.C.; Forouzesh, B.; Syed, S.; Mita, M.; Schwartz, G.; Cooper, J.; Curtright, J.; Rowinsky, E.K. A phase II and pharmacological study of the matrix metalloproteinase inhibitor (MMPI) COL-3 in patients with advanced soft tissue sarcomas. Invest. New Drugs 2007, 25, 359. [Google Scholar] [CrossRef] [PubMed]
- Baxter, B.T.; Pearce, W.H.; Waltke, E.A.; Littooy, F.N.; Hallett, J.W., Jr.; Kent, K.C.; Upchurch, G.R., Jr.; Chaikof, E.L.; Mills, J.L.; Fleckten, B.; et al. Prolonged administration of doxycycline in patients with small asymptomatic abdominal aortic aneurysms: Report of a prospective (Phase II) multicenter study. J. Vasc. Surg. 2002, 36, 1–12. [Google Scholar] [CrossRef]
- Rizvi, N.A.; Humphrey, J.S.; Ness, E.A.; Johnson, M.D.; Gupta, E.; Williams, K.; Daly, D.J.; Sonnichsen, D.; Conway, D.; Marshall, J.; et al. A phase I study of oral BMS-275291, a novel nonhydroxamate sheddase-sparing matrix metalloproteinase inhibitor, in patients with advanced or metastatic cancer. Clin. Cancer Res. 2004, 10, 1963–1970. [Google Scholar] [CrossRef]
- Miller, K.D.; Saphner, R.J.; Waterhouse, D.M.; Chen, T.-T.; Rush-Taylor, A.; Sparano, J.A.; Wolff, A.C.; Cobleigh, M.A.; Galbraith, S.; Sledge, G.W. A randomized phase II feasibility trial of BMS-275291 in patients with early stage breast cancer. Clin. Cancer Res. 2004, 10, 1971–1975. [Google Scholar] [CrossRef]
- Chiappori, A.A.; Eckhardt, S.G.; Bukowski, R.; Sullivan, D.M.; Ikeda, M.; Yano, Y.; Yamada-Sawada, T.; Kambayashi, Y.; Tanaka, K.; Javle, M.M.; et al. A phase I pharmacokinetic and pharmacodynamic study of s-3304, a novel matrix metalloproteinase inhibitor, in patients with advanced and refractory solid tumors. Clin. Cancer Res. 2007, 13, 2091–2099. [Google Scholar] [CrossRef]
- Magnussen, H.; Watz, H.; Kirsten, A.; Wang, M.; Wray, H.; Samuelsson, V.; Mo, J.; Kay, R. Safety and tolerability of an oral MMP-9 and-12 inhibitor, AZD1236, in patients with moderate-to-severe COPD: A randomised controlled 6-week trial. Pulm. Pharmacol. Ther. 2011, 24, 563–570. [Google Scholar] [CrossRef]
- Dahl, R.; Titlestad, I.; Lindqvist, A.; Wielders, P.; Wray, H.; Wang, M.; Samuelsson, V.; Mo, J.; Holt, A. Effects of an oral MMP-9 and-12 inhibitor, AZD1236, on biomarkers in moderate/severe COPD: A randomised controlled trial. Pulm. Pharmacol. Ther. 2012, 25, 169–177. [Google Scholar] [CrossRef]
- Botkjaer, K.A.; Kwok, H.F.; Terp, M.G.; Karatt-Vellatt, A.; Santamaria, S.; McCafferty, J.; Andreasen, P.A.; Itoh, Y.; Ditzel, H.J.; Murphy, G. Development of a specific affinity-matured exosite inhibitor to MT1-MMP that efficiently inhibits tumor cell invasion in vitro and metastasis in vivo. Oncotarget 2016, 7, 16773. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Inhibitor Names | Class/Structure | Selectivity | Diseases | Clinical Trial | Outcomes/ Side Effects | References |
|---|---|---|---|---|---|---|
| Batimastat | Peptidomimetic/ Hydroxamate | Broad spectrum | Malignant tumor | Phase I | Local toxicities i.e., abdominal discomfort | [81] |
| Marimastat | Peptidomimetic/ Hydroxamate | Broad spectrum | Progressive ovarian, prostatic, pancreatic and colorectal cancer | Phase III | Adverse musculoskeletal (MS) syndrome | [82] |
| Pancreatic cancer | Phase III | Musculoskeletal pain and inflammation | [83] | |||
| Pancreatic cancer | Phase III, in combination with gemcitabine | Well tolerated but no therapeutic beneficial effects | [40] | |||
| Gastric cancer | Phase III | Severe musculoskeletal (MS) syndrome | [84] | |||
| Metastatic breast cancer | Phase I | Musculoskeletal pain associated with inferior survival | [85] | |||
| MMI-270 | Hydroxamate/Small molecule | Broad spectrum | Advanced solid cancer | Phase I | Rash and musculoskeletal pain | [86] |
| Prinomastat | Hydroxamate/Small molecule | Broad spectrum | Advanced cancer | Phase I | No response in tumor growth | [36] |
| Non-small cell lung cancer (stage IIIB or IV) | Phase III | Musculoskeletal syndrome | [87] | |||
| Esophagus cancer | Phase II | Unexpected thromboembolic events | [88] | |||
| Tanomastat (Bay 12-9566) | Biphenyl, thioether zinc-binding group/small molecule | MMP2, -3 and -9 | Solid tumor | Phase I | Mild toxicity, no musculoskeletal pain. No effect on tumor | [89] |
| Pancreatic cancer without prior chemotherapy | Phase III | Poorer survival | [90] | |||
| Ovarian cancer | Phase III | Well tolerated but did not impact patients’ survival | [91] | |||
| Metastat | Tetracycline derivatives/small molecule | MMP2 and -9 | Refractory solid tumors | Phase I | Subcutaneous phototoxicity | [92] |
| AIDS related Kaposi’s sarcoma | Phase I Applied with sun protection | Photosensitivity reaction | [93] | |||
| Advanced soft tissue sarcoma | Phase II Applied with sun protection | Photosensitivity reaction | [94] | |||
| Periostat®/ Doxycycline | Tetracycline derivatives/small molecule | Broad spectrum | Periodontitis | Phase III | Well tolerated; improved outcome | [41,43] |
| Asymptomatic abdominal aortic aneurysms | Phase II | Well tolerated; but no significant therapeutic effects | [95] | |||
| Multiple sclerosis | Phase II Along with IFNß-1a | Well tolerated; improved outcome | [46] | |||
| Type II diabetes | Phase III | Reduced inflammation and better insulin sensitivity | [47] | |||
| Rebimastat (BMS- 275291) | Mercaptoacyl, thiol zinc-binding group/small molecule | MMP1, -2, -8, and MT1-MMP | Advanced cancer | Phase I | Well tolerated; no tumor response | [96] |
| Early-stage breast cancer | Phase III | Study was terminated because of toxicity | [97] | |||
| Non-small cell lung cancer | Phase II along with paclitaxel and carboplatin | Well tolerated but poor therapeutic response | [38] | |||
| Non-small cell lung cancer | Phase II along with paclitaxel and carboplatin | Increased toxicity with no improved survival | [37] | |||
| S-3304 | Sulfonamide derivatives/small molecule | MMP2 and -9 | Advanced solid tumors | Phase I | Well tolerated | [98] |
| AZD1236 | MMP9 and -12 | Moderate to severe Chronic obstructive pulmonary disease (COPD) | Phase II | Well tolerated but no therapeutic efficacy | [99,100] | |
| Neovastat (AE-941) | Mixed extract from shark cartilage | Broad spectrum | Non-small cell lung cancer (stage III) | Phase III along with chemotherapy | Well tolerated but no therapeutic effect | [39] |
| Compounds | Target | Binding Site | Mechanism of Action | Assays and Models Tested on | References |
|---|---|---|---|---|---|
| N-[4 (difluoromethoxy) phenyl] 2-[(4-oxo-6-propyl 1Hpyrimidin-2yl) sulfanyl]-acetamide | MMP9 | Hemopexin (PEX) | Interfered with homodimerization; inhibition of cell migration and proliferation | Tumor growth and metastasis (xenograft mouse model) | [16] |
| NSC405020 | MT1-MMP | Hemopexin (PEX) | Interfered with homodimerization and interaction with catalytic domain | Tumor growth (xenograft mouse model) | [24] |
| JNJ0966: (N-{2-[(2-methoxyphenyl) amino]-4-methyl-4,5-bi-1,3-thiazol-2-yl} acetamide) | MMP9 | Pro-peptide domain | Inhibit activation of MMP9 without affecting MMP1, -2, -3, -9 and MT1-MMP | Autoimmune encephalomyelitis (mouse model) | [29] |
| N-(4-fluorophenyl)-4-(4-oxo-3,4,5,6,7,8-hexahydroquinazolin-2-ylthio) butanamide | MMP9 | Hemopexin (PEX) | Inhibition of homodimerization; decreased cancer cell migration- blocks cancer cell invasion of basement membrane and angiogenesis | in vitro migration assay, Tumor growth (xenograft mouse model), angiogenesis (chicken chorioallantoic membrane) | [17] |
| Synthesized acrylamides (1) 3-bromo-N-(4-nitrophenyl) propenamide (2) 3-bromo-N-{4-[(pyrimidine-2-yl) sulfamoyl] phenyl} propenamide (3) 3-bromo-N-{4-[(4,6 dimethylpyrimidin-2-yl) sulfamoyl]-phenyl} propanamide | MMP9 | Hemopexin (PEX) | Inhibition of 4T1 breast cancer cell growth; inhibition of MMP9 gelatinolytic activity | in vitro migration assay and (xenograft mouse model) | [22] |
| Name | Antibody Type | Target | Epitope/Domains | Assays and Models Tested on | References |
|---|---|---|---|---|---|
| LEM-2/5 | Monoclonal | MT1-MMP | Surface epitope; V-P loop | Migrating cancer cells; | [59] |
| lung pathology; influenza | [48] | ||||
| SD3 | Monoclonal | MMP2 and -9 | Catalytic domain | Inflammatory bowel disease (mouse model); colitis | [55] |
| REGA-3G12 | Monoclonal | MMP-9 | Catalytic domain other than Zn2+ binding | Inhibited MMP9 proteolytic activity | [56,57] |
| DX-2400 | Fab fragment | MT1-MMP | Catalytic domain | Breast cancer | [58] |
| Multiple (A4-7 Fc-ScFv, E2_C6 Fc-ScFv) | Antibody fragments | MT1-MMP | Catalytic domain outside the active site cleft, inhibiting binding to triple helical collagen | Tumor growth and proliferation (xenograft mouse model) | [101] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Das, N.; Benko, C.; Gill, S.E.; Dufour, A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals 2021, 14, 31. https://doi.org/10.3390/ph14010031
Das N, Benko C, Gill SE, Dufour A. The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals. 2021; 14(1):31. https://doi.org/10.3390/ph14010031
Chicago/Turabian StyleDas, Nabangshu, Colette Benko, Sean E. Gill, and Antoine Dufour. 2021. "The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors" Pharmaceuticals 14, no. 1: 31. https://doi.org/10.3390/ph14010031
APA StyleDas, N., Benko, C., Gill, S. E., & Dufour, A. (2021). The Pharmacological TAILS of Matrix Metalloproteinases and Their Inhibitors. Pharmaceuticals, 14(1), 31. https://doi.org/10.3390/ph14010031