ZeOncoTest: Refining and Automating the Zebrafish Xenograft Model for Drug Discovery in Cancer

Abstract
:1. Introduction
2. Results
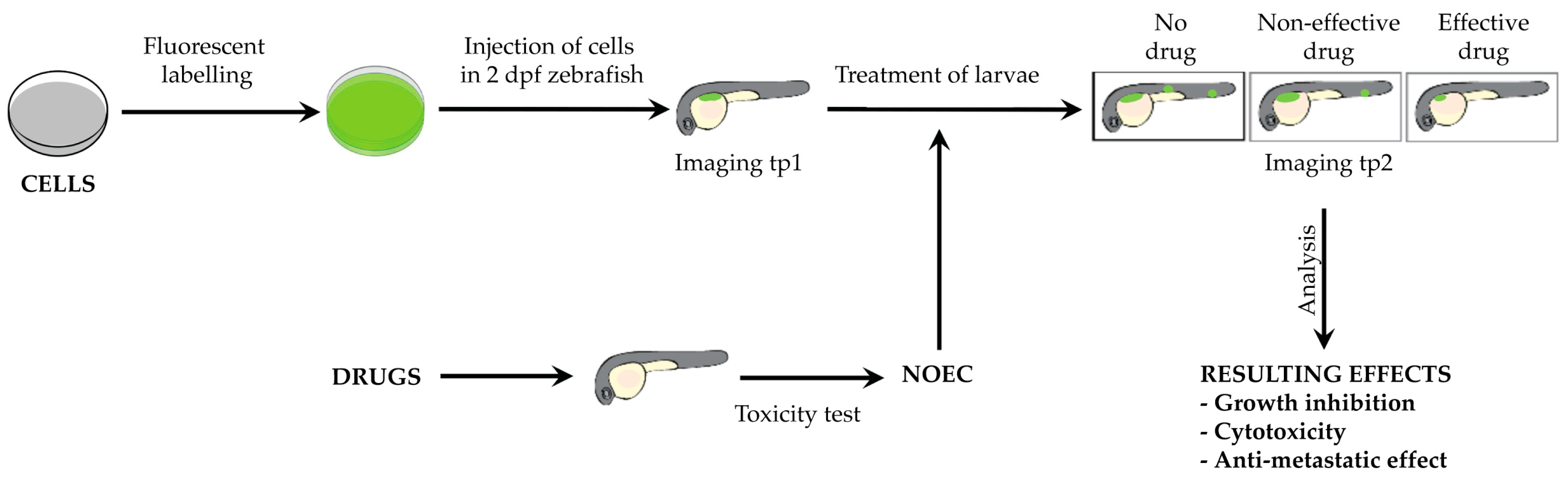
2.1. ZeOncoTest: General Experimental Setup and Workflow
2.2. Improved and New Methodologies
2.2.1. 3D Imaging, Automation, and Throughput
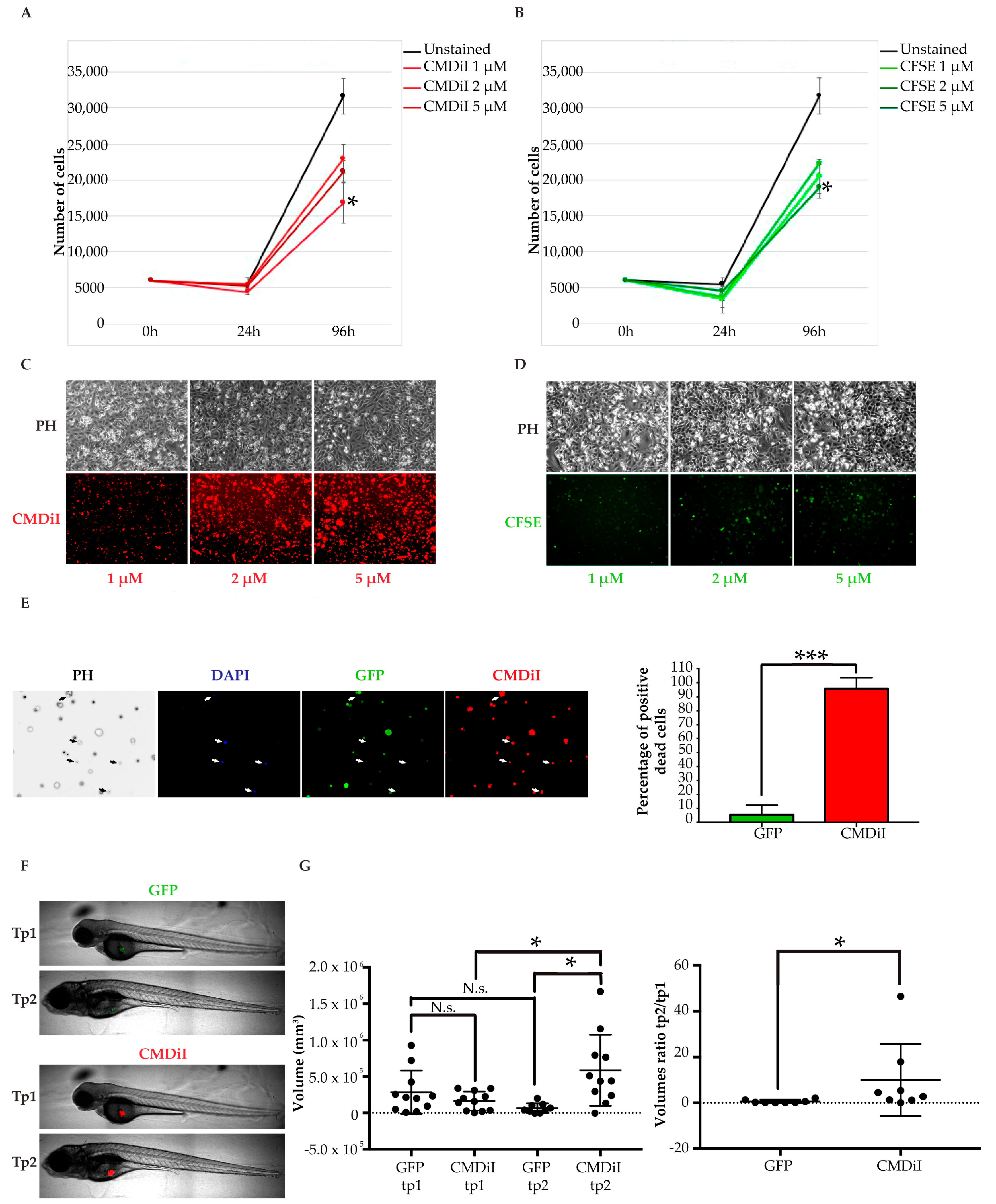
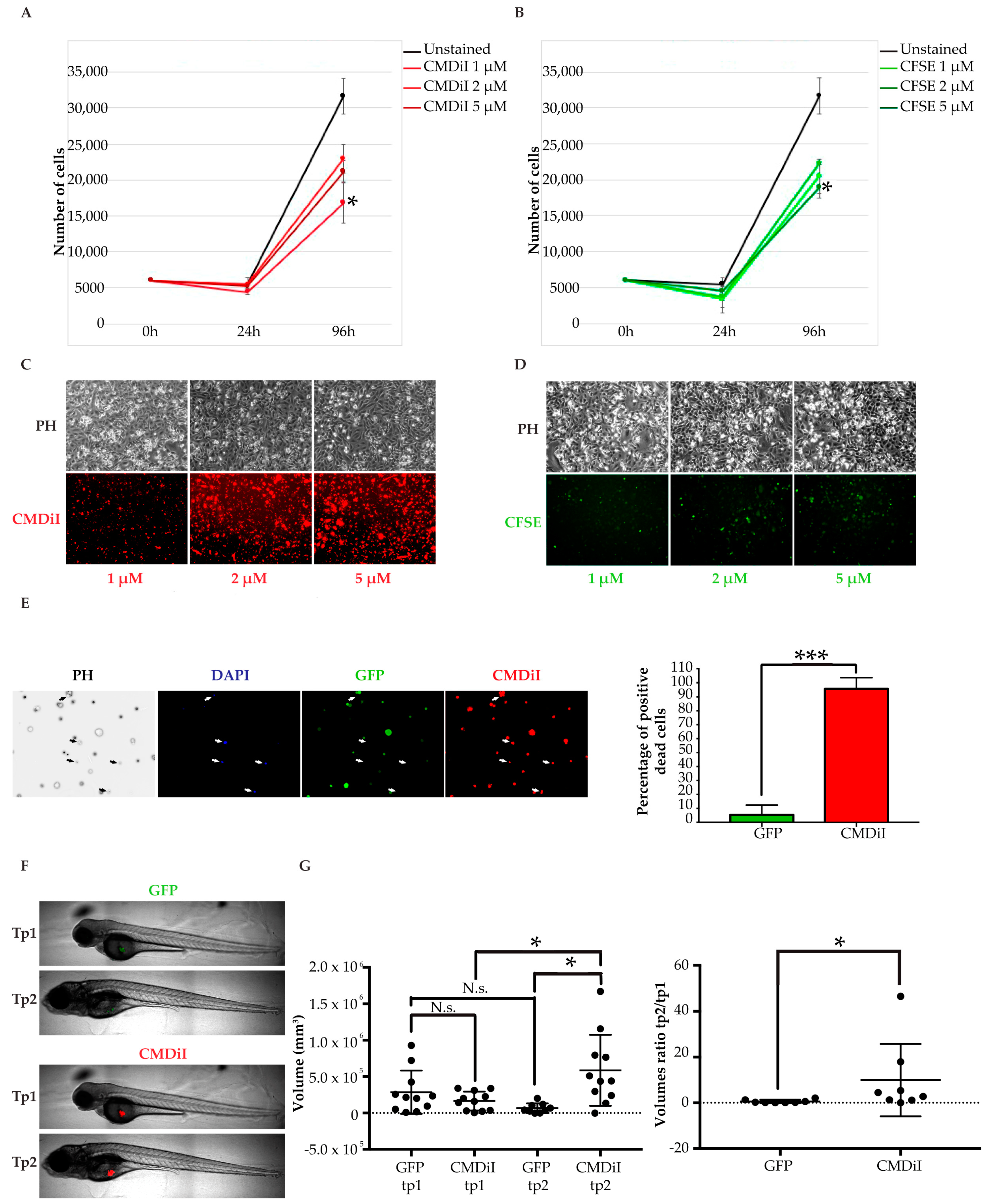
2.2.2. Choice of a Suitable Cell Labeling Method
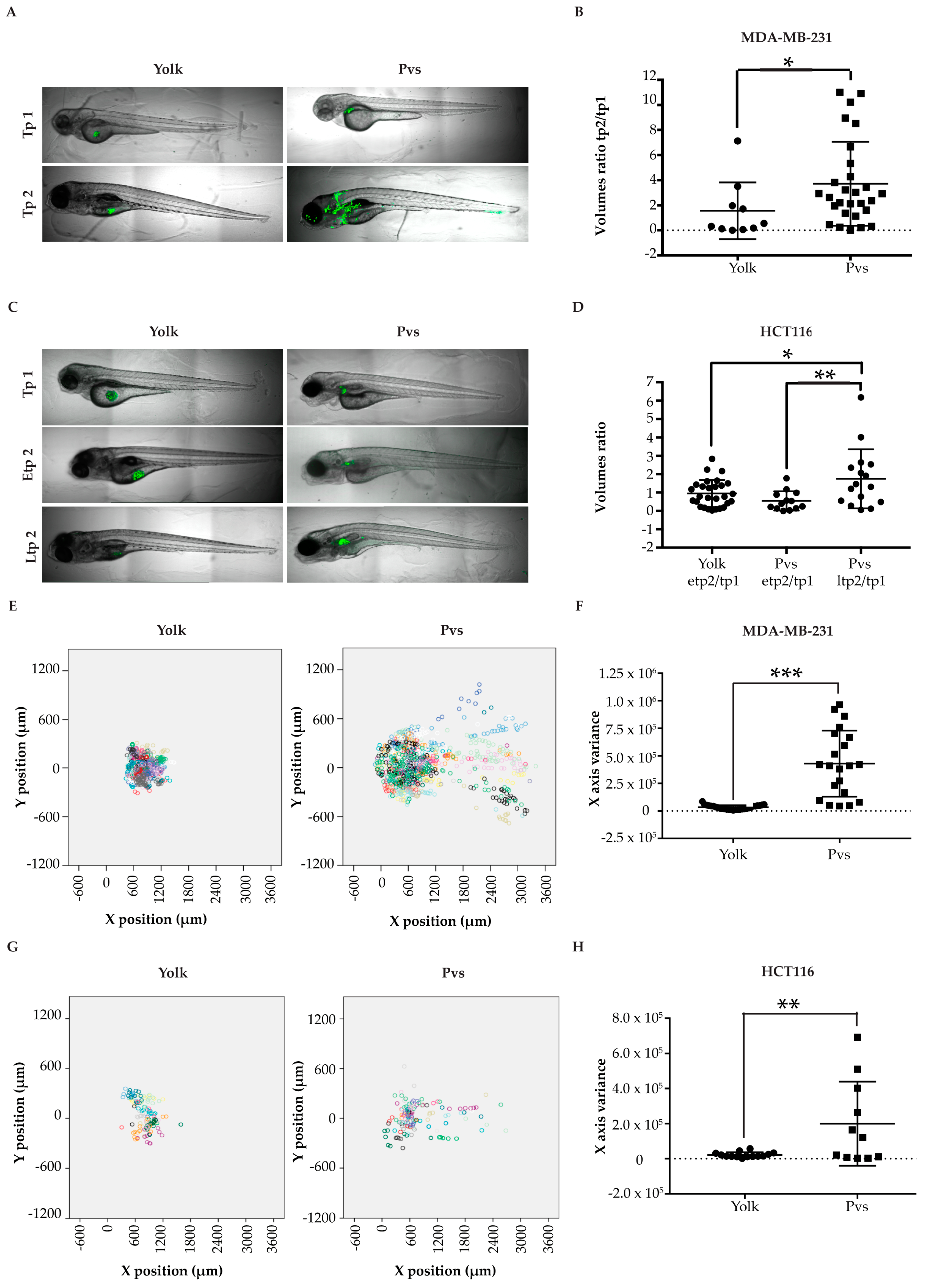
2.2.3. Establishment of an Appropriate Injection Site
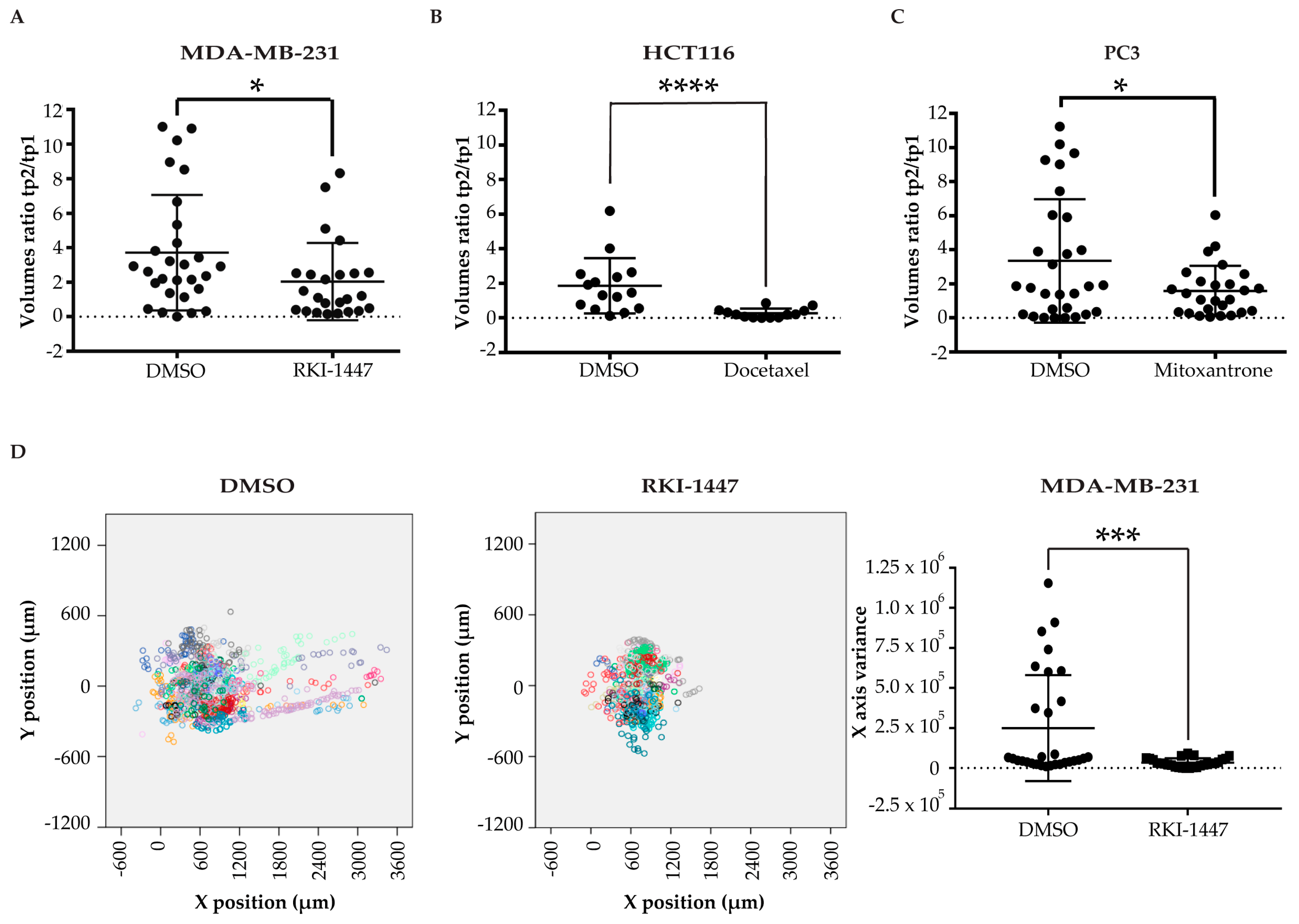
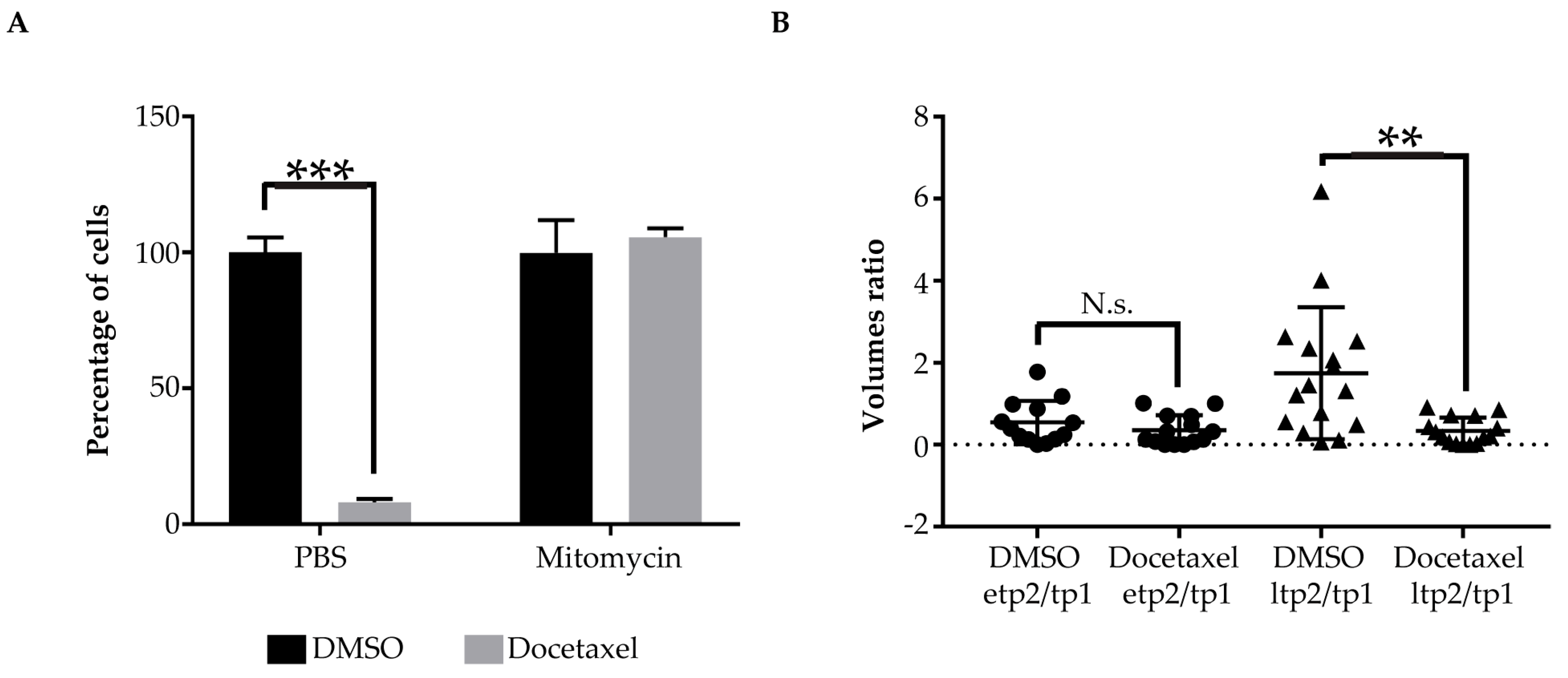
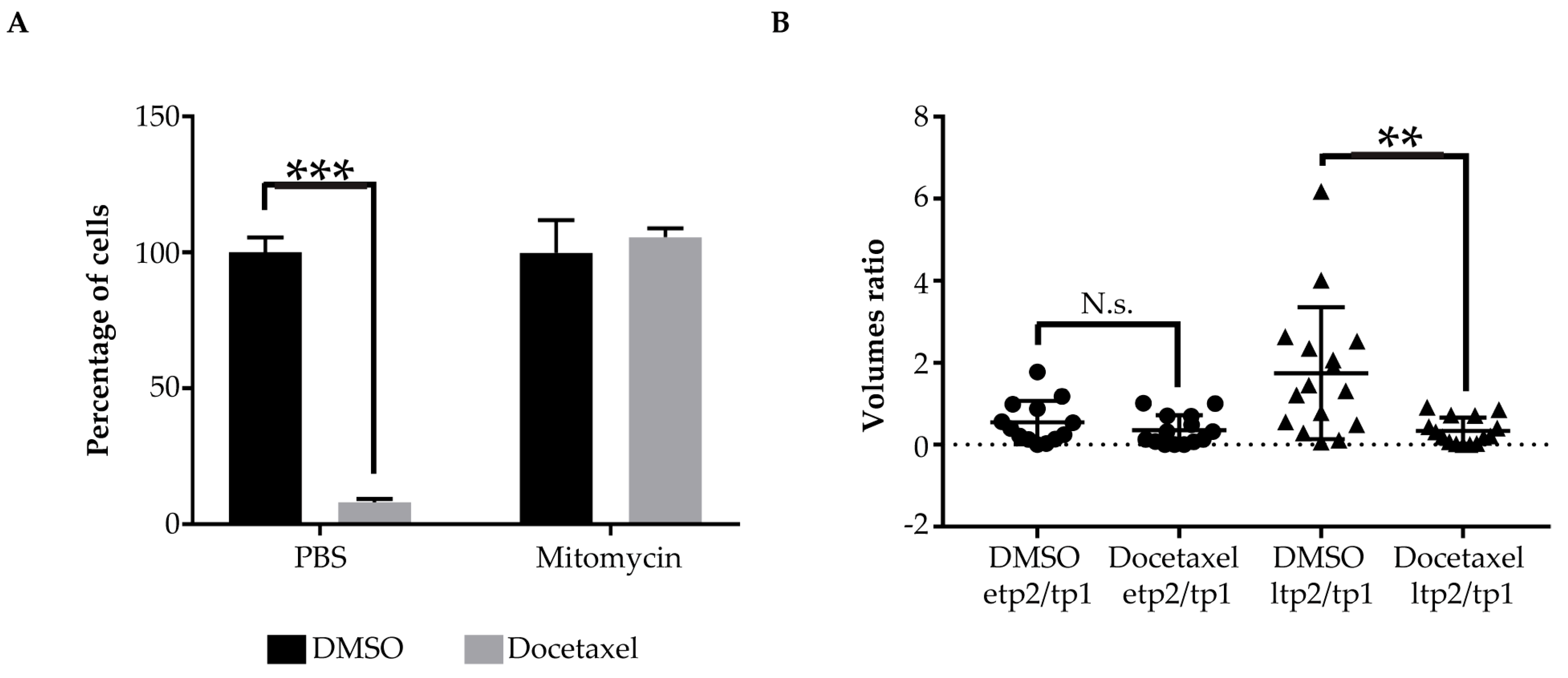
2.3. Pharmacological Validation of the ZeOncoTest
2.4. Addressing Drugs Mechanism of Action with the ZeOncoTest
3. Discussion
4. Materials and Methods
4.1. Cell Lines and Zebrafish Handling
4.2. Infection and Dye Staining of Human Cell Lines
4.3. Induction of Cell Death and Hexosaminidase Assay
4.4. Zebrafish Injection
4.5. Drug Treatment
4.6. Automated Confocal Imaging and Analysis
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibue, T.; Weinberg, R.A. EMT, CSCs, and drug resistance: The mechanistic link and clinical implications. Nat. Rev. Clin. Oncol. 2017, 14, 611–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Marion, D.M.S.; Domanska, U.M.; Timmer-Bosscha, H.; Walenkamp, A.M.E. Studying cancer metastasis: Existing models, challenges and future perspectives. Crit. Rev. Oncol. Hematol. 2016, 97, 107–117. [Google Scholar] [CrossRef]
- Sia, D.; Moeini, A.; Labgaa, I.; Villanueva, A. The future of patient-derived tumor xenografts in cancer treatment. Pharmacogenomics 2015, 16, 1671–1683. [Google Scholar] [CrossRef]
- DeBord, L.C.; Pathak, R.R.; Villaneuva, M.; Liu, H.-C.; Harrington, D.A.; Yu, W.; Lewis, M.T.; Sikora, A.G. The chick chorioallantoic membrane (CAM) as a versatile patient-derived xenograft (PDX) platform for precision medicine and preclinical research. Am. J. Cancer Res. 2018, 8, 1642–1660. [Google Scholar]
- Sánchez, N.S.; Mills, G.B.; Shaw, K.R.M. Precision oncology: Neither a silver bullet nor a dream. Pharmacogenomics 2017, 18, 1525–1539. [Google Scholar] [CrossRef]
- Moreno, L.; Pearson, A.D. How can attrition rates be reduced in cancer drug discovery? Expert Opin. Drug Discov. 2013, 8, 363–368. [Google Scholar] [CrossRef]
- Workman, P.; Draetta, G.F.; Schellens, J.H.M.; Bernards, R. How Much Longer Will We Put Up with 100,000 Cancer Drugs? Cell 2017, 168, 579–583. [Google Scholar] [CrossRef] [Green Version]
- Kari, G.; Rodeck, U.; Dicker, A.P. Zebrafish: An Emerging Model System for Human Disease and Drug Discovery. Clin. Pharmacol. Ther. 2007, 82, 70–80. [Google Scholar] [CrossRef]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Ito, T.; Ando, H.; Imamura, Y.; Yamaguchi, Y.; Handa, H.; Ito, T.; Ando, H.; Suzuki, T.; Ogura, T.; Hotta, K. Identification of a Primary Target of Thalidomide Teratogenicity Linked references are available on JSTOR for this article: Identification of a Primary Target of Thalidomide Teratogenicity. Science 2010, 327, 1345–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Terriente, J.; Pujades, C. Use of Zebrafish Embryos for Small Molecule Screening Related to Cancer. Dev. Dyn. 2013, 242, 97–107. [Google Scholar] [CrossRef] [PubMed]
- Letrado, P.; De Miguel, I.; Lamberto, I.; Díez-Martínez, R.; Oyarzabal, J. Zebrafish: Speeding up the cancer drug discovery process. Cancer Res. 2018, 78, 6048–6058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beckwith, L.G.; Moore, J.L.; Tsao-wu, G.S.; Harshbarger, J.C.; Cheng, K.C. Ethylnitrosourea Induces Neoplasia in Zebrafish (Danio rerio). Lab. Investig. 2000, 80, 379–385. [Google Scholar] [CrossRef] [Green Version]
- Kirchberger, S.; Sturtzel, C.; Pascoal, S.; Distel, M. Quo natas, Danio?—Recent Progress in Modeling Cancer in Zebrafish. Front. Oncol. 2017, 7, 186. [Google Scholar] [CrossRef]
- Ignatius, M.S.; Hayes, M.N.; Moore, F.E.; Tang, Q.; Garcia, S.P.; Blackburn, P.R.; Baxi, K.; Wang, L.; Jin, A.; Ramakrishnan, A.; et al. Tp53 Deficiency Causes a Wide Tumor Spectrum and Increases Embryonal Rhabdomyosarcoma Metastasis in Zebrafish. eLife 2018, 7, 1–19. [Google Scholar] [CrossRef]
- Howe, K.; Clark, M.D.; Torroja, C.F.; Torrance, J.; Berthelot, C.; Muffato, M.; Collins, J.E.; Humphray, S.; McLaren, K.; Matthews, L.; et al. The zebrafish reference genome sequence and its relationship to the human genome. Nature 2013, 496, 498–503. [Google Scholar] [CrossRef] [Green Version]
- Patton, E.E.; Widlund, H.R.; Kutok, J.L.; Kopani, K.R.; Amatruda, J.F.; Murphey, R.D.; Berghmans, S.; Mayhall, E.A.; Traver, D.; Fletcher, C.D.M.; et al. BRAF Mutations Are Sufficient to Promote Nevi Formation and Cooperate with p53 in the Genesis of Melanoma. Curr. Biol. 2005, 15, 249–254. [Google Scholar] [CrossRef] [Green Version]
- Mayrhofer, M.; Gourain, V.; Reischl, M.; Affaticati, P.; Jenett, A.; Joly, J.-S.; Benelli, M.; Demichelis, F.; Poliani, P.L.; Sieger, D.; et al. A novel brain tumour model in zebrafish reveals the role of YAP activation in MAPK-and PI3K-induced malignant growth. Dis. Model. Mech. 2017, 10, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Cornet, C.; Di Donato, V.; Terriente, J. Combining Zebrafish and CRISPR/Cas9: Toward a more efficient drug discovery pipeline. Front. Pharmacol. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Traver, D.; Herbomel, P.; Patton, E.E.; Murphey, R.D.; Yoder, J.A.; Litman, G.W.; Catic, A.; Amemiya, C.T.; Zon, L.I.; Trede, N.S. The zebrafish as a model organism to study development of the immune system. Adv. Immunol. 2003, 81, 253–330. [Google Scholar] [PubMed]
- Zhang, B.; Shimada, Y.; Hirota, T.; Ariyoshi, M.; Kuroyanagi, J.; Nishimura, Y.; Tanaka, T. Novel immunologic tolerance of human cancer cell xenotransplants in zebrafish. Transl. Res. 2016, 170, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jarque, S.; Ibarra, J.; Rubio-Brotons, M.; García-Fernández, J.; Terriente, J. Multiplex analysis platform for endocrine disruption prediction using zebrafish. Int. J. Mol. Sci. 2019, 20, 1739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Cuadrado, L.; Tracey, N.; Ma, R.; Qian, B.; Brunton, V.G. Mouse models of metastasis: Progress and prospects. Dis. Model. Mech. 2017, 10, 1061–1074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veinotte, C.J.; Dellaire, G.; Berman, J.N. Hooking the big one: The potential of zebrafish xenotransplantation to reform cancer drug screening in the genomic era. Dis. Model. Mech. 2014, 7, 745–754. [Google Scholar] [CrossRef] [Green Version]
- Bentley, V.L.; Veinotte, C.J.; Corkery, D.P.; Pinder, J.B.; Leblanc, M.A.; Bedard, K.; Weng, A.P.; Berman, J.N.; Dellaire, G. Focused chemical genomics using zebrafish xenotransplantation as a pre-clinical therapeutic platform for T-cell acute lymphoblastic leukemia. Haematologica 2015, 100, 70–76. [Google Scholar] [CrossRef]
- Wertman, J.; Veinotte, C.J.; Dellaire, G.; Berman, J.N. Cancer and Zebrafish; Springer: Cham, Switzerland, 2016; pp. 289–314. [Google Scholar]
- Mercatali, L.; La Manna, F.; Groenewoud, A.; Casadei, R.; Recine, F.; Miserocchi, G.; Pieri, F.; Liverani, C.; Bongiovanni, A.; Spadazzi, C.; et al. Development of a Patient-Derived Xenograft (PDX) of Breast Cancer Bone Metastasis in a Zebrafish Model. Int. J. Mol. Sci. 2016, 17, 1375. [Google Scholar] [CrossRef]
- Fior, R.; Póvoa, V.; Mendes, R.V.; Carvalho, T.; Gomes, A.; Figueiredo, N.; Ferreira, M.G. Single-cell functional and chemosensitive profiling of combinatorial colorectal therapy in zebrafish xenografts. Proc. Natl. Acad. Sci. USA 2017, 114, E8234–E8243. [Google Scholar] [CrossRef] [Green Version]
- Taylor, A.M.; Zon, L.I. Zebrafish Tumor Assays: The State of Transplantation. Zebrafish 2009, 6, 339–346. [Google Scholar] [CrossRef]
- Gedye, C.; Sirskyj, D.; Lobo, N.C.; Meens, J.; Hyatt, E.; Robinette, M.; Fleshner, N.; Hamilton, R.J.; Kulkarni, G.; Zlotta, A.; et al. Cancer stem cells are underestimated by standard experimental methods in clear cell renal cell carcinoma. Sci. Rep. 2016, 6, 25220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Youn, H.; Hong, K.-J. In Vivo Non Invasive Molecular Imaging for Immune Cell Tracking in Small Animals. Immune Netw. 2012, 12, 223. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, A.K.; Bulte, J.W.M. Seeing Stem Cells at Work in Vivo. Stem Cell Rev. Rep. 2014, 10, 127–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanna, C.; Kwok, L.; Finlay-Schultz, J.; Sartorius, C.A.; Cittelly, D.M. Labeling of Breast Cancer Patient-derived Xenografts with Traceable Reporters for Tumor Growth and Metastasis Studies. J. Vis. Exp. 2016, 30, e54944. [Google Scholar] [CrossRef] [PubMed]
- Schlegel, A.; Stainier, D.Y.R. Microsomal triglyceride transfer protein is required for yolk lipid utilization and absorption of dietary lipids in zebrafish larvae. Biochemistry 2006, 45, 15179–15187. [Google Scholar] [CrossRef] [PubMed]
- Patel, R.A.; Forinash, K.D.; Pireddu, R.; Sun, Y.; Sun, N.; Martin, M.P.; Schönbrunn, E.; Lawrence, N.J.; Sebti, S.M. RKI-1447 is a potent inhibitor of the Rho-associated ROCK kinases with anti-invasive and antitumor activities in breast cancer. Cancer Res. 2012, 72, 5025–5034. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Schaefer, T.; Konantz, M.; Braun, M.; Varga, Z.; Paczulla, A.M.; Reich, S.; Jacob, F.; Perner, S.; Moch, H.; et al. Prominent oncogenic roles of EVI1 in breast carcinoma. Cancer Res. 2017, 77, 2148–2160. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Zhou, A.W.; Fu, Y.C.; Verma, U.N.; Tripathy, D.; Pfrenkel, E.; Rbecerra, C. Efficacy of sequential treatment of HCT116 colon cancer monolayers and xenografts with docetaxel, flavopiridol, and 5-fluorouracil. Acta Pharmacol. Sin. 2006, 27, 1375–1381. [Google Scholar] [CrossRef] [Green Version]
- Ban, J.O.; Lee, H.S.; Jeong, H.-S.; Song, S.; Hwang, B.Y.; Moon, D.C.; Yoon, D.Y.; Han, S.B.; Hong, J.T. Thiacremonone Augments Chemotherapeutic Agent-Induced Growth Inhibition in Human Colon Cancer Cells through Inactivation of Nuclear Factor-B. Mol. Cancer Res. 2009, 7, 870–879. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.Y.; Park, S.Y.; Kang, Y.; Thapa, D.; Choi, H.G.; Kim, J.A. Role of nonsteroidal anti-inflammatory drug-activated gene-1 in docetaxel-induced cell death of human colorectal cancer cells with different p53 status. Arch. Pharm. Res. 2011, 34, 323–330. [Google Scholar] [CrossRef]
- Thomadaki, H.; Mavridis, K.; Talieri, M.; Scorilas, A. Treatment of PC3 prostate cancer cells with mitoxantrone, etoposide, doxorubicin and carboplatin induces distinct alterations in the expression of kallikreins 5 and 11. Thromb. Haemost. 2009, 101, 373–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Li, R.; Ma, Y.; Zhang, C.; Huang, T.; Zhu, S. Transcriptome analysis of differentially expressed genes and pathways associated with mitoxantrone treatment prostate cancer. J. Cell. Mol. Med. 2019, 23, 1987–2000. [Google Scholar] [CrossRef] [PubMed]
- Yvon, A.-M.C.; Wadsworth, P.; Jordan, M.A. Taxol Suppresses Dynamics of Individual Microtubules in Living Human Tumor Cells. Mol. Biol. Cell 1999, 10, 947–959. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herbst, R.S.; Khuri, F.R. Mode of action of docetaxel—A basis for combination with novel anticancer agents. Cancer Treat. Rev. 2003, 29, 407–415. [Google Scholar] [CrossRef]
- Lam, S.H.; Chua, H.L.; Gong, Z.; Lam, T.J.; Sin, Y.M. Development and maturation of the immune system in zebrafish, Danio rerio: A gene expression profiling, in situ hybridization and immunological study. Dev. Comp. Immunol. 2004, 28, 9–28. [Google Scholar] [CrossRef]
- Haldi, M.; Ton, C.; Seng, W.L.; McGrath, P. Human melanoma cells transplanted into zebrafish proliferate, migrate, produce melanin, form masses and stimulate angiogenesis in zebrafish. Angiogenesis 2006, 9, 139–151. [Google Scholar] [CrossRef]
- Corkery, D.P.; Dellaire, G.; Berman, J.N. Leukaemia xenotransplantation in zebrafish-chemotherapy response assay in vivo. Br. J. Haematol. 2011, 153, 786–789. [Google Scholar] [CrossRef]
- Yang, X.J.; Cui, W.; Gu, A.; Xu, C.; Yu, S.C.; Li, T.T.; Cui, Y.H.; Zhang, X.; Bian, X.W. A Novel Zebrafish Xenotransplantation Model for Study of Glioma Stem Cell Invasion. PLoS ONE 2013, 8, e61801. [Google Scholar] [CrossRef] [Green Version]
- Hong, H.; Yang, Y.; Zhang, Y.; Cai, W. Non-Invasive Cell Tracking in Cancer and Cancer Therapy. Curr. Top. Med. Chem. 2010, 10, 1237–1248. [Google Scholar] [CrossRef]
- Pardo-Martin, C.; Chang, T.Y.; Koo, B.K.; Gilleland, C.L.; Wasserman, S.C.; Yanik, M.F. High-throughput in vivo vertebrate screening. Nat. Methods 2010, 7, 634–636. [Google Scholar] [CrossRef] [Green Version]
- Di Giacomo, V.; Tian, T.V.; Mas, A.; Pecoraro, M.; Batlle-Morera, L.; Noya, L.; Martín-Caballero, J.; Ruberte, J.; Keyes, W.M. ΔNp63α promotes adhesion of metastatic prostate cancer cells to the bone through regulation of CD82. Oncogene 2017, 36, 4381–4392. [Google Scholar] [CrossRef] [PubMed]
- Fleten, K.G.; Bakke, K.M.; Mælandsmo, G.M.; Abildgaard, A.; Redalen, K.R.; Flatmark, K. Use of non-invasive imaging to monitor response to aflibercept treatment in murine models of colorectal cancer liver metastases. Clin. Exp. Metastasis 2017, 34, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kuruppu, D.; Bhere, D.; Farrar, C.T.; Shah, K.; Brownell, A.L.; Tanabe, K.K. A model of breast cancer meningeal metastases: Characterization with in vivo molecular imaging. Cancer Gene Ther. 2019, 26, 145–156. [Google Scholar] [CrossRef] [PubMed]
- Park, J.Y.; Jeong, A.L.; Joo, H.J.; Han, S.; Kim, S.-H.; Kim, H.-Y.; Lim, J.-S.; Lee, M.-S.; Choi, H.-K.; Yang, Y. Development of suspension cell culture model to mimic circulating tumor cells. Oncotarget 2018, 9, 622–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.J.; Zon, L.I. The “definitive” (and ’primitive’) guide to zebrafish hematopoiesis. Oncogene 2004, 23, 7233–7246. [Google Scholar] [CrossRef] [Green Version]
- Reichardt, P.; Tabone, M.D.; Mora, J.; Morland, B.; Jones, R.L. Risk-benefit of dexrazoxane for preventing anthracycline-related cardiotoxicity: Re-evaluating the European labeling. Futur. Oncol. 2018, 14, 2663–2676. [Google Scholar] [CrossRef]
- Haslam, A.; Prasad, V. Confirmatory Trials for Drugs Approved on a Single Trial. Circ. Cardiovasc. Qual. Outcomes 2019, 12, e005494. [Google Scholar] [CrossRef]
- Zhang, Z.; Yu, X.; Wang, Z.; Wu, P.; Huang, J. Anthracyclines potentiate anti-tumor immunity: A new opportunity for chemoimmunotherapy. Cancer Lett. 2015, 369, 331–335. [Google Scholar] [CrossRef]
- Brown, J.S.; Sundar, R.; Lopez, J. Combining DNA damaging therapeutics with immunotherapy: More haste, less speed. Br. J. Cancer 2018, 118, 312–324. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Shalabi, A.; Hubbard-Lucey, V.M. Comprehensive analysis of the clinical immuno-oncology landscape. Ann. Oncol. 2018, 29, 84–91. [Google Scholar] [CrossRef]
- Stoletov, K.; Montel, V.; Lester, R.D.; Gonias, S.L.; Klemke, R. High-resolution imaging of the dynamic tumor cell vascular interface in transparent zebrafish. Proc. Natl. Acad. Sci. USA 2007, 104, 17406–17411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoli, S.; Ribatti, D.; Cotelli, F.; Presta, M. Mammalian tumor xenografts induce neovascularization in zebrafish embryos. Cancer Res. 2007, 67, 2927–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoli, S.; Presta, M. The zebrafish/tumor xenograft angiogenesis assay. Nat. Protoc. 2007, 2, 2918–2923. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.L.C.; Rouhi, P.; Jensen, L.D.; Zhang, D.; Ji, H.; Hauptmann, G.; Ingham, P.; Cao, Y. Hypoxia-induced pathological angiogenesis mediates tumor cell dissemination, invasion, and metastasis in a zebrafish tumor model. Proc. Natl. Acad. Sci. USA 2009, 106, 19485–19490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Wang, X.; Zhao, Y.; Li, Z.; Lin, S.; Wei, Y.; Yang, H. A novel xenograft model in zebrafish for high-resolution investigating dynamics of neovascularization in tumors. PLoS ONE 2011, 6, e217368. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Lamers, G.E.M.; Beenakker, J.W.M.; Cui, C.; Ghotra, V.P.S.; Danen, E.H.J.; Meijer, A.H.; Spaink, H.P.; Snaar-Jagalska, B.E. Neutrophil-mediated experimental metastasis is enhanced by VEGFR inhibition in a zebrafish xenograft model. J. Pathol. 2012, 227, 431–445. [Google Scholar] [CrossRef] [Green Version]
- Tulotta, C.; He, S.; van der Ent, W.; Chen, L.; Groenewoud, A.; Spaink, H.P.; Snaar-Jagalska, B.E. Imaging Cancer Angiogenesis and Metastasis in a Zebrafish Embryo Model. In Cancer and Zebrafish; Springer: Cham, Switzerland, 2016; Volume 916, pp. 239–263. [Google Scholar]
- Gabellini, C.; Gómez-Abenza, E.; Ibáñez-Molero, S.; Tupone, M.G.; Pérez-Oliva, A.B.; de Oliveira, S.; Del Bufalo, D.; Mulero, V. Interleukin 8 mediates bcl-xL-induced enhancement of human melanoma cell dissemination and angiogenesis in a zebrafish xenograft model. Int. J. Cancer 2018, 142, 584–596. [Google Scholar] [CrossRef]
- Westerfield, M. The Principles of Humane Experimental Technique, 4th ed.; Eugene, United States; Special Edition Published by Universities Federation for Animal Welfare (UFAW); Methuen & Co.: London, UK, 1992; ISBN 0900767782. [Google Scholar]
- Landegren, U. Measurement of cell numbers by means of the endogenous enzyme hexosaminidase. Applications to detection of lymphokines and cell surface antigens. J. Immunol. Methods 1984, 67, 379–388. [Google Scholar] [CrossRef]
- Westhoff, J.H.; Giselbrecht, S.; Schmidts, M.; Schindler, S.; Beales, P.L.; Tönshoff, B.; Liebel, U.; Gehrig, J. Development of an automated imaging pipeline for the analysis of the zebrafish larval kidney. PLoS ONE 2013, 8, e82137. [Google Scholar] [CrossRef] [Green Version]
- Wittbrodt, J.N.; Liebel, U.; Gehrig, J. Generation of orientation tools for automated zebrafish screening assays using desktop 3D printing. BMC Biotechnol. 2014, 14, 36. [Google Scholar] [CrossRef] [Green Version]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug | NOEC |
|---|---|
| Docetaxel | 10 µM |
| Mitoxantrone | 3 µM |
| RKI-1447 | 10 µM |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cornet, C.; Dyballa, S.; Terriente, J.; Di Giacomo, V. ZeOncoTest: Refining and Automating the Zebrafish Xenograft Model for Drug Discovery in Cancer. Pharmaceuticals 2020, 13, 1. https://doi.org/10.3390/ph13010001
Cornet C, Dyballa S, Terriente J, Di Giacomo V. ZeOncoTest: Refining and Automating the Zebrafish Xenograft Model for Drug Discovery in Cancer. Pharmaceuticals. 2020; 13(1):1. https://doi.org/10.3390/ph13010001
Chicago/Turabian StyleCornet, Carles, Sylvia Dyballa, Javier Terriente, and Valeria Di Giacomo. 2020. "ZeOncoTest: Refining and Automating the Zebrafish Xenograft Model for Drug Discovery in Cancer" Pharmaceuticals 13, no. 1: 1. https://doi.org/10.3390/ph13010001
APA StyleCornet, C., Dyballa, S., Terriente, J., & Di Giacomo, V. (2020). ZeOncoTest: Refining and Automating the Zebrafish Xenograft Model for Drug Discovery in Cancer. Pharmaceuticals, 13(1), 1. https://doi.org/10.3390/ph13010001