Rosmarinic Acid Methyl Ester Regulates Ovarian Cancer Cell Migration and Reverses Cisplatin Resistance by Inhibiting the Expression of Forkhead Box M1


 , ,
, ,
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
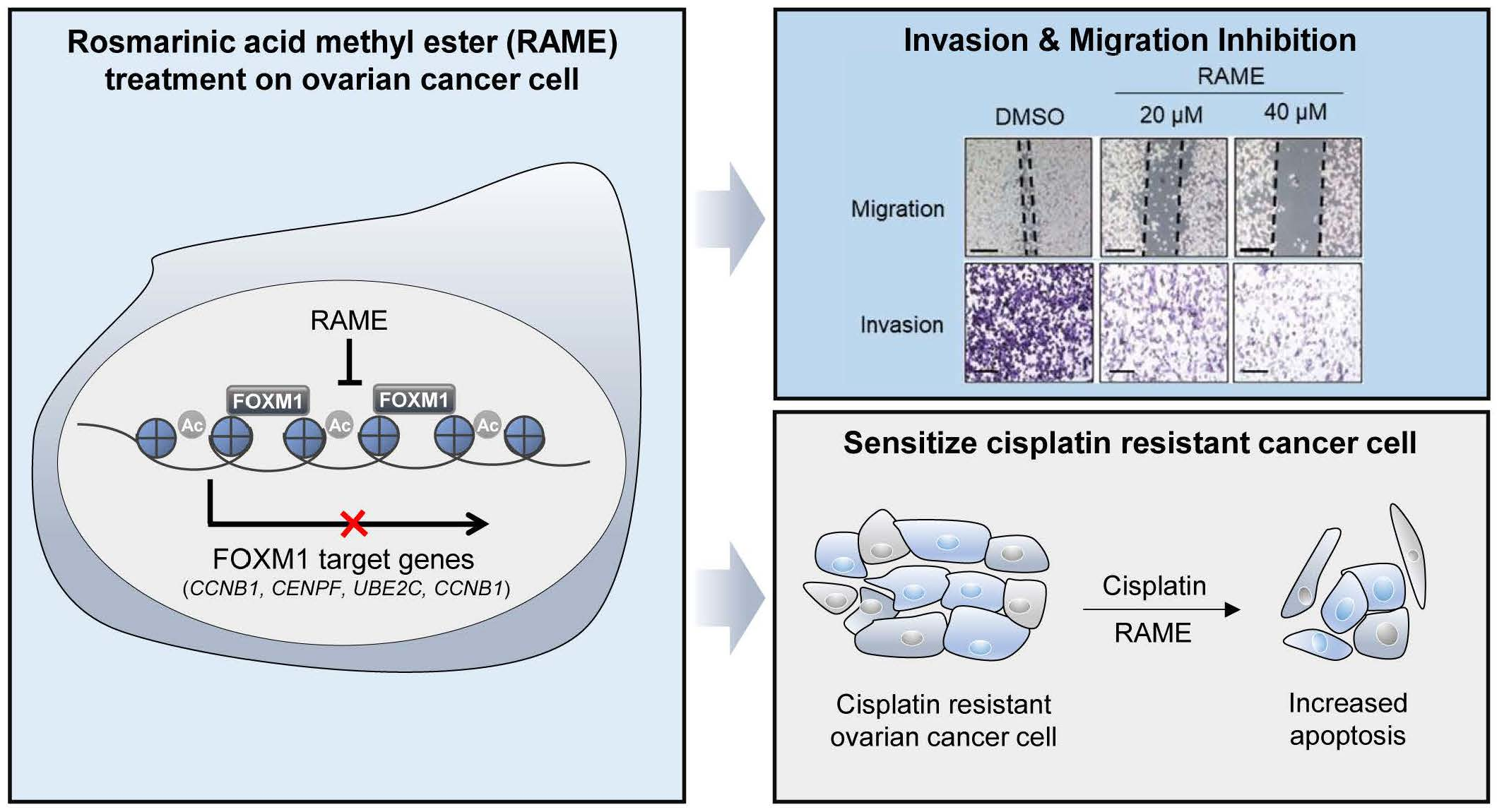
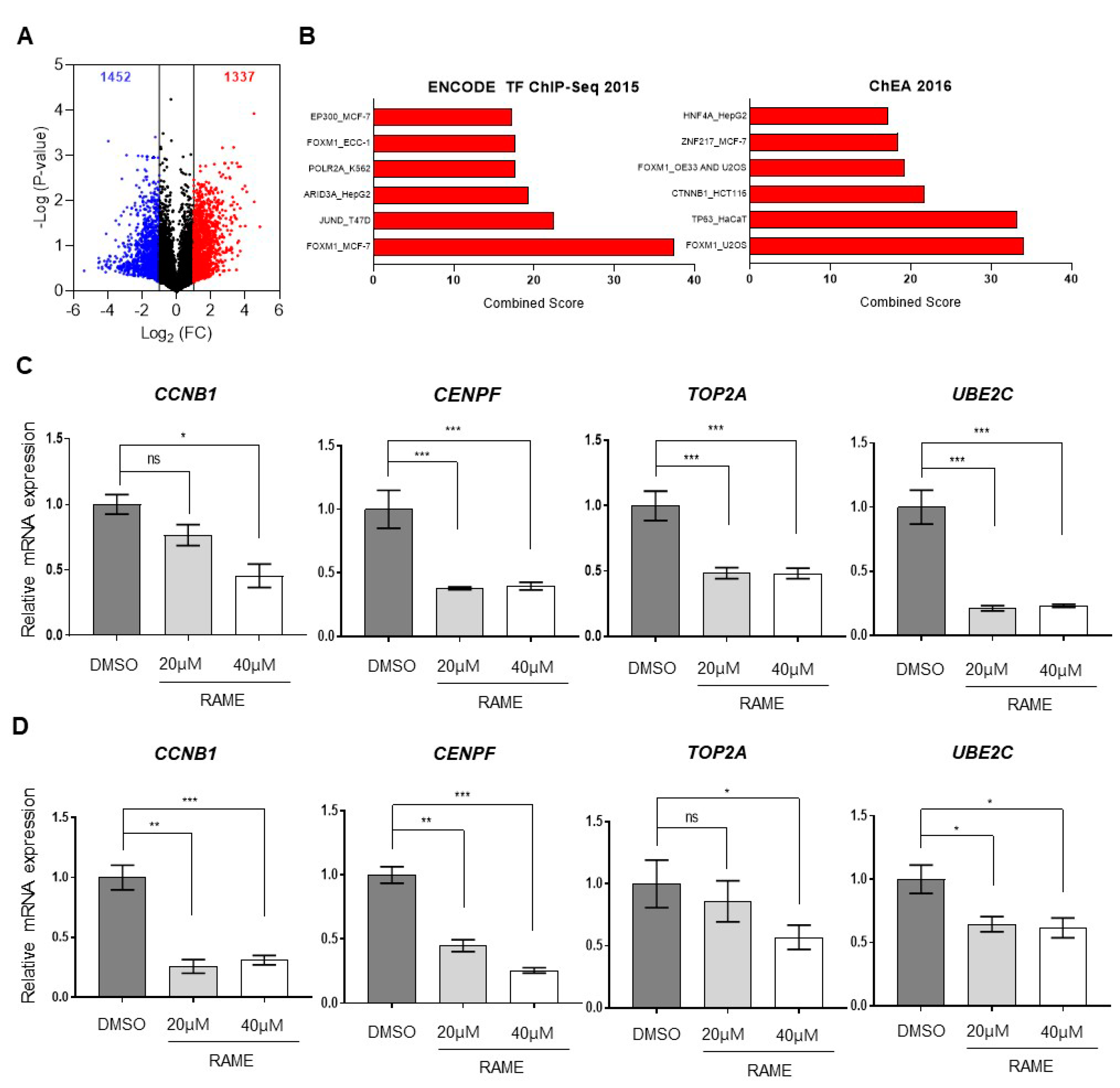
2.1. Transcriptome Analysis of Ovarian Cancer Cells Treated with RAME Shows that FOXM1 Target Genes are Downregulated
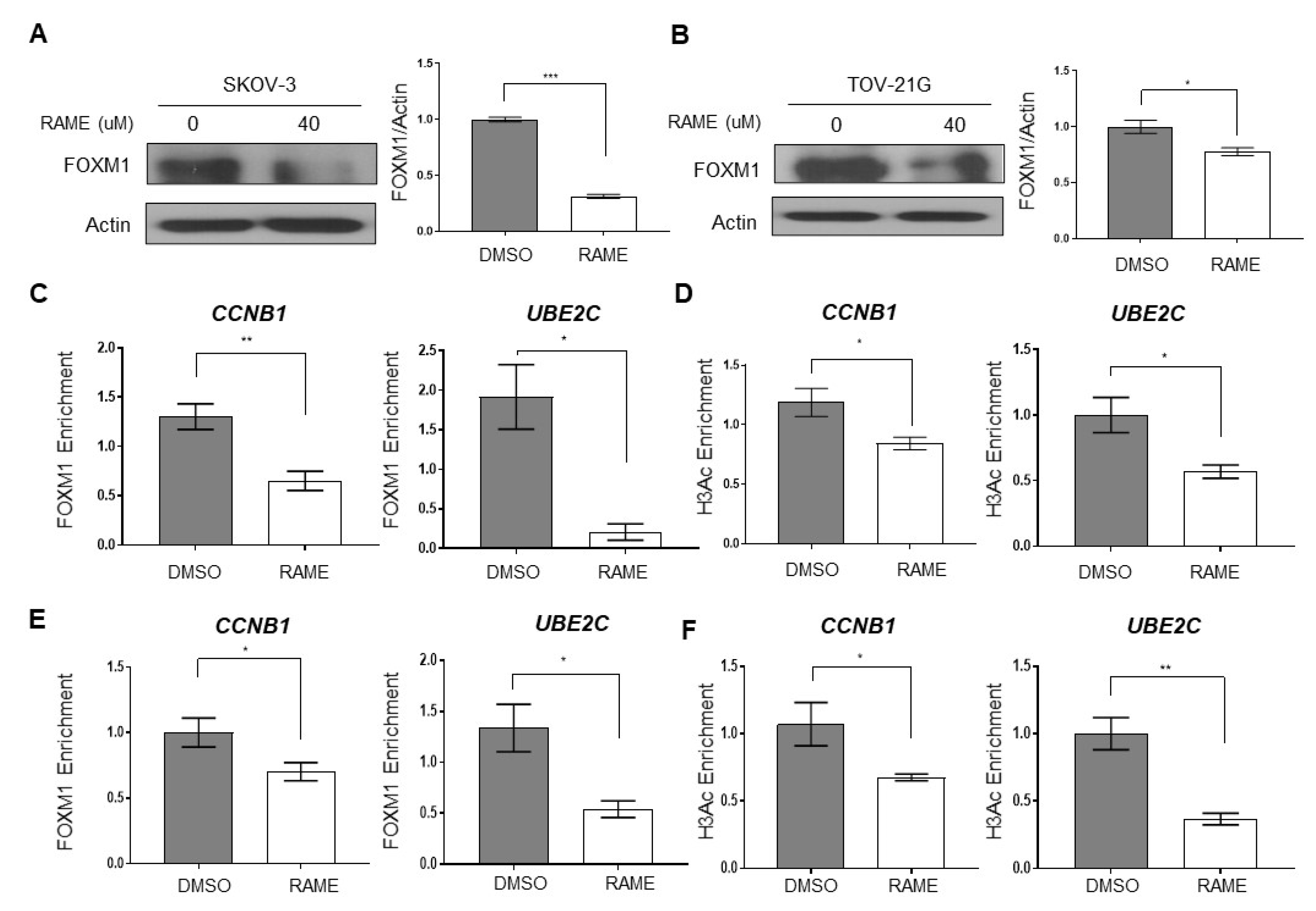
2.2. RAME Inhibits Expression of FOXM1 and its Interaction with Target Genes
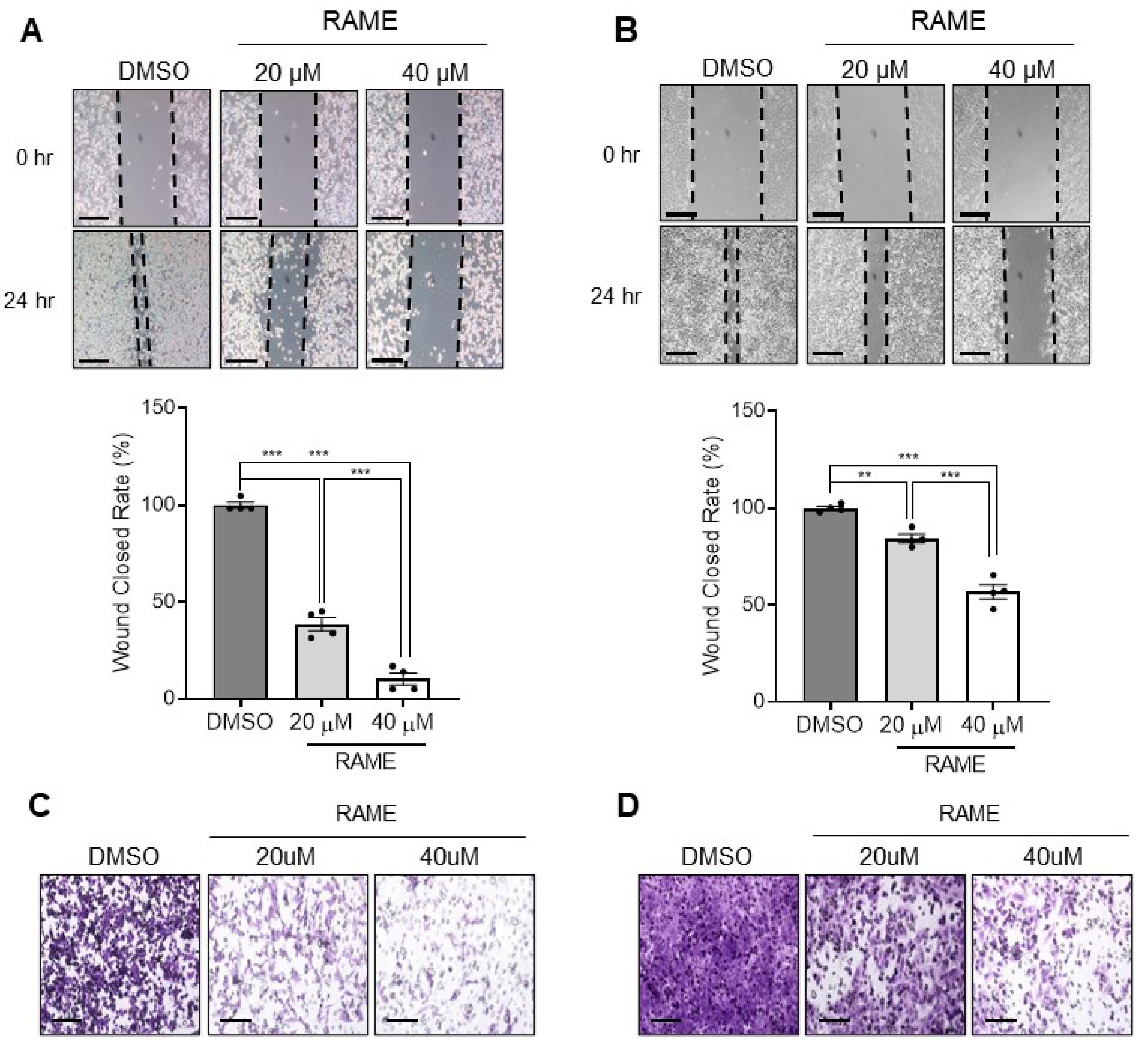
2.3. RAME Inhibits Cell Migration and Invasion in Ovarian Cancer Cell Line
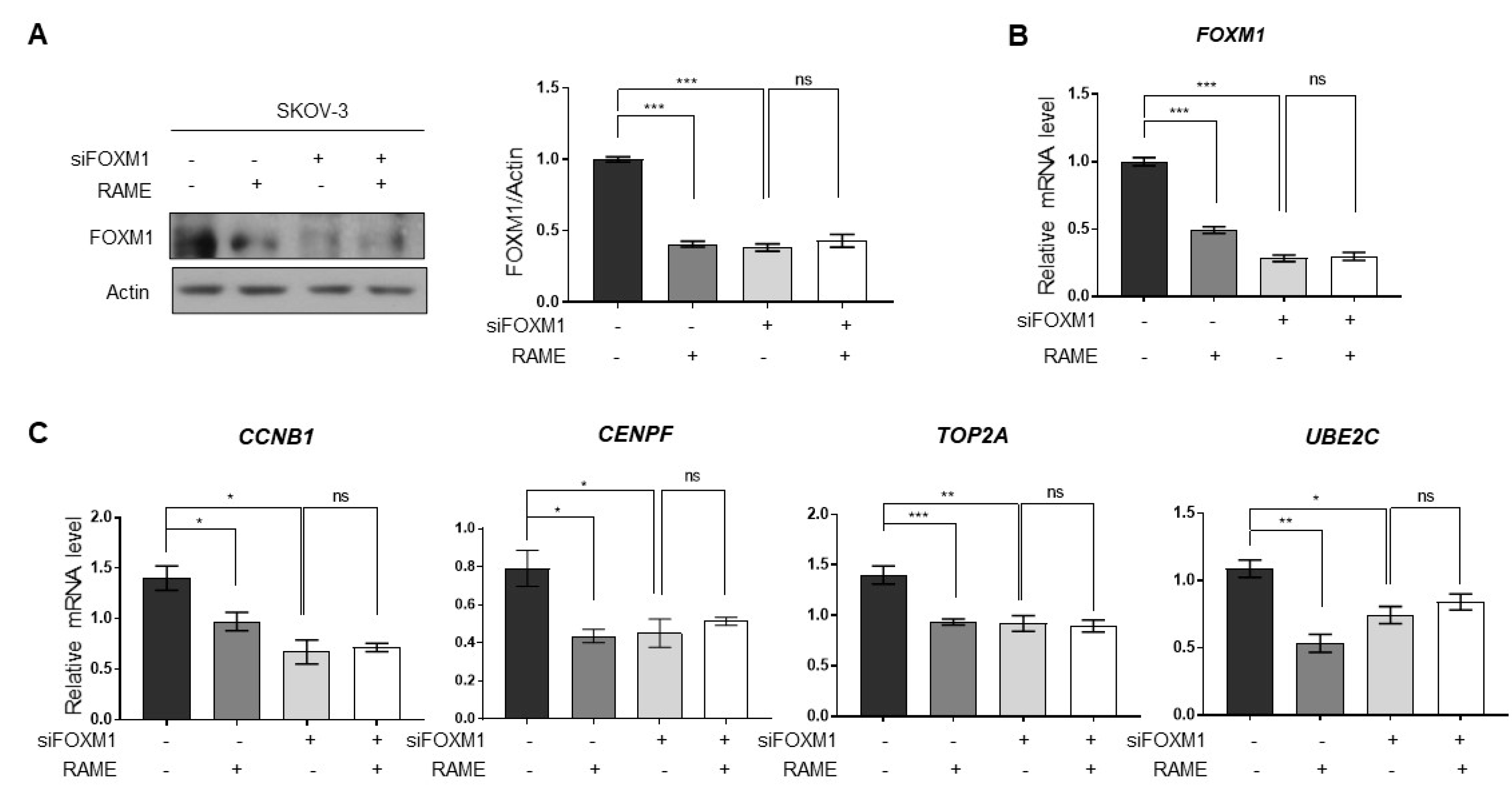
2.4. RAME Regulates Target Gene Expression via FOXM1
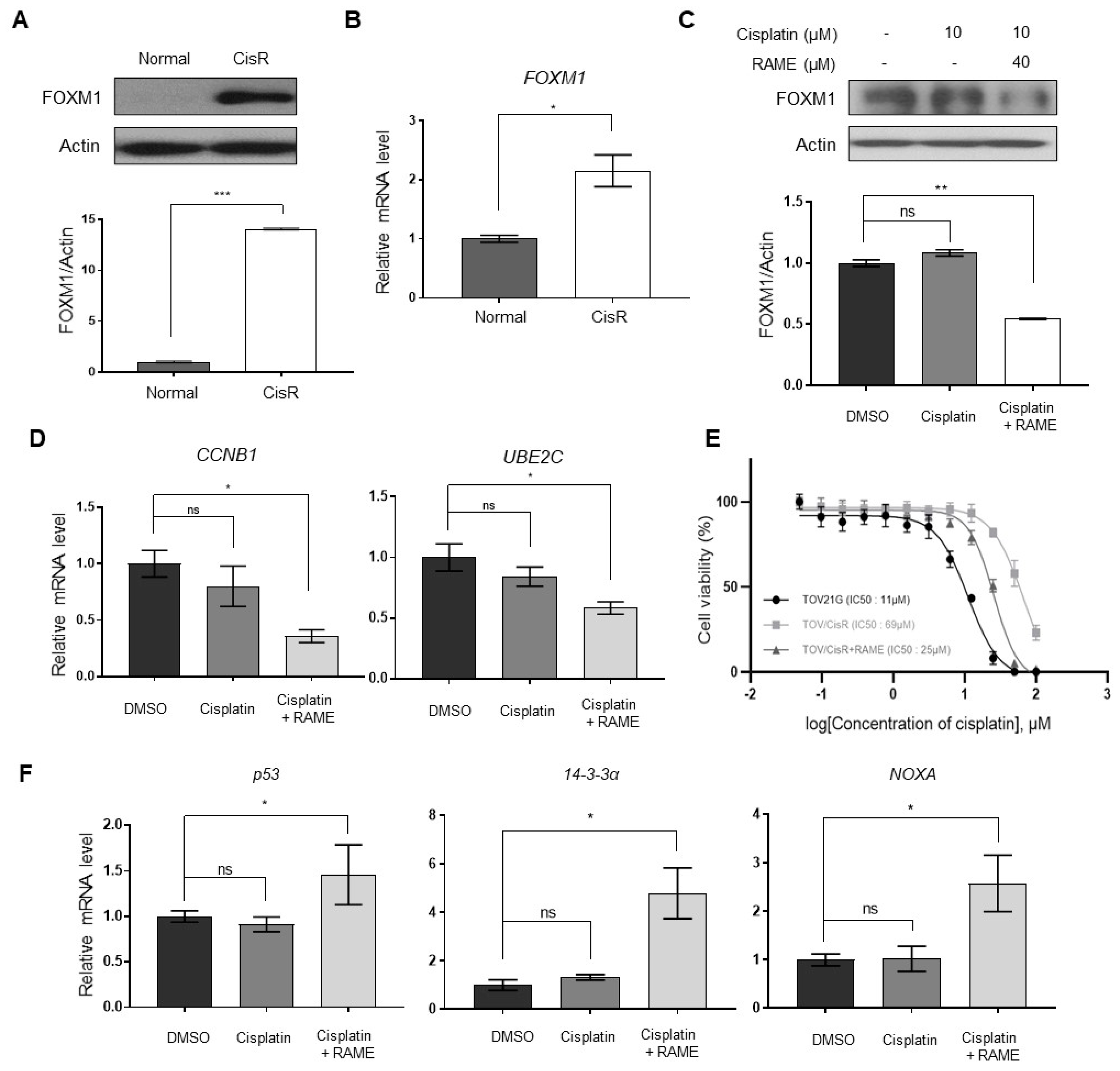
2.5. RAME Accelerates Anticancer Drug Effects in Cisplatin Resistant Ovarian Cancer Cell Line
3. Discussion
4. Materials and Methods
4.1. Antibodies and Reagents
4.2. Cell Culture and Establishment of Cisplatin-Resistant TOV-21G (TOV/CisR) Cells
4.3. RNA Isolation, Library Preparation, and RNA-Sequencing
4.4. RNA Extraction and Quantative Real-Time PCR (qPCR)
4.5. Protein Extraction and Immunoblotting
4.6. Chromatin Immnoprecipitation (ChIP)-qPCR
4.7. Wound Healing Assay
4.8. Transwell Migration Assay
4.9. Knockdown of FOXM1
4.10. Cell Viability Assay
4.11. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Stewart, C.; Ralyea, C.; Lockwood, S. Ovarian cancer: An integrated review. Semin. Oncol. Nurs. 2019, 35, 151–156. [Google Scholar] [CrossRef]
- Chang, L.-C.; Huang, C.-F.; Lai, M.-S.; Shen, L.-J.; Wu, F.-L.L.; Cheng, W.-F. Prognostic factors in epithelial ovarian cancer: A population-based study. PLoS ONE 2018, 13, e0194993. [Google Scholar] [CrossRef]
- Kurosaki, A.; Hasegawa, K.; Kato, T.; Abe, K.; Hanaoka, T.; Miyara, A.; O’Shannessy, D.J.; Somers, E.B.; Yasuda, M.; Sekino, T.; et al. Serum folate receptor alpha as a biomarker for ovarian cancer: Implications for diagnosis, prognosis and predicting its local tumor expression. Int. J. Cancer 2016, 138, 1994–2002. [Google Scholar] [CrossRef]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef]
- Cui, J.; Shi, M.; Xie, D.; Wei, D.; Jia, Z.; Zheng, S.; Gao, Y.; Huang, S.; Xie, K. FOXM1 promotes the warburg effect and pancreatic cancer progression via transactivation of LDHA expression. Clin. Cancer Res. 2014, 20, 2595–2606. [Google Scholar] [CrossRef]
- Milewski, D.; Balli, D.; Ustiyan, V.; Le, T.; Dienemann, H.; Warth, A.; Breuhahn, K.; Whitsett, J.A.; Kalinichenko, V.V.; Kalin, T.V. FOXM1 activates AGR2 and causes progression of lung adenomas into invasive mucinous adenocarcinomas. PLoS Genetics 2017, 13, e1007097. [Google Scholar] [CrossRef]
- Ziegler, Y.; Laws, M.J.; Sanabria Guillen, V.; Kim, S.H.; Dey, P.; Smith, B.P.; Gong, P.; Bindman, N.; Zhao, Y.; Carlson, K.; et al. Suppression of FOXM1 activities and breast cancer growth in vitro and in vivo by a new class of compounds. NPJ Breast Cancer 2019, 5, 1–11. [Google Scholar] [CrossRef]
- Chen, X.; Müller, G.A.; Quaas, M.; Fischer, M.; Han, N.; Stutchbury, B.; Sharrocks, A.D.; Engeland, K. The forkhead transcription factor FOXM1 controls cell cycle-dependent gene expression through an atypical chromatin binding mechanism. Mol. Cell. Biol. 2013, 33, 227–236. [Google Scholar] [CrossRef]
- Laoukili, J.; Kooistra, M.R.H.; Brás, A.; Kauw, J.; Kerkhoven, R.M.; Morrison, A.; Clevers, H.; Medema, R.H. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat. Cell Biol. 2005, 7, 126–136. [Google Scholar] [CrossRef]
- Wang, I.-C.; Chen, Y.-J.; Hughes, D.E.; Ackerson, T.; Major, M.L.; Kalinichenko, V.V.; Costa, R.H.; Raychaudhuri, P.; Tyner, A.L.; Lau, L.F. FoxM1 regulates transcription of JNK1 to promote the G1/S transition and tumor cell invasiveness. J. Biol. Chem. 2008, 283, 20770–20778. [Google Scholar] [CrossRef]
- Gartel, A.L. FOXM1 in Cancer: Interactions and Vulnerabilities. Cancer Res. 2017, 77, 3135–3139. [Google Scholar] [CrossRef]
- He, S.; Shen, H.-W.; Xu, L.; Zhao, X.-H.; Yuan, L.; Niu, G.; You, Z.; Shuzhong, Y. FOXM1 promotes tumor cell invasion and correlates with poor prognosis in early-stage cervical cancer. Gynecol. Oncol. 2012, 127, 601–610. [Google Scholar] [CrossRef]
- Luo, X.-Y.; Yao, J.; Nie, P.; Yang, Z.; Feng, H.; Chen, P.; Shi, X.; Zou, Z. FOXM1 promotes invasion and migration of colorectal cancer cells partially dependent on HSPA5 transactivation. Oncotarget 2016, 7, 26480–26495. [Google Scholar] [CrossRef]
- Luo, W.; Gao, F.; Li, S.; Liu, L. FoxM1 promotes cell proliferation, invasion, and stem cell properties in nasopharyngeal carcinoma. Front. Oncol. 2018, 8, 8. [Google Scholar] [CrossRef]
- Zhang, N.; Wei, P.; Gong, A.; Chiu, W.-T.; Lee, H.-T.; Colman, H.; Huang, H.; Xue, J.; Liu, M.; Wang, Y.; et al. FoxM1 promotes β-catenin nuclear localization and controls wnt target-gene expression and glioma tumorigenesis. Cancer Cell 2011, 20, 427–442. [Google Scholar] [CrossRef]
- Abedini, A.; Roumy, V.; Mahieux, S.; Biabiany, M.; Standaert-Vitse, A.; Rivière, C.; Sahpaz, S.; Bailleul, F.; Neut, C.; Hennebelle, T. Rosmarinic acid and its methyl ester as antimicrobial components of the hydromethanolic extract ofhyptis atrorubenspoit. (lamiaceae). Evidence-Based Complement. Altern. Med. 2013, 2013, 1–11. Available online: https://www.hindawi.com/journals/ecam/2013/604536/ (accessed on 9 July 2020). [CrossRef]
- So, Y.; Lee, S.Y.; Han, A.-R.; Kim, J.-B.; Jeong, H.G.; Jin, C.H. Rosmarinic acid methyl ester inhibits LPS-induced NO production via suppression of MyD88- dependent and -independent pathways and induction of HO-1 in RAW 264.7 Cells. Molecules 2016, 21, 1083. [Google Scholar] [CrossRef]
- Zhu, F.; Xu, Z.; Yonekura, L.; Yang, R.; Tamura, H. Antiallergic activity of rosmarinic acid esters is modulated by hydrophobicity, and bulkiness of alkyl side chain. Biosci. Biotechnol. Biochem. 2015, 79, 1178–1182. [Google Scholar] [CrossRef]
- Ding, H.-Y.; Chou, T.-H.; Liang, C.-H. Antioxidant and antimelanogenic properties of rosmarinic acid methyl ester from Origanum vulgare. Food Chem. 2010, 123, 254–262. [Google Scholar] [CrossRef]
- Nam, K.H.; Yi, S.A.; Nam, G.; Noh, J.S.; Park, J.W.; Lee, M.G.; Park, J.H.; Oh, H.; Lee, J.; Lee, K.R.; et al. Identification of a novel S6K1 inhibitor, rosmarinic acid methyl ester, for treating cisplatin-resistant cervical cancer. BMC Cancer 2019, 19, 773. [Google Scholar] [CrossRef]
- Li, B.; Zhu, H.-B.; Song, G.-D.; Cheng, J.-H.; Li, C.-Z.; Zhang, Y.-Z.; Zhao, P. Regulating the CCNB1 gene can affect cell proliferation and apoptosis in pituitary adenomas and activate epithelial-to-mesenchymal transition. Oncol. Lett. 2019, 18, 4651–4658. [Google Scholar] [CrossRef] [PubMed]
- Chai, N.; Xie, H.-H.; Yin, J.-P.; Sa, K.-D.; Guo, Y.; Wang, M.; Liu, J.; Zhang, X.-F.; Zhang, X.; Yin, H.; et al. FOXM1 promotes proliferation in human hepatocellular carcinoma cells by transcriptional activation of CCNB1. Biochem. Biophys. Res. Commun. 2018, 500, 924–929. [Google Scholar] [CrossRef] [PubMed]
- Lokody, I. FOXM1 and CENPF: Co-pilots driving prostate cancer. Nat. Rev. Cancer 2014, 14, 451. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Lu, J.; Fang, Q.; Lu, Y.; Xie, C.; Wu, H.; Yin, Z. UBE2C functions as a potential oncogene by enhancing cell proliferation, migration, invasion, and drug resistance in hepatocellular carcinoma cells. Biosci. Rep. 2019, 39. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.; Ding, Z.; Huang, N.; Huang, Z.; Zhang, N.; Xia, Z. Forkhead Box M1 positively regulates UBE2C and protects glioma cells from autophagic death. Cell Cycle 2017, 16, 1705–1718. [Google Scholar] [CrossRef] [PubMed]
- Pei, Y.-F.; Yin, X.-M.; Liu, X.-Q. TOP2A induces malignant character of pancreatic cancer through activating β-catenin signaling pathway. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 197–207. [Google Scholar] [CrossRef] [PubMed]
- Vasudevan, H.N.; Braunstein, S.E.; Phillips, J.J.; Pekmezci, M.; Tomlin, B.A.; Wu, A.; Reis, G.F.; Magill, S.T.; Zhang, J.; Feng, F.Y.; et al. Comprehensive molecular profiling identifies FOXM1 as a key transcription factor for meningioma proliferation. Cell Rep. 2018, 22, 3672–3683. [Google Scholar] [CrossRef]
- Zhao, F.; Siu, M.K.Y.; Jiang, L.; Tam, K.F.; Ngan, H.Y.S.; Le, X.F.; Wong, O.G.W.; Wong, E.S.Y.; Gomes, A.R.; Bella, L.; et al. Overexpression of forkhead box protein M1 (FOXM1) in ovarian cancer correlates with poor patient survival and contributes to paclitaxel resistance. PLoS ONE 2014, 9, e113478. [Google Scholar] [CrossRef]
- Wen, N.; Wang, Y.; Wen, L.; Zhao, S.-H.; Ai, Z.-H.; Wang, Y.; Wu, B.; Lu, H.-X.; Yang, H.; Liu, W.-C.; et al. Overexpression of FOXM1 predicts poor prognosis and promotes cancer cell proliferation, migration and invasion in epithelial ovarian cancer. J. Transl. Med. 2014, 12, 134. [Google Scholar] [CrossRef]
- Barger, C.J.; Zhang, W.; Hillman, J.; Stablewski, A.B.; Higgins, M.J.; Vanderhyden, B.C.; Odunsi, K.; Karpf, A.R. Genetic determinants of FOXM1 overexpression in epithelial ovarian cancer and functional contribution to cell cycle progression. Oncotarget 2015, 6, 27613–27627. [Google Scholar] [CrossRef]
- Bektas, N.; Haaf, A.T.; Veeck, J.; Wild, P.J.; Lüscher-Firzlaff, J.; Hartmann, A.; Knüchel, R.; Dahl, E. Tight correlation between expression of the Forkhead transcription factor FOXM1 and HER2 in human breast cancer. BMC Cancer 2008, 8, 42. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Qiao, W.-B.; Shan, L. Expression and functional characterization of FOXM1 in non-small cell lung cancer. OncoTargets Ther. 2018, 11, 3385–3393. [Google Scholar] [CrossRef] [PubMed]
- Wierstra, I.; Alves, J. FOXM1, a typical proliferation-associated transcription factor. Biol. Chem. 2007, 388, 1257–1274. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, S.; Hirano, I.; Okinaka, K.; Takemura, T.; Yokota, D.; Ono, T.; Shigeno, K.; Shibata, K.; Fujisawa, S.; Ohnishi, K. The FOXM1 transcriptional factor promotes the proliferation of leukemia cells through modulation of cell cycle progression in acute myeloid leukemia. Carcinogenesis 2010, 31, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Tassi, R.A.; Todeschini, P.; Siegel, E.R.; Calza, S.; Cappella, P.; Ardighieri, L.; Cadei, M.; Bugatti, M.; Romani, C.; Bandiera, E.; et al. FOXM1 expression is significantly associated with chemotherapy resistance and adverse prognosis in non-serous epithelial ovarian cancer patients. J. Exp. Clin. Cancer Res. 2017, 36, 63. [Google Scholar] [CrossRef] [PubMed]
- Gartel, A.L. FoxM1 inhibitors as potential anticancer drugs. Expert Opin. Ther. Targets 2008, 12, 663–665. [Google Scholar] [CrossRef] [PubMed]
- Halasi, M.; Hitchinson, B.; Shah, B.N.; Váraljai, R.; Khan, I.; Benevolenskaya, E.V.; Gaponenko, V.; Arbiser, J.L.; Gartel, A.L. Honokiol is a FOXM1 antagonist. Cell Death Dis. 2018, 9, 1–8. [Google Scholar] [CrossRef]
- Lheureux, S.; Braunstein, M.; Oza, A.M. Epithelial ovarian cancer: Evolution of management in the era of precision medicine. CA A Cancer J. Clin. 2019, 69, 280–304. [Google Scholar] [CrossRef]
- Torre, L.A.; Trabert, B.; DeSantis, C.E.; Miller, K.D.; Samimi, G.; Runowicz, C.D.; Gaudet, M.M.; Jemal, A.; Siegel, R.L. Ovarian cancer statistics, 2018. CA A Cancer J. Clin. 2018, 68, 284–296. [Google Scholar] [CrossRef]
- Brain, K.E.; Smits, S.; Simon, A.E.; Forbes, L.J.; Roberts, C.; Robbé, I.J.; Steward, J.; White, C.; Neal, R.D.; Hanson, J. Ovarian cancer symptom awareness and anticipated delayed presentation in a population sample. BMC Cancer 2014, 14, 171. [Google Scholar] [CrossRef]
- DiSilvestro, P.; Secord, A.A. Maintenance treatment of recurrent ovarian cancer: Is it ready for prime time? Cancer Treat. Rev. 2018, 69, 53–65. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Landen, C.N.; Li, Y.; Alvarez, R.D.; Tollefsbol, T.O. Enhancement of Cisplatin-Mediated Apoptosis in Ovarian Cancer Cells through Potentiating G2/M Arrest and p21 Upregulation by Combinatorial Epigallocatechin Gallate and Sulforaphane. Available online: https://www.hindawi.com/journals/jo/2013/872957/ (accessed on 13 July 2020).
- Dasari, S.; Tchounwou, P.B. Cisplatin in cancer therapy: Molecular mechanisms of action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [PubMed]
- Köberle, B.; Tomicic, M.T.; Usanova, S.; Kaina, B. Cisplatin resistance: Preclinical findings and clinical implications. Biochim. Biophys. Acta Bioenerg. 2010, 1806, 172–182. [Google Scholar] [CrossRef] [PubMed]
- Shen, D.-W.; Pouliot, L.M.; Hall, M.D.; Gottesman, M.M. Cisplatin resistance: A cellular self-defense mechanism resulting from multiple epigenetic and genetic changes. Pharmacol. Rev. 2012, 64, 706–721. [Google Scholar] [CrossRef]
- Chen, S.-H.; Chang, J.-Y. New insights into mechanisms of cisplatin resistance: From tumor cell to microenvironment. Int. J. Mol. Sci. 2019, 20, 4136. [Google Scholar] [CrossRef]
- Sun, X.; Wang, S.; Gai, J.; Guan, J.; Li, J.; Li, Y.; Zhao, J.; Zhao, C.; Fu, L.; Li, Q. SIRT5 promotes cisplatin resistance in ovarian cancer by suppressing DNA damage in a ROS-dependent manner via regulation of the Nrf2/HO-1 pathway. Front. Oncol. 2019, 9, 754. [Google Scholar] [CrossRef]
- Zhou, J.; Wang, Y.; Wang, Y.; Yin, X.; He, Y.; Chen, L.; Wang, W.; Liu, T.; Di, W. FOXM1 modulates cisplatin sensitivity by regulating EXO1 in ovarian cancer. PLoS ONE 2014, 9, e96989. [Google Scholar] [CrossRef]
- Kwok, J.M.-M.; Peck, B.; Monteiro, L.J.; Schwenen, H.D.C.; Millour, J.; Coombes, R.C.; Myatt, S.S.; Lam, E.W.-F. FOXM1 confers acquired cisplatin resistance in breast cancer cells. Mol. Cancer Res. 2010, 8, 24–34. [Google Scholar] [CrossRef]
- Wang, Y.; Wen, L.; Zhao, S.-H.; Ai, Z.-H.; Guo, J.-Z.; Liu, W.-C. FoxM1 expression is significantly associated with cisplatin-based chemotherapy resistance and poor prognosis in advanced non-small cell lung cancer patients. Lung Cancer 2013, 79, 173–179. [Google Scholar] [CrossRef]
- Okada, T.; Murata, K.; Hirose, R.; Matsuda, C.; Komatsu, T.; Ikekita, M.; Nakawatari, M.; Nakayama, F.; Wakatsuki, M.; Ohno, T.; et al. Upregulated expression of FGF13/FHF2 mediates resistance to platinum drugs in cervical cancer cells. Sci. Rep. 2013, 3, 2899. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Chen, E.Y.; Tan, C.M.; Kou, Y.; Duan, Q.; Wang, Z.; Meirelles, G.V.; Clark, N.R.; Ma’Ayan, A. Enrichr: Interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinform. 2013, 14, 128. [Google Scholar] [CrossRef] [PubMed]
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Liao, G.-B.; Li, X.-Z.; Zeng, S.; Liu, C.; Yang, S.-M.; Yang, L.; Hu, C.-J.; Bai, J.-Y. Regulation of the master regulator FOXM1 in cancer. Cell Commun. Signal. 2018, 16, 57. [Google Scholar] [CrossRef]
- Gartel, A.L. Targeting FOXM1 auto-regulation in cancer. Cancer Biol. Ther. 2015, 16, 185–186. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Halasi, M.; Gartel, A.L. A novel mode of FoxM1 regulation: Positive auto-regulatory loop. Cell Cycle 2009, 8, 1966–1967. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, S.H.; Nam, K.H.; Kim, K.; Yi, S.A.; Lee, J.; Han, J.-W. Rosmarinic Acid Methyl Ester Regulates Ovarian Cancer Cell Migration and Reverses Cisplatin Resistance by Inhibiting the Expression of Forkhead Box M1. Pharmaceuticals 2020, 13, 302. https://doi.org/10.3390/ph13100302
Lim SH, Nam KH, Kim K, Yi SA, Lee J, Han J-W. Rosmarinic Acid Methyl Ester Regulates Ovarian Cancer Cell Migration and Reverses Cisplatin Resistance by Inhibiting the Expression of Forkhead Box M1. Pharmaceuticals. 2020; 13(10):302. https://doi.org/10.3390/ph13100302
Chicago/Turabian StyleLim, Soo Hyun, Ki Hong Nam, Kyungtae Kim, Sang Ah Yi, Jaecheol Lee, and Jeung-Whan Han. 2020. "Rosmarinic Acid Methyl Ester Regulates Ovarian Cancer Cell Migration and Reverses Cisplatin Resistance by Inhibiting the Expression of Forkhead Box M1" Pharmaceuticals 13, no. 10: 302. https://doi.org/10.3390/ph13100302
APA StyleLim, S. H., Nam, K. H., Kim, K., Yi, S. A., Lee, J., & Han, J.-W. (2020). Rosmarinic Acid Methyl Ester Regulates Ovarian Cancer Cell Migration and Reverses Cisplatin Resistance by Inhibiting the Expression of Forkhead Box M1. Pharmaceuticals, 13(10), 302. https://doi.org/10.3390/ph13100302