Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections

 ,
, 
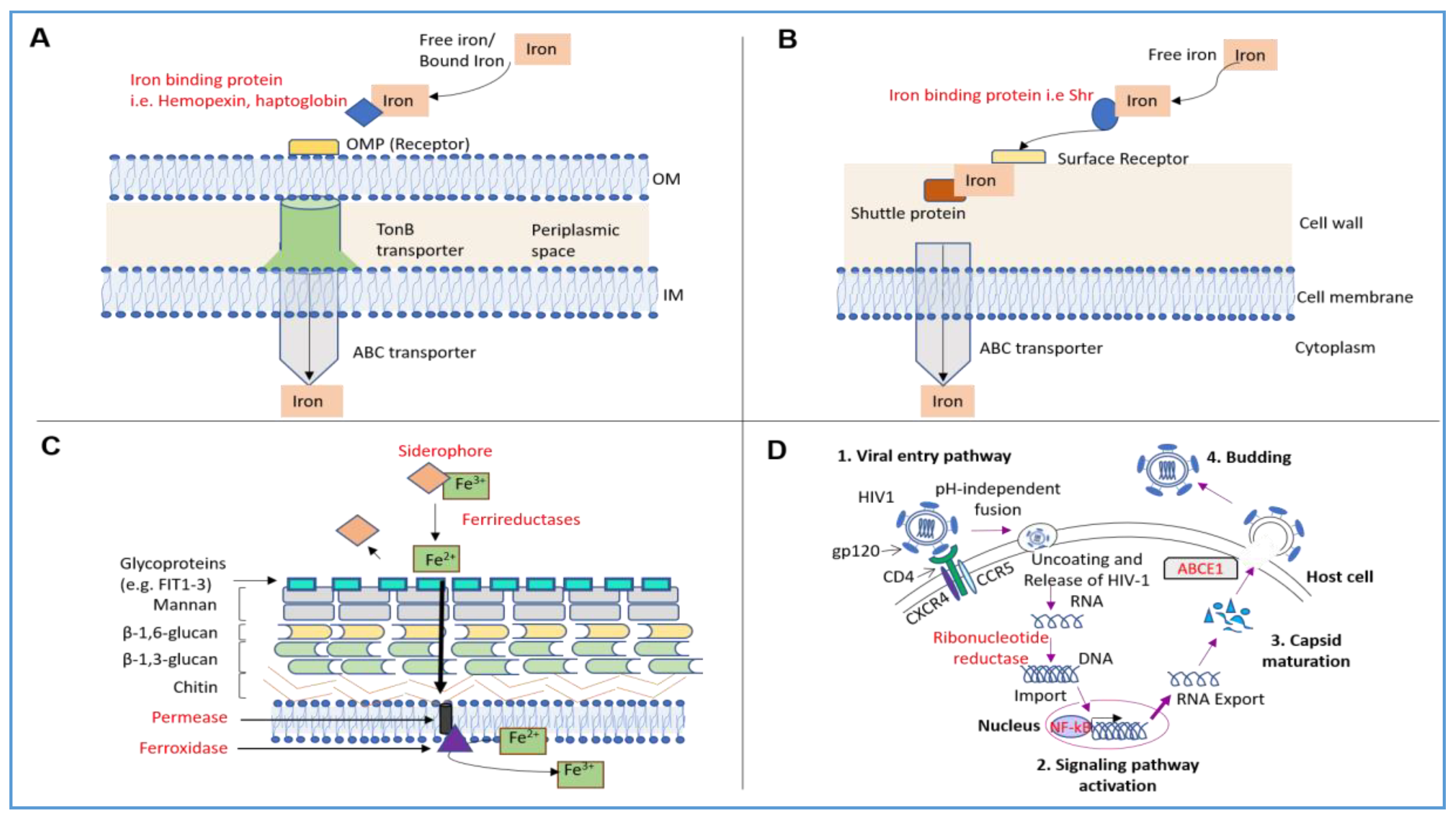{kind=link}
Abstract
1. Introduction
1.1. Iron, an Essential Element for Survival of Both Hosts and Microorganisms
1.2. Host Cell Iron Metabolic Pathway
1.3. Iron Chelation and Associated Risks
2. RNA-Based Viral Infections
2.1. Hepatitis C Virus (HCV)
2.2. Human Immunodeficiency Virus (HIV)
3. Bacterial Infections
3.1. Pseudomonas Aeruginosa: A Gram-Negative Microbe Associated with Wound Infections and Cystic Fibrosis
3.2. Porphyromonas Gingivalis, Prevotella Intermedia, and Fusobacterium Nucleatum: Bacteria Associated with Periodontitis
3.3. Streptococcus Pneumoniae: A Gram-Positive Bacteria
3.4. Mycobacterium Tuberculosis
4. Fungal Infections
4.1. Cryptococcus Neoformans
4.2. Aspergillus Fumigatus
4.3. Rhizopus Oryzae
5. Concluding Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABC | ATP-binding cassette |
| AIDS | acquired immune deficiency syndrome |
| ALT | alanine aminotransferase |
| ART | anti-retroviral therapy |
| ATPase ABCE1 | ATP binding cassette subfamily E member 1 |
| bLF | bovine lactoferrin |
| NRAMP1 | natural resistance-associated macrophage protein 1 |
| BLM | bleomycin |
| BPS | bathophenanthroline disulfonate |
| bZIP | basic leucine zipper containing domain |
| CD81 | Cluster of Differentiation 81 |
| CDK | cyclin dependent kinases |
| CFEM | Common in Fungal Extracellular Membrane |
| CLDN1 | Claudin 1 |
| DCYTB | duodenal cytochrome b |
| DFO | Desferioxamine or deferoxamine |
| DFP or L1 | Deferiprone |
| DFRA or DFX | Deferasirox |
| DFT | Desferrithiocin |
| DMT1 | Divalent Metal Transporter 1 |
| DNA | deoxyribonucleic acid |
| EGCG | Epigallocatechin-3-gallate |
| EGFR | epidermal growth factor receptor |
| eIF3 | Eukaryotic initiation factor 3 |
| FDA | Food and Drug Administration |
| Fe-SIH | iron-salicylaldehyde isonicotinoyl hydrazine |
| FeSO4 | iron (II) sulfate |
| FPN1 | ferroportin |
| FsC | fusarinine C |
| FTN | ferritin |
| Fur | ferric uptake regulator |
| GATA | Globin Transcription Factor |
| HAMP | hepcidin |
| HasA | heme acquisition system A |
| HCP1 | Heme Carrier Protein |
| HCV | Hepatitis C Virus |
| HIV | Human Immunodeficiency Virus |
| HO-1 | heme oxygenase 1 |
| HO-2 | heme oxygenase 2 |
| IFN | Interferon |
| IRES | internal ribosome entry site |
| JAK | Janus Kinase |
| LIP | labile iron pool |
| NF-kB | nuclear factor kappa-light-chain-enhancer of activated B cells |
| NS3 | non-structural protein 3 |
| NTBI | non-transferring bound iron |
| OCLN | Occludin |
| OMP | outer membrane protein |
| PBL | peripheral blood lymphocytes |
| PCFT | proton-coupled folate transporter |
| Pch | pyochelin |
| Pvd | pyoverdine |
| PZP | pyrazolopyridinone |
| RBC | red blood cells |
| RIA | reductive iron assimilation |
| RNA | ribonucleic acid |
| RNR | ribonucleotide reductase |
| ROS | reactive oxygen species |
| SIT | siderophore iron transporter |
| SLC39A8 | Solute carrier family 39 member 8 |
| SRB1 | Scavenger receptor class B type 1 |
| STAT | Signal Transducer and Activator of Transcription |
| STEAP3 | Six Transmembrane Epithelial Antigen of Prostate 3 |
| TAFC | triacetylfusarinine C |
| TBI | transferrin-bound iron |
| TGF-β | transforming growth factor beta |
References
- Abbaspour, N.; Hurrell, R.; Kelishadi, R. Review on iron and its importance for human health. J. Res. Med. Sci. 2014, 19, 164–174. [Google Scholar]
- Bogdan, A.R.; Miyazawa, M.; Hashimoto, K.; Tsuji, Y. Regulators of iron homeostasis: New players in metabolism, cell death, and disease. Trends Biochem. Sci. 2016, 41, 274–286. [Google Scholar] [CrossRef] [PubMed]
- Cassat, J.E.; Skaar, E.P. Iron in infection and immunity. Cell Host Microbe 2013, 13, 509–519. [Google Scholar] [CrossRef] [PubMed]
- Drakesmith, H.; Prentice, A. Viral infection and iron metabolism. Nat. Rev. Microbiol. 2008, 6, 541–552. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T. Systemic iron homeostasis. Physiol. Rev. 2013, 93, 1721–1741. [Google Scholar] [CrossRef]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef]
- Ji, C.; Kosman, D.J. Molecular mechanisms of non-transferrin-bound and transferring-bound iron uptake in primary hippocampal neurons. J. Neurochem. 2015, 133, 668–683. [Google Scholar] [CrossRef]
- Silva, B.; Faustino, P. An overview of molecular basis of iron metabolism regulation and the associated pathologies. Biochim. Biophys. Acta 2015, 1852, 1347–1359. [Google Scholar] [CrossRef] [PubMed]
- Knutson, M.D. Iron transport proteins: Gateways of cellular and systemic iron homeostasis. J. Biol. Chem. 2017, 292, 12735–12743. [Google Scholar] [CrossRef]
- Sangkhae, V.; Nemeth, E. Regulation of the iron homeostatic hormone hepcidin. Adv. Nutr. 2017, 8, 126–136. [Google Scholar] [CrossRef]
- Dev, S.; Babitt, J.L. Overview of iron metabolism in health and disease. Hemodial. Int. 2017, 21 (Suppl. 1), S6–S20. [Google Scholar]
- Yoshida, T.; Noguchi, M.; Kikuchi, G. Oxygenated form of heme. Heme oxygenase complex and requirement for second electron to initiate heme degradation from the oxygenated complex. J. Biol. Chem. 1980, 255, 4418–4420. [Google Scholar] [PubMed]
- Kakhlon, O.; Cabantchik, Z.I. The labile iron pool: Characterization, measurement, and participation in cellular processes (1). Free Radic. Biol. Med. 2002, 33, 1037–1046. [Google Scholar] [PubMed]
- MacKenzie, E.L.; Iwasaki, K.; Tsuji, Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid. Redox Signal. 2008, 10, 997–1030. [Google Scholar] [CrossRef]
- Coffey, R.; Ganz, T. Iron homeostasis: An anthropocentric perspective. J. Biol. Chem. 2017, 292, 12727–12734. [Google Scholar]
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2019, 133, 46–54. [Google Scholar] [CrossRef]
- Sendamarai, A.K.; Ohgami, R.S.; Fleming, M.D.; Lawrence, C.M. Structure of the membrane proximal oxidoreductase domain of human steap3, the dominant ferrireductase of the erythroid transferrin cycle. Proc. Natl. Acad. Sci. USA 2008, 105, 7410–7415. [Google Scholar] [CrossRef]
- Lane, D.J.; Bae, D.H.; Merlot, A.M.; Sahni, S.; Richardson, D.R. Duodenal cytochrome b (dcytb) in iron metabolism: An update on function and regulation. Nutrients 2015, 7, 2274–2296. [Google Scholar]
- van Raaij, S.E.G.; Srai, S.K.S.; Swinkels, D.W.; van Swelm, R.P.L. Iron uptake by zip8 and zip14 in human proximal tubular epithelial cells. Biometals 2019, 32, 211–226. [Google Scholar]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- Mackenzie, B.; Hediger, M.A. Slc11 family of h+-coupled metal-ion transporters nramp1 and dmt1. Pflug. Arch. Eur. J. Physiol. 2004, 447, 571–579. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Wang, J.; Pantopoulos, K. Regulation of cellular iron metabolism. Biochem. J. 2011, 434, 365–381. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Nairz, M.; Schroll, A.; Sonnweber, T.; Weiss, G. The struggle for iron-a metal at the host-pathogen interface. Cell. Microbiol. 2010, 12, 1691–1702. [Google Scholar] [CrossRef]
- Mobarra, N.; Shanaki, M.; Ehteram, H.; Nasiri, H.; Sahmani, M.; Saeidi, M.; Goudarzi, M.; Pourkarim, H.; Azad, M. A review on iron chelators in treatment of iron overload syndromes. Int. J. Hematol. Oncol. Stem Cell Res. 2016, 10, 239–247. [Google Scholar]
- Nunez, M.T.; Chana-Cuevas, P. New perspectives in iron chelation therapy for the treatment of neurodegenerative diseases. Pharmaceuticals 2018, 11, 109. [Google Scholar] [CrossRef]
- Hatcher, H.C.; Singh, R.N.; Torti, F.M.; Torti, S.V. Synthetic and natural iron chelators: Therapeutic potential and clinical use. Future Med. Chem. 2009, 1, 1643–1670. [Google Scholar] [CrossRef]
- Chiani, M.; Akbarzadeh, A.; Farhangi, A.; Mazinani, M.; Saffari, Z.; Emadzadeh, K.; Mehrabi, M.R. Optimization of culture medium to increase the production of desferrioxamine b (desferal) in streptomyces pilosus. Pak. J. Biol. Sci. 2010, 13, 546–550. [Google Scholar] [CrossRef]
- Bergeron, R.J.; Wiegand, J.; McManis, J.S.; Bharti, N. Desferrithiocin: A search for clinically effective iron chelators. J. Med. Chem. 2014, 57, 9259–9291. [Google Scholar] [CrossRef]
- Kontoghiorghe, C.N.; Kolnagou, A.; Kontoghiorghes, G.J. Phytochelators intended for clinical use in iron overload, other diseases of iron imbalance and free radical pathology. Molecules 2015, 20, 20841–20872. [Google Scholar] [CrossRef]
- Kontoghiorghes, G.J.; Kontoghiorghe, C.N. Iron and chelation in biochemistry and medicine: New approaches to controlling iron metabolism and treating related diseases. Cells 2020, 9, 1456. [Google Scholar] [CrossRef]
- Rosa, L.; Cutone, A.; Lepanto, M.S.; Paesano, R.; Valenti, P. Lactoferrin: A natural glycoprotein involved in iron and inflammatory homeostasis. Int. J. Mol. Sci. 2017, 18, 1985. [Google Scholar] [CrossRef]
- Wang, B.; Timilsena, Y.P.; Blanch, E.; Adhikari, B. Lactoferrin: Structure, function, denaturation and digestion. Crit. Rev. Food Sci. Nutr. 2019, 59, 580–596. [Google Scholar] [CrossRef]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar]
- Stojiljkovic, I.; Hantke, K. Hemin uptake system of yersinia enterocolitica: Similarities with other tonb-dependent systems in gram-negative bacteria. EMBO J. 1992, 11, 4359–4367. [Google Scholar] [CrossRef]
- Tong, Y.; Guo, M. Bacterial heme-transport proteins and their heme-coordination modes. Arch. Biochem. Biophys. 2009, 481, 1–15. [Google Scholar] [CrossRef]
- Sook, B.R.; Block, D.R.; Sumithran, S.; Montanez, G.E.; Rodgers, K.R.; Dawson, J.H.; Eichenbaum, Z.; Dixon, D.W. Characterization of siaa, a streptococcal heme-binding protein associated with a heme abc transport system. Biochemistry 2008, 47, 2678–2688. [Google Scholar] [CrossRef]
- Zhu, H.; Liu, M.; Lei, B. The surface protein shr of streptococcus pyogenes binds heme and transfers it to the streptococcal heme-binding protein shp. BMC Microbiol. 2008, 8, 15. [Google Scholar] [CrossRef]
- Philpott, C.C. Iron uptake in fungi: A system for every source. Biochim. Biophys. Acta 2006, 1763, 636–645. [Google Scholar] [CrossRef]
- Lesuisse, E.; Labbe, P. Reductive and non-reductive mechanisms of iron assimilation by the yeast saccharomyces cerevisiae. J. Gen. Microbiol. 1989, 135, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Marsh, M. The cell biology of receptor-mediated virus entry. J. Cell Biol. 2011, 195, 1071–1082. [Google Scholar] [PubMed]
- Fanales-Belasio, E.; Raimondo, M.; Suligoi, B.; Butto, S. Hiv virology and pathogenetic mechanisms of infection: A brief overview. Annali Dell’istituto Superiore di Sanita 2010, 46, 5–14. [Google Scholar] [CrossRef]
- Choe, H.; Jemielity, S.; Abraham, J.; Radoshitzky, S.R.; Farzan, M. Transferrin receptor 1 in the zoonosis and pathogenesis of new world hemorrhagic fever arenaviruses. Curr. Opin. Microbiol. 2011, 14, 476–482. [Google Scholar] [CrossRef]
- Romeo, A.M.; Christen, L.; Niles, E.G.; Kosman, D.J. Intracellular chelation of iron by bipyridyl inhibits DNA virus replication: Ribonucleotide reductase maturation as a probe of intracellular iron pools. J. Biol. Chem. 2001, 276, 24301–24308. [Google Scholar] [CrossRef]
- Sappey, C.; Boelaert, J.R.; Legrand-Poels, S.; Forceille, C.; Favier, A.; Piette, J. Iron chelation decreases nf-kappa b and hiv type 1 activation due to oxidative stress. AIDS Res. Hum. Retrovir. 1995, 11, 1049–1061. [Google Scholar]
- Dooher, J.E.; Schneider, B.L.; Reed, J.C.; Lingappa, J.R. Host abce1 is at plasma membrane hiv assembly sites and its dissociation from gag is linked to subsequent events of virus production. Traffic 2007, 8, 195–211. [Google Scholar]
- Dubuisson, J.; Cosset, F.L. Virology and cell biology of the hepatitis c virus life cycle: An update. J. Hepatol. 2014, 61, S3–S13. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar]
- Chevaliez, S.; Pawlotsky, J.M. Hcv genome and life cycle. In Hepatitis C Viruses: Genomes and Molecular Biology; Tan, S.L., Ed.; Horizon Bioscience: Norfolk, UK, 2006. [Google Scholar]
- Wong, R.J.; Gish, R.G. Metabolic manifestations and complications associated with chronic hepatitis c virus infection. Gastroenterol. Hepatol. 2016, 12, 293–299. [Google Scholar]
- Herrera, J.L. Iron depletion is not effective in inducing a virologic response in patients with chronic hepatitis c who failed to respond to interferon therapy. Am. J. Gastroenterol. 1999, 94, 3571–3575. [Google Scholar] [CrossRef] [PubMed]
- Stevenson, N.J.; Murphy, A.G.; Bourke, N.M.; Keogh, C.A.; Hegarty, J.E.; O’Farrelly, C. Ribavirin enhances ifn-alpha signalling and mxa expression: A novel immune modulation mechanism during treatment of hcv. PLoS ONE 2011, 6, e27866. [Google Scholar]
- McHutchison, J.G.; Gordon, S.C.; Schiff, E.R.; Shiffman, M.L.; Lee, W.M.; Rustgi, V.K.; Goodman, Z.D.; Ling, M.H.; Cort, S.; Albrecht, J.K. Interferon alfa-2b alone or in combination with ribavirin as initial treatment for chronic hepatitis c. Hepatitis interventional therapy group. N. Engl. J. Med. 1998, 339, 1485–1492. [Google Scholar]
- Goto, K.; Roca Suarez, A.A.; Wrensch, F.; Baumert, T.F.; Lupberger, J. Hepatitis c virus and hepatocellular carcinoma: When the host loses its grip. Int. J. Mol. Sci. 2020, 21, 3057. [Google Scholar] [CrossRef] [PubMed]
- Theurl, I.; Zoller, H.; Obrist, P.; Datz, C.; Bachmann, F.; Elliott, R.M.; Weiss, G. Iron regulates hepatitis c virus translation via stimulation of expression of translation initiation factor 3. J. Infect. Dis. 2004, 190, 819–825. [Google Scholar] [CrossRef]
- Sartori, M.; Andorno, S.; Rossini, A.; Boldorini, R.; Bozzola, C.; Carmagnola, S.; Del Piano, M.; Albano, E. Phlebotomy improves histology in chronic hepatitis c males with mild iron overload. World J. Gastroenterol. 2010, 16, 596–602. [Google Scholar] [CrossRef]
- Akkaya, O.; Kiyici, M.; Yilmaz, Y.; Ulukaya, E.; Yerci, O. Clinical significance of activity of alt enzyme in patients with hepatitis c virus. World J. Gastroenterol. 2007, 13, 5481–5485. [Google Scholar] [CrossRef]
- Hayashi, H.; Takikawa, T.; Nishimura, N.; Yano, M.; Isomura, T.; Sakamoto, N. Improvement of serum aminotransferase levels after phlebotomy in patients with chronic active hepatitis c and excess hepatic iron. Am. J. Gastroenterol. 1994, 89, 986–988. [Google Scholar]
- Kakizaki, S.; Takagi, H.; Horiguchi, N.; Toyoda, M.; Takayama, H.; Nagamine, T.; Mori, M.; Kato, N. Iron enhances hepatitis c virus replication in cultured human hepatocytes. Liver 2000, 20, 125–128. [Google Scholar] [CrossRef]
- Ali, N.; Siddiqui, A. The la antigen binds 5’ noncoding region of the hepatitis c virus rna in the context of the initiator aug codon and stimulates internal ribosome entry site-mediated translation. Proc. Natl. Acad. Sci. USA 1997, 94, 2249–2254. [Google Scholar] [CrossRef]
- Wolin, S.L.; Cedervall, T. The la protein. Annu. Rev. Biochem. 2002, 71, 375–403. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Liu, Y.; An, D.; Diao, H.; Xu, W.; He, X.; Sun, R.; Wei, L.; Li, L. Regulation of hepatitis c virus translation initiation by iron: Role of eif3 and la protein. Virus Res. 2012, 167, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Fillebeen, C.; Pantopoulos, K. Iron inhibits replication of infectious hepatitis c virus in permissive huh7.5.1 cells. J. Hepatol. 2010, 53, 995–999. [Google Scholar] [CrossRef] [PubMed]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of hiv-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. Hiv latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef]
- Justiz Vaillant, A.A.; Gulick, P.G. HIV Disease; Statpearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Ford, E.S.; Puronen, C.E.; Sereti, I. Immunopathogenesis of asymptomatic chronic hiv infection: The calm before the storm. Curr. Opin. HIV AIDS 2009, 4, 206–214. [Google Scholar] [CrossRef]
- Kappe, R.; Levitz, S.; Harrison, T.S.; Ruhnke, M.; Ampel, N.M.; Just-Nubling, G. Recent advances in cryptococcosis, candidiasis and coccidioidomycosis complicating hiv infection. Med. Mycol. 1998, 36 (Suppl. 1), 207–215. [Google Scholar]
- Sadowski, I.; Hashemi, F.B. Strategies to eradicate hiv from infected patients: Elimination of latent provirus reservoirs. Cell. Mol. Life Sci. 2019, 76, 3583–3600. [Google Scholar] [CrossRef]
- Kemnic, T.R.; Gulick, P.G. HIV Antiretroviral Therapy; Statpearls: Treasure Island, FL, USA, 2020. [Google Scholar]
- Riera, A.; Gimferrer, E.; Cadafalch, J.; Remacha, A.; Martin, S. Prevalence of high serum and red cell ferritin levels in hiv-infected patients. Haematologica 1994, 79, 165–167. [Google Scholar]
- Diebold, J.; Tabbara, W.; Marche, C.; Audouin, J.; Le Tourneau, A. Bone marrow changes at several stages of hiv infection, studied on bone marrow biopsies in 85 patients. Arch. Anat. Cytol. Pathol. 1991, 39, 137–146. [Google Scholar]
- Geremia, G.K.; McCluney, K.W.; Adler, S.S.; Charletta, D.A.; Hoile, R.D.; Huckman, M.S.; Ramsey, R.G. The magnetic resonance hypointense spine of aids. J. Comput. Assist. Tomogr. 1990, 14, 785–789. [Google Scholar] [CrossRef]
- Gelman, B.B.; Rodriguez-Wolf, M.G.; Wen, J.; Kumar, S.; Campbell, G.R.; Herzog, N. Siderotic cerebral macrophages in the acquired immunodeficiency syndrome. Arch. Pathol. Lab. Med. 1992, 116, 509–516. [Google Scholar] [PubMed]
- Smith, T.W.; DeGirolami, U.; Henin, D.; Bolgert, F.; Hauw, J.J. Human immunodeficiency virus (hiv) leukoencephalopathy and the microcirculation. J. Neuropathol. Exp. Neurol. 1990, 49, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, R.K.; Mhiri, C.; Baudrimont, M.; Roullet, E.; Berry, J.P.; Poirier, J. Iron pigment deposits, small vessel vasculitis, and erythrophagocytosis in the muscle of human immunodeficiency virus-infected patients. Hum. Pathol. 1991, 22, 1187–1194. [Google Scholar] [CrossRef]
- Savarino, A.; Pescarmona, G.P.; Boelaert, J.R. Iron metabolism and hiv infection: Reciprocal interactions with potentially harmful consequences? Cell Biochem. Funct. 1999, 17, 279–287. [Google Scholar] [CrossRef]
- Greaves, D.E.; Griffiths, W.J.; Lever, A.M. Does venesection reduce hiv viral load in patients with hereditary haemochromatosis? Antivir. Ther. 2013, 18, 135–138. [Google Scholar] [CrossRef]
- Xu, M.; Kashanchi, F.; Foster, A.; Rotimi, J.; Turner, W.; Gordeuk, V.R.; Nekhai, S. Hepcidin induces hiv-1 transcription inhibited by ferroportin. Retrovirology 2010, 7, 104. [Google Scholar] [CrossRef]
- Whalan, R.H.; Funnell, S.G.; Bowler, L.D.; Hudson, M.J.; Robinson, A.; Dowson, C.G. Distribution and genetic diversity of the abc transporter lipoproteins piua and piaa within streptococcus pneumoniae and related streptococci. J. Bacteriol. 2006, 188, 1031–1038. [Google Scholar] [CrossRef]
- Traore, H.N.; Meyer, D. The effect of iron overload on in vitro hiv-1 infection. J. Clin. Virol. 2004, 31 (Suppl. 1), S92–S98. [Google Scholar] [CrossRef]
- Nabel, G.; Baltimore, D. An inducible transcription factor activates expression of human immunodeficiency virus in t cells. Nature 1987, 326, 711–713. [Google Scholar] [CrossRef]
- Hoffbrand, A.V.; Ganeshaguru, K.; Hooton, J.W.; Tattersall, M.H. Effect of iron deficiency and desferrioxamine on DNA synthesis in human cells. Br. J. Haematol. 1976, 33, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.A.; van der Bruggen, T.; Oudshoorn, M.; Nottet, H.S.; Marx, J.J.; van Asbeck, B.S. Inhibition of human immunodeficiency virus type 1 replication in human mononuclear blood cells by the iron chelators deferoxamine, deferiprone, and bleomycin. J. Infect. Dis. 2000, 181, 484–490. [Google Scholar] [CrossRef] [PubMed]
- Georgiou, N.A.; van der Bruggen, T.; Oudshoorn, M.; Hider, R.C.; Marx, J.J.; van Asbeck, B.S. Human immunodeficiency virus type 1 replication inhibition by the bidentate iron chelators cp502 and cp511 is caused by proliferation inhibition and the onset of apoptosis. Eur. J. Clin. Investig. 2002, 32 (Suppl. 1), 91–96. [Google Scholar] [CrossRef] [PubMed]
- Debebe, Z.; Ammosova, T.; Jerebtsova, M.; Kurantsin-Mills, J.; Niu, X.; Charles, S.; Richardson, D.R.; Ray, P.E.; Gordeuk, V.R.; Nekhai, S. Iron chelators icl670 and 311 inhibit hiv-1 transcription. Virology 2007, 367, 324–333. [Google Scholar] [CrossRef] [PubMed]
- Kumari, N.; Iordanskiy, S.; Kovalskyy, D.; Breuer, D.; Niu, X.; Lin, X.; Xu, M.; Gavrilenko, K.; Kashanchi, F.; Dhawan, S.; et al. Phenyl-1-pyridin-2yl-ethanone-based iron chelators increase ikappab-alpha expression, modulate cdk2 and cdk9 activities, and inhibit hiv-1 transcription. Antimicrob. Agents Chemother. 2014, 58, 6558–6571. [Google Scholar] [CrossRef] [PubMed]
- Caza, M.; Kronstad, J.W. Shared and distinct mechanisms of iron acquisition by bacterial and fungal pathogens of humans. Front. Cell. Infect. Microbiol. 2013, 3, 80. [Google Scholar] [CrossRef]
- Navarre, W.W.; Schneewind, O. Surface proteins of gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol. Mol. Biol. Rev. 1999, 63, 174–229. [Google Scholar] [CrossRef]
- Garmory, H.S.; Titball, R.W. Atp-binding cassette transporters are targets for the development of antibacterial vaccines and therapies. Infect. Immun. 2004, 72, 6757–6763. [Google Scholar] [CrossRef]
- Radkov, A.D.; Hsu, Y.P.; Booher, G.; VanNieuwenhze, M.S. Imaging bacterial cell wall biosynthesis. Annu. Rev. Biochem. 2018, 87, 991–1014. [Google Scholar] [CrossRef]
- Delepelaire, P. Bacterial abc transporters of iron containing compounds. Res. Microbiol. 2019, 170, 345–357. [Google Scholar] [CrossRef]
- Runyen-Janecky, L.J. Role and regulation of heme iron acquisition in gram-negative pathogens. Front. Cell. Infect. Microbiol. 2013, 3, 55. [Google Scholar] [CrossRef] [PubMed]
- Schalk, I.J.; Yue, W.W.; Buchanan, S.K. Recognition of iron-free siderophores by tonb-dependent iron transporters. Mol. Microbiol. 2004, 54, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Larsen, R.A.; Foster-Hartnett, D.; McIntosh, M.A.; Postle, K. Regions of escherichia coli tonb and fepa proteins essential for in vivo physical interactions. J. Bacteriol. 1997, 179, 3213–3221. [Google Scholar] [CrossRef] [PubMed]
- Letoffe, S.; Redeker, V.; Wandersman, C. Isolation and characterization of an extracellular haem-binding protein from pseudomonas aeruginosa that shares function and sequence similarities with the serratia marcescens hasa haemophore. Mol. Microbiol. 1998, 28, 1223–1234. [Google Scholar] [CrossRef] [PubMed]
- Cope, L.D.; Thomas, S.E.; Hrkal, Z.; Hansen, E.J. Binding of heme-hemopexin complexes by soluble hxua protein allows utilization of this complexed heme by haemophilus influenzae. Infect. Immun. 1998, 66, 4511–4516. [Google Scholar] [CrossRef] [PubMed]
- Ochsner, U.A.; Vasil, M.L. Gene repression by the ferric uptake regulator in pseudomonas aeruginosa: Cycle selection of iron-regulated genes. Proc. Natl. Acad. Sci. USA 1996, 93, 4409–4414. [Google Scholar] [CrossRef] [PubMed]
- Schmitt, M.P.; Drazek, E.S. Construction and consequences of directed mutations affecting the hemin receptor in pathogenic corynebacterium species. J. Bacteriol. 2001, 183, 1476–1481. [Google Scholar] [CrossRef]
- Mazmanian, S.K.; Skaar, E.P.; Gaspar, A.H.; Humayun, M.; Gornicki, P.; Jelenska, J.; Joachmiak, A.; Missiakas, D.M.; Schneewind, O. Passage of heme-iron across the envelope of staphylococcus aureus. Science 2003, 299, 906–909. [Google Scholar] [CrossRef]
- Klockgether, J.; Cramer, N.; Wiehlmann, L.; Davenport, C.F.; Tummler, B. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2011, 2, 150. [Google Scholar] [CrossRef]
- Moradali, M.F.; Ghods, S.; Rehm, B.H. Pseudomonas aeruginosa lifestyle: A paradigm for adaptation, survival, and persistence. Front. Cell. Infect. Microbiol. 2017, 7, 39. [Google Scholar] [CrossRef]
- Ruffin, M.; Brochiero, E. Repair process impairment by pseudomonas aeruginosa in epithelial tissues: Major features and potential therapeutic avenues. Front. Cell. Infect. Microbiol. 2019, 9, 182. [Google Scholar] [CrossRef] [PubMed]
- Minandri, F.; Imperi, F.; Frangipani, E.; Bonchi, C.; Visaggio, D.; Facchini, M.; Pasquali, P.; Bragonzi, A.; Visca, P. Role of iron uptake systems in pseudomonas aeruginosa virulence and airway infection. Infect. Immun. 2016, 84, 2324–2335. [Google Scholar] [CrossRef] [PubMed]
- Brandel, J.; Humbert, N.; Elhabiri, M.; Schalk, I.J.; Mislin, G.L.; Albrecht-Gary, A.M. Pyochelin, a siderophore of pseudomonas aeruginosa: Physicochemical characterization of the iron(iii), copper(ii) and zinc(ii) complexes. Dalton Trans. 2012, 41, 2820–2834. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D.; Graham, R. Isolation of an iron-binding compound from pseudomonas aeruginosa. J. Bacteriol. 1979, 137, 357–364. [Google Scholar] [CrossRef]
- Mislin, G.L.; Hoegy, F.; Cobessi, D.; Poole, K.; Rognan, D.; Schalk, I.J. Binding properties of pyochelin and structurally related molecules to fpta of pseudomonas aeruginosa. J. Mol. Biol. 2006, 357, 1437–1448. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.D. Effect of pyochelin on the virulence of pseudomonas aeruginosa. Infect. Immun. 1982, 36, 17–23. [Google Scholar] [CrossRef]
- Takase, H.; Nitanai, H.; Hoshino, K.; Otani, T. Impact of siderophore production on pseudomonas aeruginosa infections in immunosuppressed mice. Infect. Immun. 2000, 68, 1834–1839. [Google Scholar] [CrossRef]
- Kim, M.; Christley, S.; Khodarev, N.N.; Fleming, I.; Huang, Y.; Chang, E.; Zaborina, O.; Alverdy, J.C. Pseudomonas aeruginosa wound infection involves activation of its iron acquisition system in response to fascial contact. J. Trauma Acute Care Surg. 2015, 78, 823–829. [Google Scholar] [CrossRef]
- Marshall, B.; Stintzi, A.; Gilmour, C.; Meyer, J.M.; Poole, K. Citrate-mediated iron uptake in pseudomonas aeruginosa: Involvement of the citrate-inducible feca receptor and the feob ferrous iron transporter. Microbiology 2009, 155, 305–315. [Google Scholar] [CrossRef]
- Ochsner, U.A.; Johnson, Z.; Vasil, M.L. Genetics and regulation of two distinct haem-uptake systems, phu and has, in pseudomonas aeruginosa. Microbiology 2000, 146 Pt 1, 185–198. [Google Scholar] [CrossRef]
- Bhagirath, A.Y.; Li, Y.; Somayajula, D.; Dadashi, M.; Badr, S.; Duan, K. Cystic fibrosis lung environment and pseudomonas aeruginosa infection. BMC Pulm. Med. 2016, 16, 174. [Google Scholar] [CrossRef] [PubMed]
- Aali, M.; Caldwell, A.; House, K.; Zhou, J.; Chappe, V.; Lehmann, C. Iron chelation as novel treatment for lung inflammation in cystic fibrosis. Med. Hypotheses 2017, 104, 86–88. [Google Scholar] [CrossRef] [PubMed]
- Ankley, L.M.; Monteiro, M.P.; Camp, K.M.; O’Quinn, R.; Castillo, A.R. Manuka honey chelates iron and impacts iron regulation in key bacterial pathogens. J. Appl. Microbiol. 2020, 128, 1015–1024. [Google Scholar] [CrossRef] [PubMed]
- Valm, A.M. The structure of dental plaque microbial communities in the transition from health to dental caries and periodontal disease. J. Mol. Biol. 2019, 431, 2957–2969. [Google Scholar] [CrossRef]
- Naito, M.; Ogura, Y.; Itoh, T.; Shoji, M.; Okamoto, M.; Hayashi, T.; Nakayama, K. The complete genome sequencing of prevotella intermedia strain oma14 and a subsequent fine-scale, intra-species genomic comparison reveal an unusual amplification of conjugative and mobile transposons and identify a novel prevotella-lineage-specific repeat. DNA Res. 2016, 23, 11–19. [Google Scholar]
- Ang, M.Y.; Dutta, A.; Wee, W.Y.; Dymock, D.; Paterson, I.C.; Choo, S.W. Comparative genome analysis of fusobacterium nucleatum. Genome Biol. Evol. 2016, 8, 2928–2938. [Google Scholar] [CrossRef]
- Kabwe, M.; Brown, T.L.; Dashper, S.; Speirs, L.; Ku, H.; Petrovski, S.; Chan, H.T.; Lock, P.; Tucci, J. Genomic, morphological and functional characterisation of novel bacteriophage fnu1 capable of disrupting fusobacterium nucleatum biofilms. Sci. Rep. 2019, 9, 9107. [Google Scholar] [CrossRef]
- Subbarao, K.C.; Nattuthurai, G.S.; Sundararajan, S.K.; Sujith, I.; Joseph, J.; Syedshah, Y.P. Gingival crevicular fluid: An overview. J. Pharm. Bioallied Sci. 2019, 11, S135–S139. [Google Scholar] [CrossRef]
- Duchesne, P.; Grenier, D.; Mayrand, D. Binding and utilization of human transferrin by prevotella nigrescens. Infect. Immun. 1999, 67, 576–580. [Google Scholar] [CrossRef]
- Nelson, K.E.; Fleischmann, R.D.; DeBoy, R.T.; Paulsen, I.T.; Fouts, D.E.; Eisen, J.A.; Daugherty, S.C.; Dodson, R.J.; Durkin, A.S.; Gwinn, M.; et al. Complete genome sequence of the oral pathogenic bacterium porphyromonas gingivalis strain w83. J. Bacteriol. 2003, 185, 5591–5601. [Google Scholar] [CrossRef]
- Bramanti, T.E.; Holt, S.C. Roles of porphyrins and host iron transport proteins in regulation of growth of porphyromonas gingivalis w50. J. Bacteriol. 1991, 173, 7330–7339. [Google Scholar] [CrossRef] [PubMed]
- Wyss, C. Growth of porphyromonas gingivalis, treponema denticola, t. Pectinovorum, t. Socranskii, and t. Vincentii in a chemically defined medium. J. Clin. Microbiol. 1992, 30, 2225–2229. [Google Scholar] [CrossRef] [PubMed]
- Shoji, M.; Shibata, Y.; Shiroza, T.; Yukitake, H.; Peng, B.; Chen, Y.Y.; Sato, K.; Naito, M.; Abiko, Y.; Reynolds, E.C.; et al. Characterization of hemin-binding protein 35 (hbp35) in porphyromonas gingivalis: Its cellular distribution, thioredoxin activity and role in heme utilization. BMC Microbiol. 2010, 10, 152. [Google Scholar] [CrossRef] [PubMed]
- Schifferle, R.E.; Shostad, S.A.; Bayers-Thering, M.T.; Dyer, D.W.; Neiders, M.E. Effect of protoporphyrin ix limitation on porphyromonas gingivalis. J. Endod. 1996, 22, 352–355. [Google Scholar] [CrossRef]
- Olczak, T.; Simpson, W.; Liu, X.; Genco, C.A. Iron and heme utilization in porphyromonas gingivalis. FEMS Microbiol. Rev. 2005, 29, 119–144. [Google Scholar] [CrossRef]
- Bramanti, T.E.; Holt, S.C. Hemin uptake in porphyromonas gingivalis: Omp26 is a hemin-binding surface protein. J. Bacteriol. 1993, 175, 7413–7420. [Google Scholar] [CrossRef][Green Version]
- Sroka, A.; Sztukowska, M.; Potempa, J.; Travis, J.; Genco, C.A. Degradation of host heme proteins by lysine-and arginine-specific cysteine proteinases (gingipains) of porphyromonas gingivalis. J. Bacteriol. 2001, 183, 5609–5616. [Google Scholar] [CrossRef]
- Lewis, J.P. Metal uptake in host-pathogen interactions: Role of iron in porphyromonas gingivalis interactions with host organisms. Periodontol. 2000 2010, 52, 94–116. [Google Scholar] [CrossRef]
- Shi, Y.; Ratnayake, D.B.; Okamoto, K.; Abe, N.; Yamamoto, K.; Nakayama, K. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutination of porphyromonas gingivalis. Construction of mutants with a combination of rgpa, rgpb, kgp, and haga. J. Biol. Chem. 1999, 274, 17955–17960. [Google Scholar] [CrossRef]
- Byrne, D.P.; Potempa, J.; Olczak, T.; Smalley, J.W. Evidence of mutualism between two periodontal pathogens: Co-operative haem acquisition by the hmuy haemophore of porphyromonas gingivalis and the cysteine protease interpain a (inpa) of prevotella intermedia. Mol. Oral Microbiol. 2013, 28, 219–229. [Google Scholar] [CrossRef]
- Okamoto, M.; Maeda, N.; Kondo, K.; Leung, K.P. Hemolytic and hemagglutinating activities of prevotella intermedia and prevotella nigrescens. FEMS Microbiol. Lett. 1999, 178, 299–304. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Byrne, D.P.; Wawrzonek, K.; Jaworska, A.; Birss, A.J.; Potempa, J.; Smalley, J.W. Role of the cysteine protease interpain a of prevotella intermedia in breakdown and release of haem from haemoglobin. Biochem. J. 2009, 425, 257–264. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moon, J.H.; Herr, Y.; Kim, S.W.; Lee, J.Y. In vitro activity of deferoxamine against porphyromonas gingivalis. FEMS Microbiol. Lett. 2011, 323, 61–67. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Moon, J.H.; Kim, C.; Lee, H.S.; Kim, S.W.; Lee, J.Y. Antibacterial and antibiofilm effects of iron chelators against prevotella intermedia. J. Med. Microbiol. 2013, 62, 1307–1316. [Google Scholar] [CrossRef]
- Ben Lagha, A.; Dudonne, S.; Desjardins, Y.; Grenier, D. Wild blueberry (vaccinium angustifolium ait.) polyphenols target fusobacterium nucleatum and the host inflammatory response: Potential innovative molecules for treating periodontal diseases. J. Agric. Food Chem. 2015, 63, 6999–7008. [Google Scholar] [CrossRef]
- Ben Lagha, A.; Haas, B.; Grenier, D. Tea polyphenols inhibit the growth and virulence properties of fusobacterium nucleatum. Sci. Rep. 2017, 7, 44815. [Google Scholar] [CrossRef]
- Ardila, C.M.; Granada, M.I.; Guzman, I.C. Antibiotic resistance of subgingival species in chronic periodontitis patients. J. Periodontal Res. 2010, 45, 557–563. [Google Scholar] [CrossRef]
- Walker, C.B.; Bueno, L.C. Antibiotic resistance in an oral isolate of prevotella intermedia. Clin. Infect. Dis. 1997, 25 (Suppl. 2), S281–S283. [Google Scholar] [CrossRef]
- Bostanci, N.; Belibasakis, G.N. Porphyromonas gingivalis: An invasive and evasive opportunistic oral pathogen. FEMS Microbiol. Lett. 2012, 333, 1–9. [Google Scholar] [CrossRef]
- Donati, C.; Hiller, N.L.; Tettelin, H.; Muzzi, A.; Croucher, N.J.; Angiuoli, S.V.; Oggioni, M.; Dunning Hotopp, J.C.; Hu, F.Z.; Riley, D.R.; et al. Structure and dynamics of the pan-genome of streptococcus pneumoniae and closely related species. Genome Biol. 2010, 11, R107. [Google Scholar] [CrossRef]
- Hoyer, J.; Bartel, J.; Gomez-Mejia, A.; Rohde, M.; Hirschfeld, C.; Hess, N.; Sura, T.; Maass, S.; Hammerschmidt, S.; Becher, D. Proteomic response of streptococcus pneumoniae to iron limitation. Int. J. Med. Microbiol. 2018, 308, 713–721. [Google Scholar] [CrossRef] [PubMed]
- Tai, S.S.; Lee, C.J.; Winter, R.E. Hemin utilization is related to virulence of streptococcus pneumoniae. Infect. Immun. 1993, 61, 5401–5405. [Google Scholar] [CrossRef] [PubMed]
- Whatmore, A.M.; Dowson, C.G. The autolysin-encoding gene (lyta) of streptococcus pneumoniae displays restricted allelic variation despite localized recombination events with genes of pneumococcal bacteriophage encoding cell wall lytic enzymes. Infect. Immun. 1999, 67, 4551–4556. [Google Scholar] [CrossRef] [PubMed]
- Steinfort, C.; Wilson, R.; Mitchell, T.; Feldman, C.; Rutman, A.; Todd, H.; Sykes, D.; Walker, J.; Saunders, K.; Andrew, P.W.; et al. Effect of streptococcus pneumoniae on human respiratory epithelium in vitro. Infect. Immun. 1989, 57, 2006–2013. [Google Scholar] [CrossRef]
- Brown, J.S.; Gilliland, S.M.; Holden, D.W. A streptococcus pneumoniae pathogenicity island encoding an abc transporter involved in iron uptake and virulence. Mol. Microbiol. 2001, 40, 572–585. [Google Scholar] [CrossRef]
- Brown, J.S.; Gilliland, S.M.; Ruiz-Albert, J.; Holden, D.W. Characterization of pit, a streptococcus pneumoniae iron uptake abc transporter. Infect. Immun. 2002, 70, 4389–4398. [Google Scholar] [CrossRef] [PubMed]
- Ehrt, S.; Schnappinger, D.; Rhee, K.Y. Metabolic principles of persistence and pathogenicity in mycobacterium tuberculosis. Nat. Rev. Microbiol. 2018, 16, 496–507. [Google Scholar] [CrossRef]
- Chao, A.; Sieminski, P.J.; Owens, C.P.; Goulding, C.W. Iron acquisition in mycobacterium tuberculosis. Chem. Rev. 2019, 119, 1193–1220. [Google Scholar] [CrossRef]
- Olakanmi, O.; Kesavalu, B.; Abdalla, M.Y.; Britigan, B.E. Iron acquisition by mycobacterium tuberculosis residing within myeloid dendritic cells. Microb. Pathog. 2013, 65, 21–28. [Google Scholar] [CrossRef]
- Dragset, M.S.; Poce, G.; Alfonso, S.; Padilla-Benavides, T.; Ioerger, T.R.; Kaneko, T.; Sacchettini, J.C.; Biava, M.; Parish, T.; Arguello, J.M.; et al. A novel antimycobacterial compound acts as an intracellular iron chelator. Antimicrob. Agents Chemother. 2015, 59, 2256–2264. [Google Scholar] [CrossRef]
- Lima, S.L.; Colombo, A.L.; de Almeida Junior, J.N. Fungal cell wall: Emerging antifungals and drug resistance. Front. Microbiol. 2019, 10, 2573. [Google Scholar] [CrossRef] [PubMed]
- Gerwien, F.; Skrahina, V.; Kasper, L.; Hube, B.; Brunke, S. Metals in fungal virulence. FEMS Microbiol. Rev. 2018, 42. [Google Scholar] [CrossRef] [PubMed]
- Outten, C.E.; Albetel, A.N. Iron sensing and regulation in saccharomyces cerevisiae: Ironing out the mechanistic details. Curr. Opin. Microbiol. 2013, 16, 662–668. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.H. Acquisition, transport, and storage of iron by pathogenic fungi. Clin. Microbiol. Rev. 1999, 12, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Howard, D.H. Iron gathering by zoopathogenic fungi. FEMS Immunol. Med. Microbiol. 2004, 40, 95–100. [Google Scholar] [CrossRef][Green Version]
- Bairwa, G.; Hee Jung, W.; Kronstad, J.W. Iron acquisition in fungal pathogens of humans. Met. Integr. Biometal Sci. 2017, 9, 215–227. [Google Scholar] [CrossRef]
- Kim, J.; Cho, Y.J.; Do, E.; Choi, J.; Hu, G.; Cadieux, B.; Chun, J.; Lee, Y.; Kronstad, J.W.; Jung, W.H. A defect in iron uptake enhances the susceptibility of cryptococcus neoformans to azole antifungal drugs. Fungal Genet. Biol. 2012, 49, 955–966. [Google Scholar] [CrossRef]
- Holz, R.W. The effects of the polyene antibiotics nystatin and amphotericin b on thin lipid membranes. Ann. N. Y. Acad. Sci. 1974, 235, 469–479. [Google Scholar] [CrossRef]
- Thamban Chandrika, N.; Shrestha, S.K.; Ngo, H.X.; Howard, K.C.; Garneau-Tsodikova, S. Novel fluconazole derivatives with promising antifungal activity. Bioorgan. Med. Chem. 2018, 26, 573–580. [Google Scholar] [CrossRef]
- Jung, W.H.; Sham, A.; Lian, T.; Singh, A.; Kosman, D.J.; Kronstad, J.W. Iron source preference and regulation of iron uptake in cryptococcus neoformans. PLoS Pathog. 2008, 4, e45. [Google Scholar] [CrossRef]
- Jung, W.H.; Hu, G.; Kuo, W.; Kronstad, J.W. Role of ferroxidases in iron uptake and virulence of cryptococcus neoformans. Eukaryot. Cell 2009, 8, 1511–1520. [Google Scholar] [PubMed]
- Ibrahim, A.S.; Gebremariam, T.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. The iron chelator deferasirox enhances liposomal amphotericin b efficacy in treating murine invasive pulmonary aspergillosis. J. Antimicrob. Chemother. 2010, 65, 289–292. [Google Scholar] [PubMed]
- Jenks, J.D.; Hoenigl, M. Treatment of aspergillosis. J. Fungi 2018, 4, 98. [Google Scholar]
- Haas, H. Fungal siderophore metabolism with a focus on aspergillus fumigatus. Nat. Prod. Rep. 2014, 31, 1266–1276. [Google Scholar]
- Blatzer, M.; Binder, U.; Haas, H. The metalloreductase freb is involved in adaptation of aspergillus fumigatus to iron starvation. Fungal Genet. Biol. 2011, 48, 1027–1033. [Google Scholar]
- Schrettl, M.; Bignell, E.; Kragl, C.; Joechl, C.; Rogers, T.; Arst, H.N., Jr.; Haynes, K.; Haas, H. Siderophore biosynthesis but not reductive iron assimilation is essential for aspergillus fumigatus virulence. J. Exp. Med. 2004, 200, 1213–1219. [Google Scholar]
- Schrettl, M.; Kim, H.S.; Eisendle, M.; Kragl, C.; Nierman, W.C.; Heinekamp, T.; Werner, E.R.; Jacobsen, I.; Illmer, P.; Yi, H.; et al. Srea-mediated iron regulation in aspergillus fumigatus. Mol. Microbiol. 2008, 70, 27–43. [Google Scholar]
- Schrettl, M.; Beckmann, N.; Varga, J.; Heinekamp, T.; Jacobsen, I.D.; Jochl, C.; Moussa, T.A.; Wang, S.; Gsaller, F.; Blatzer, M.; et al. Hapx-mediated adaption to iron starvation is crucial for virulence of aspergillus fumigatus. PLoS Pathog. 2010, 6, e1001124. [Google Scholar]
- Artis, W.M.; Fountain, J.A.; Delcher, H.K.; Jones, H.E. A mechanism of susceptibility to mucormycosis in diabetic ketoacidosis: Transferrin and iron availability. Diabetes 1982, 31, 1109–1114. [Google Scholar]
- Yamauchi, T.; Misaki, H.; Arai, H.; Iwasaki, H.; Naiki, H.; Ueda, T. An autopsy case of disseminated mucormycosis in a neutropenic patient receiving chemotherapy for the underlying solid malignancy. J. Infect. Chemother. 2002, 8, 103–105. [Google Scholar]
- Singh, N.; Aguado, J.M.; Bonatti, H.; Forrest, G.; Gupta, K.L.; Safdar, N.; John, G.T.; Pursell, K.J.; Munoz, P.; Patel, R.; et al. Zygomycosis in solid organ transplant recipients: A prospective, matched case-control study to assess risks for disease and outcome. J. Infect. Dis. 2009, 200, 1002–1011. [Google Scholar] [CrossRef]
- Almyroudis, N.G.; Sutton, D.A.; Linden, P.; Rinaldi, M.G.; Fung, J.; Kusne, S. Zygomycosis in solid organ transplant recipients in a tertiary transplant center and review of the literature. Am. J. Transpl. 2006, 6, 2365–2374. [Google Scholar] [CrossRef] [PubMed]
- Sawaya, B.P.; Weihprecht, H.; Campbell, W.R.; Lorenz, J.N.; Webb, R.C.; Briggs, J.P.; Schnermann, J. Direct vasoconstriction as a possible cause for amphotericin b-induced nephrotoxicity in rats. J. Clin. Investig. 1991, 87, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Spellberg, B.; Edwards, J., Jr. Iron acquisition: A novel perspective on mucormycosis pathogenesis and treatment. Curr. Opin. Infect. Dis. 2008, 21, 620–625. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.J.; Ibrahim, A.S.; Skory, C.; Grabherr, M.G.; Burger, G.; Butler, M.; Elias, M.; Idnurm, A.; Lang, B.F.; Sone, T.; et al. Genomic analysis of the basal lineage fungus rhizopus oryzae reveals a whole-genome duplication. PLoS Genet. 2009, 5, e1000549. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, A.S.; Spellberg, B.; Walsh, T.J.; Kontoyiannis, D.P. Pathogenesis of mucormycosis. Clin. Infect. Dis. 2012, 54 (Suppl. 1), S16–S22. [Google Scholar] [CrossRef]
- Liu, M.; Lin, L.; Gebremariam, T.; Luo, G.; Skory, C.D.; French, S.W.; Chou, T.F.; Edwards, J.E., Jr.; Ibrahim, A.S. Fob1 and fob2 proteins are virulence determinants of rhizopus oryzae via facilitating iron uptake from ferrioxamine. PLoS Pathog. 2015, 11, e1004842. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Gebremariam, T.; Lin, L.; Luo, G.; Husseiny, M.I.; Skory, C.D.; Fu, Y.; French, S.W.; Edwards, J.E., Jr.; Spellberg, B. The high affinity iron permease is a key virulence factor required for rhizopus oryzae pathogenesis. Mol. Microbiol. 2010, 77, 587–604. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Gebermariam, T.; Fu, Y.; Lin, L.; Husseiny, M.I.; French, S.W.; Schwartz, J.; Skory, C.D.; Edwards, J.E., Jr.; Spellberg, B.J. The iron chelator deferasirox protects mice from mucormycosis through iron starvation. J. Clin. Investig. 2007, 117, 2649–2657. [Google Scholar] [CrossRef]
- Ibrahim, A.S.; Edwards, J.E., Jr.; Fu, Y.; Spellberg, B. Deferiprone iron chelation as a novel therapy for experimental mucormycosis. J. Antimicrob. Chemother. 2006, 58, 1070–1073. [Google Scholar] [CrossRef]
- Boelaert, J.R.; de Locht, M.; Van Cutsem, J.; Kerrels, V.; Cantinieaux, B.; Verdonck, A.; Van Landuyt, H.W.; Schneider, Y.J. Mucormycosis during deferoxamine therapy is a siderophore-mediated infection. In vitro and in vivo animal studies. J. Clin. Investig. 1993, 91, 1979–1986. [Google Scholar] [CrossRef] [PubMed]
- Boelaert, J.R.; Van Cutsem, J.; de Locht, M.; Schneider, Y.J.; Crichton, R.R. Deferoxamine augments growth and pathogenicity of rhizopus, while hydroxypyridinone chelators have no effect. Kidney Int. 1994, 45, 667–671. [Google Scholar] [CrossRef] [PubMed]
- Reed, C.; Ibrahim, A.; Edwards, J.E., Jr.; Walot, I.; Spellberg, B. Deferasirox, an iron-chelating agent, as salvage therapy for rhinocerebral mucormycosis. Antimicrob. Agents Chemother. 2006, 50, 3968–3969. [Google Scholar] [CrossRef] [PubMed]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chhabra, R.; Saha, A.; Chamani, A.; Schneider, N.; Shah, R.; Nanjundan, M. Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals 2020, 13, 275. https://doi.org/10.3390/ph13100275
Chhabra R, Saha A, Chamani A, Schneider N, Shah R, Nanjundan M. Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals. 2020; 13(10):275. https://doi.org/10.3390/ph13100275
Chicago/Turabian StyleChhabra, Ravneet, Aishwarya Saha, Ashkon Chamani, Nicole Schneider, Riya Shah, and Meera Nanjundan. 2020. "Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections" Pharmaceuticals 13, no. 10: 275. https://doi.org/10.3390/ph13100275
APA StyleChhabra, R., Saha, A., Chamani, A., Schneider, N., Shah, R., & Nanjundan, M. (2020). Iron Pathways and Iron Chelation Approaches in Viral, Microbial, and Fungal Infections. Pharmaceuticals, 13(10), 275. https://doi.org/10.3390/ph13100275