Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents



 ,
, 
 and
and
Abstract
1. Introduction
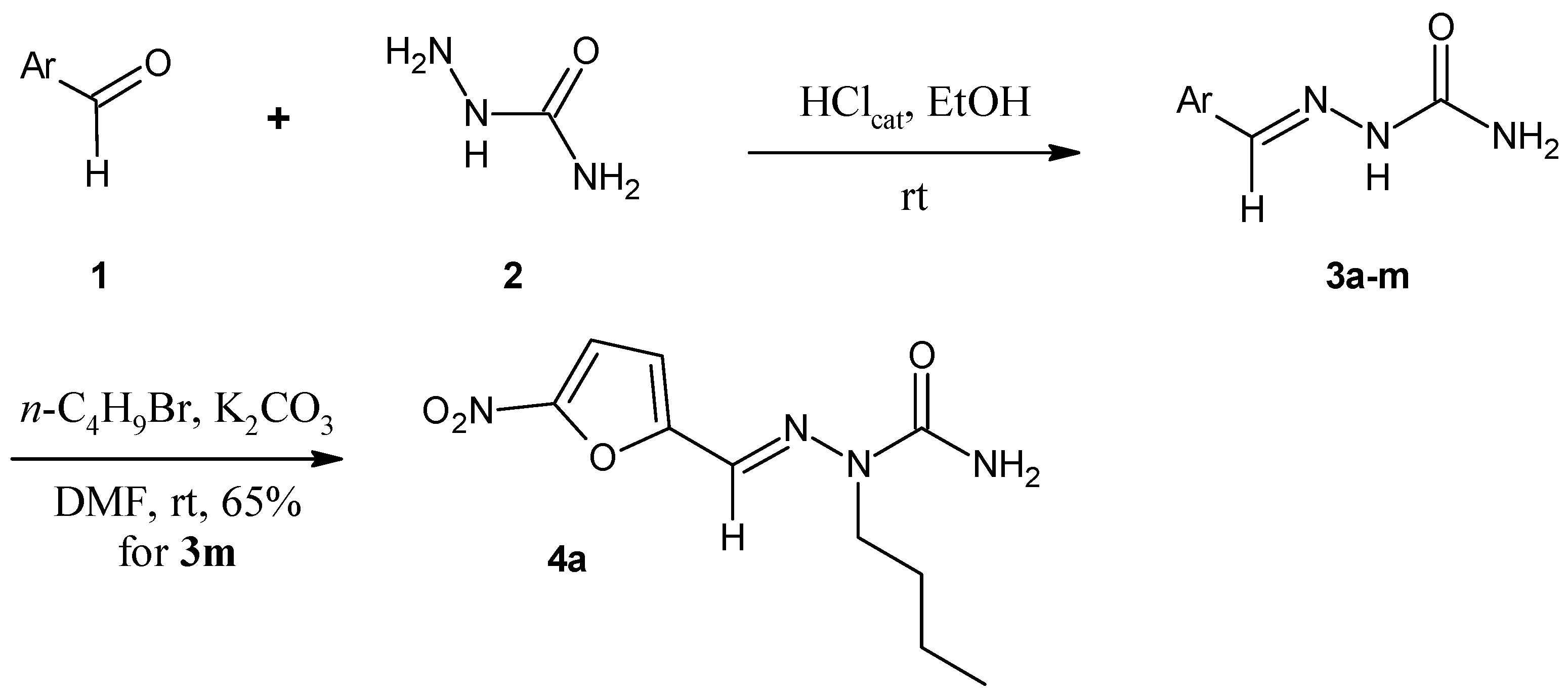
2. Results and Discussion
2.1. Chemistry
2.2. Cytotoxicity Assays
2.3. Inhibition of Protein Kinases
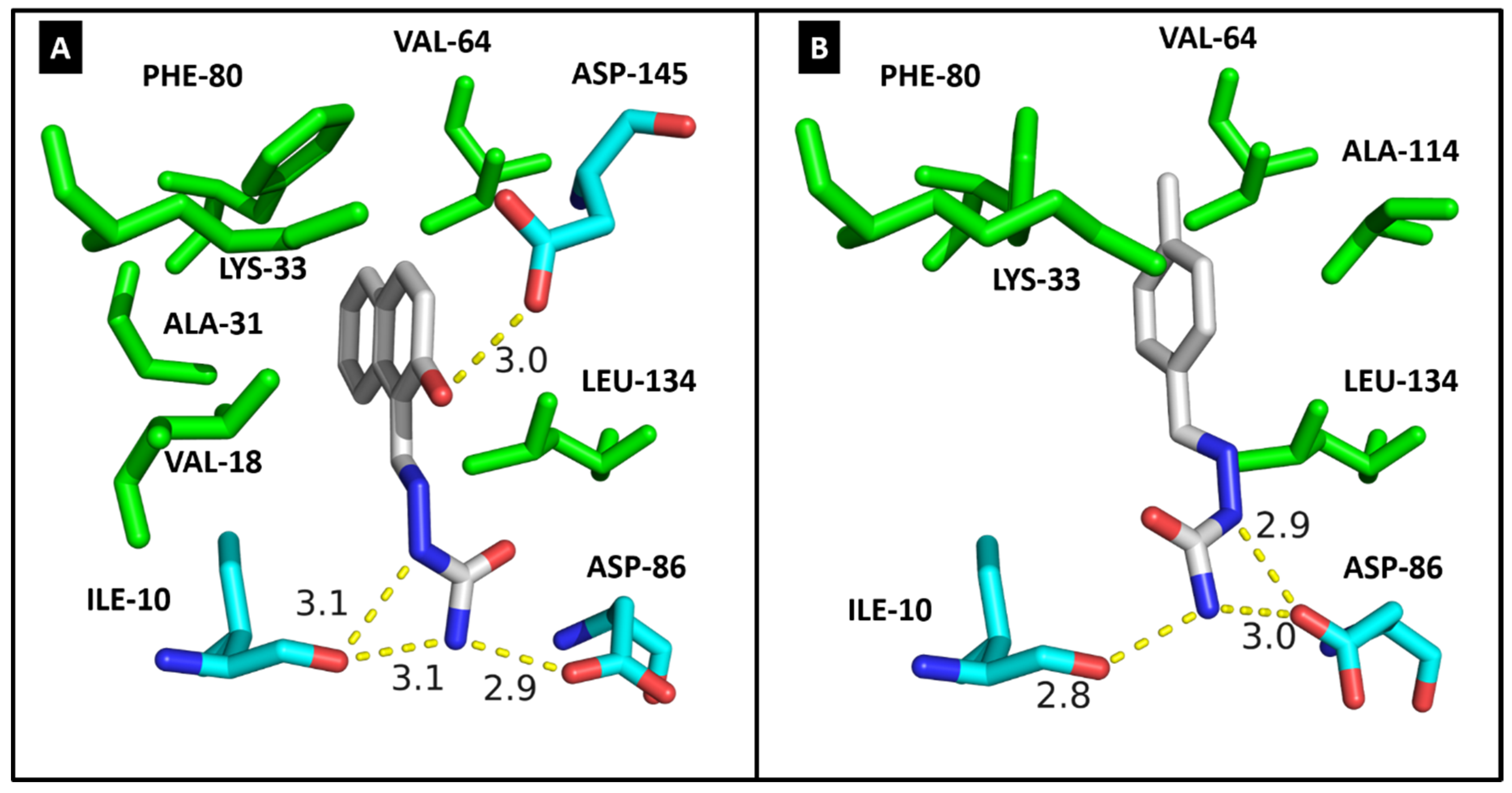
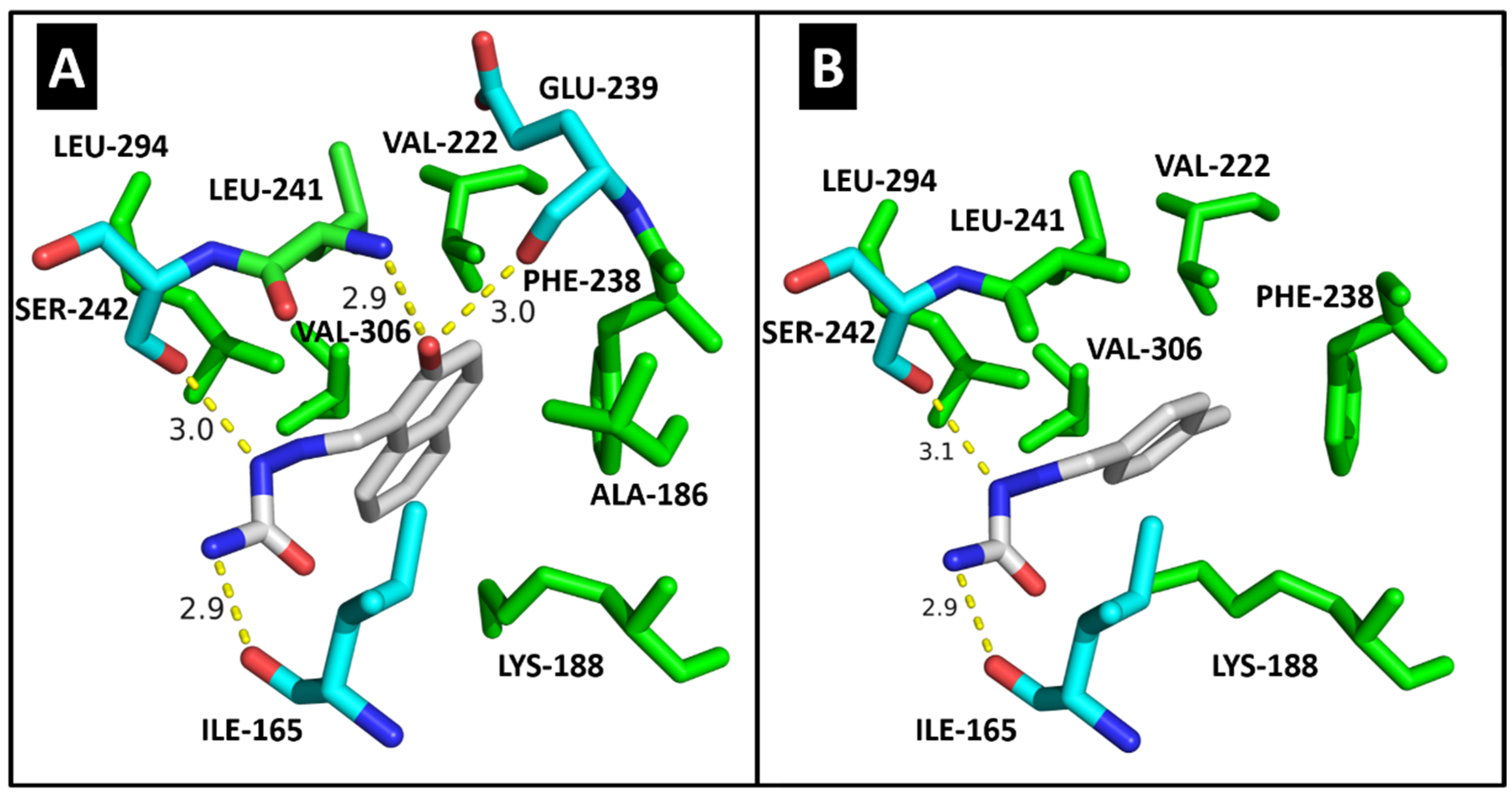
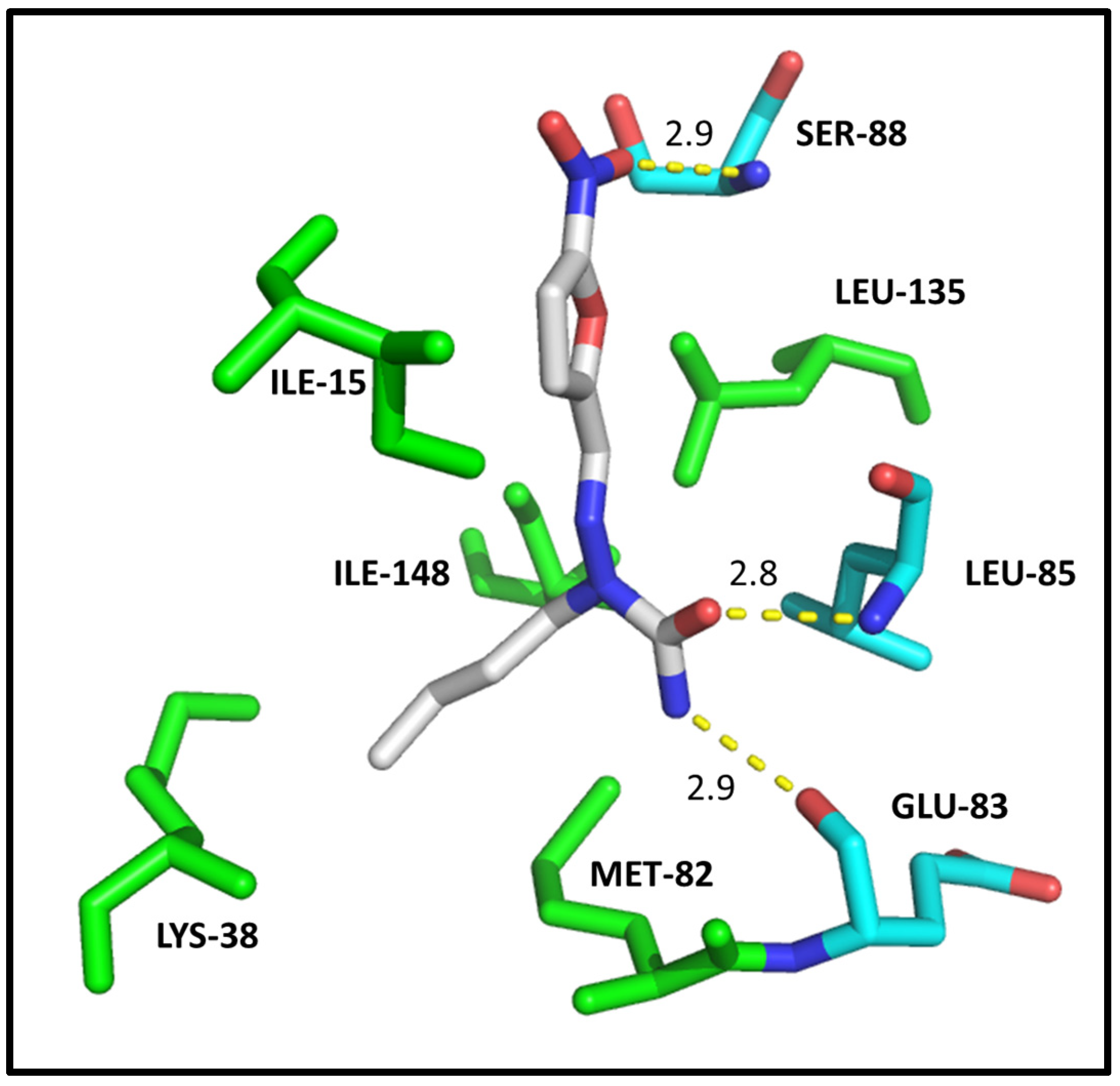
2.4. Molecular Modeling
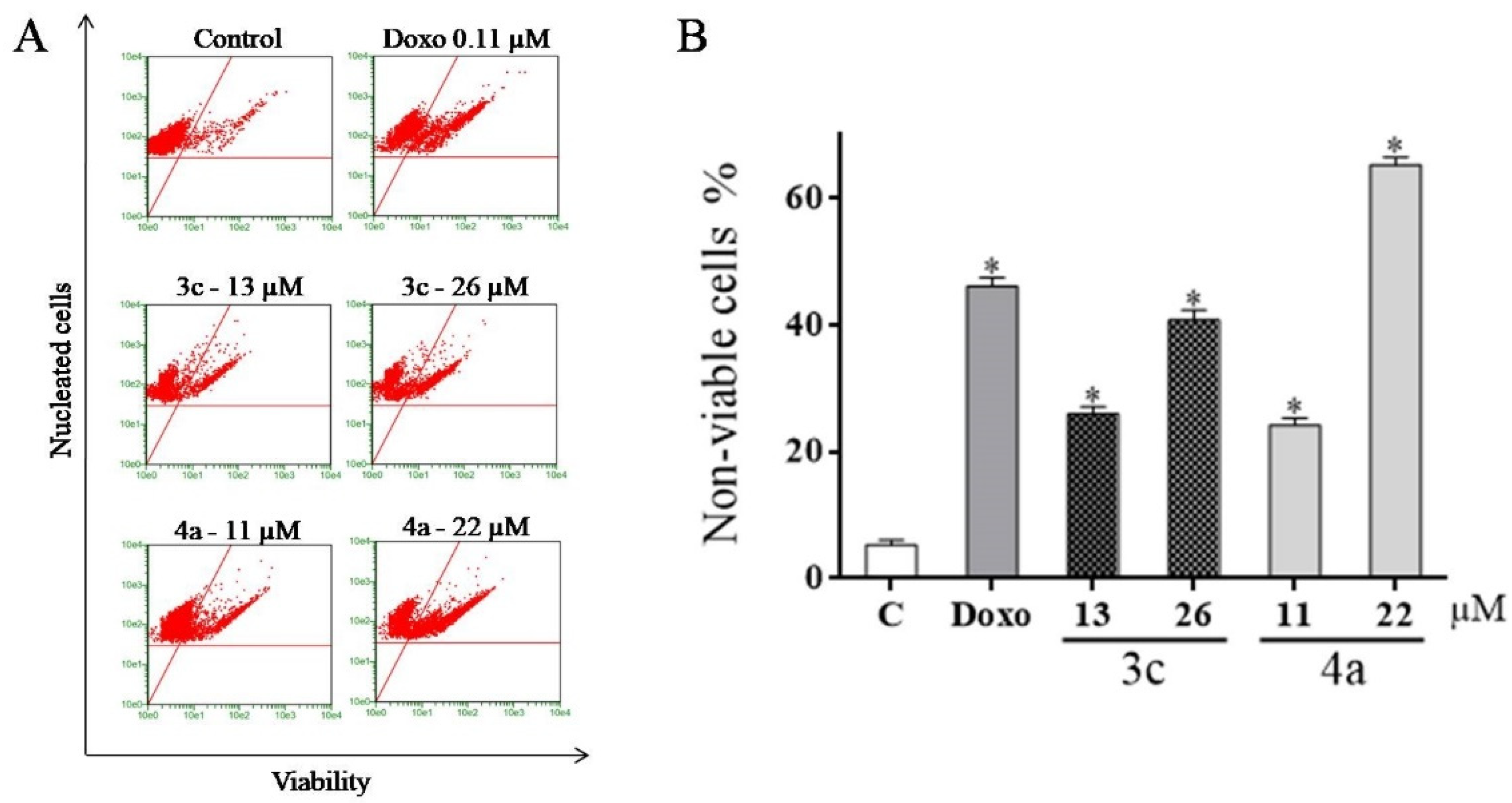
2.5. Detection of Cell Viability Using the Guava ViaCount Kit
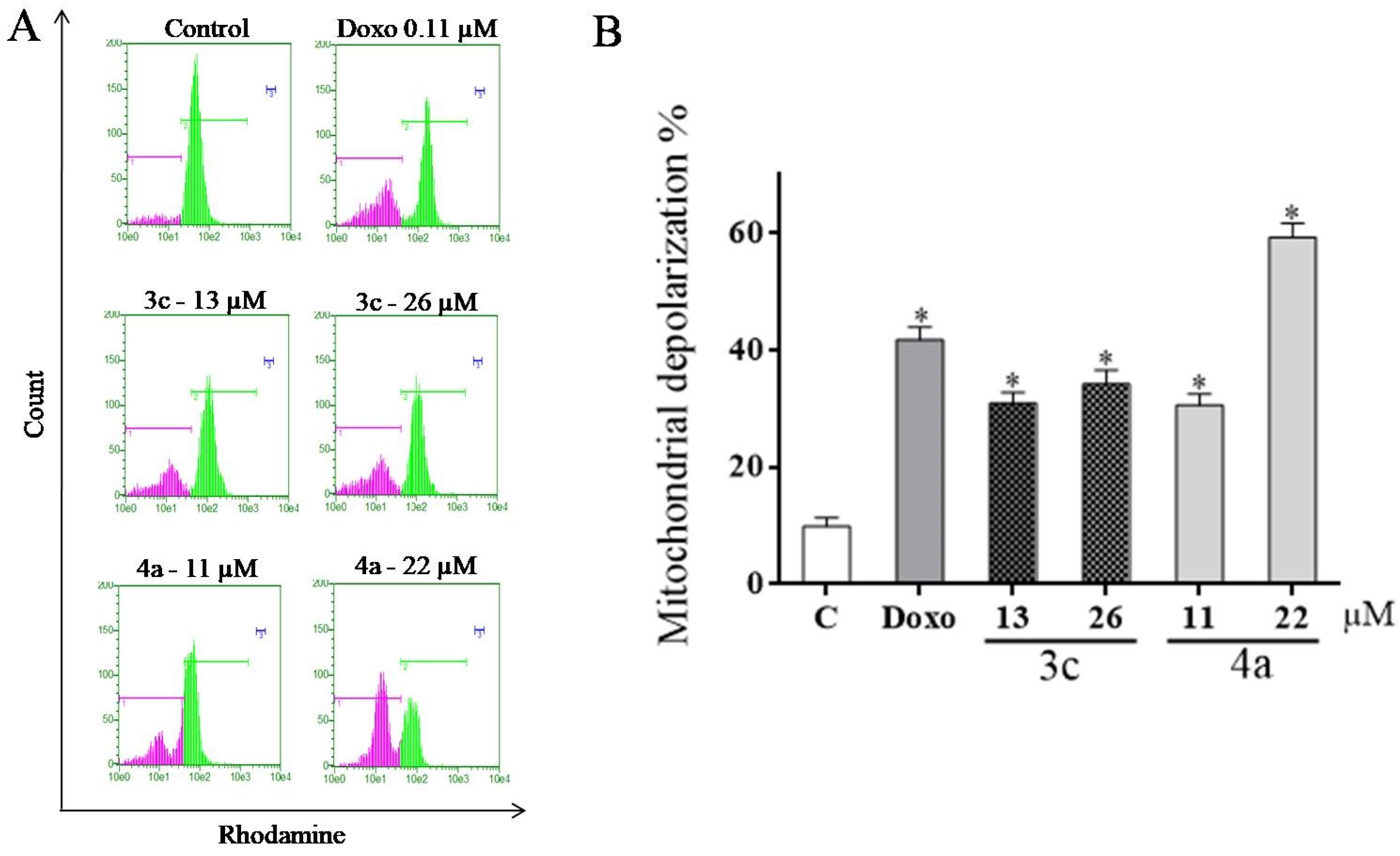
2.6. Evaluation of Mitochondrial Transmembrane Potential (ΔΨm)
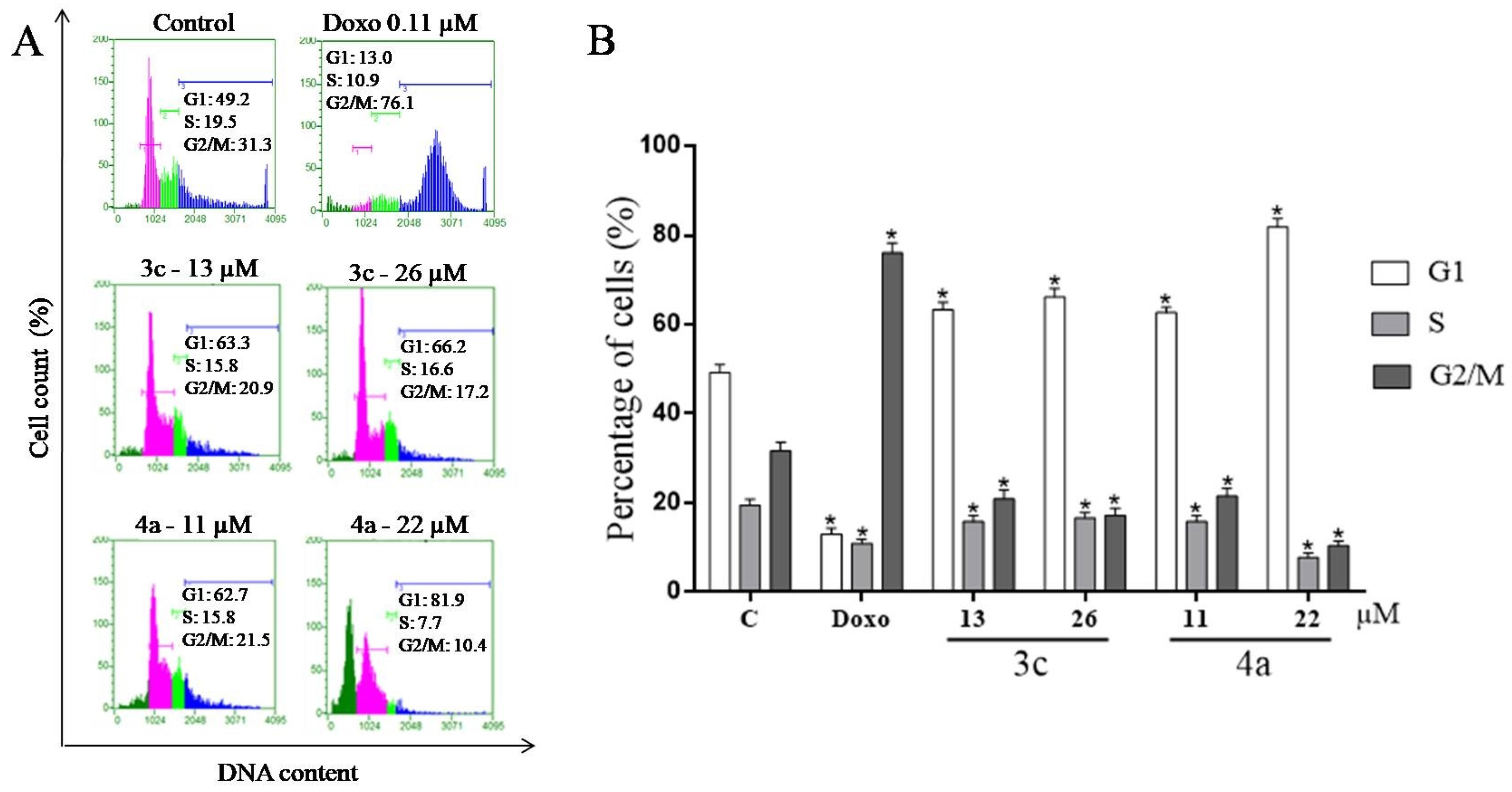
2.7. Cell Cycle Assay
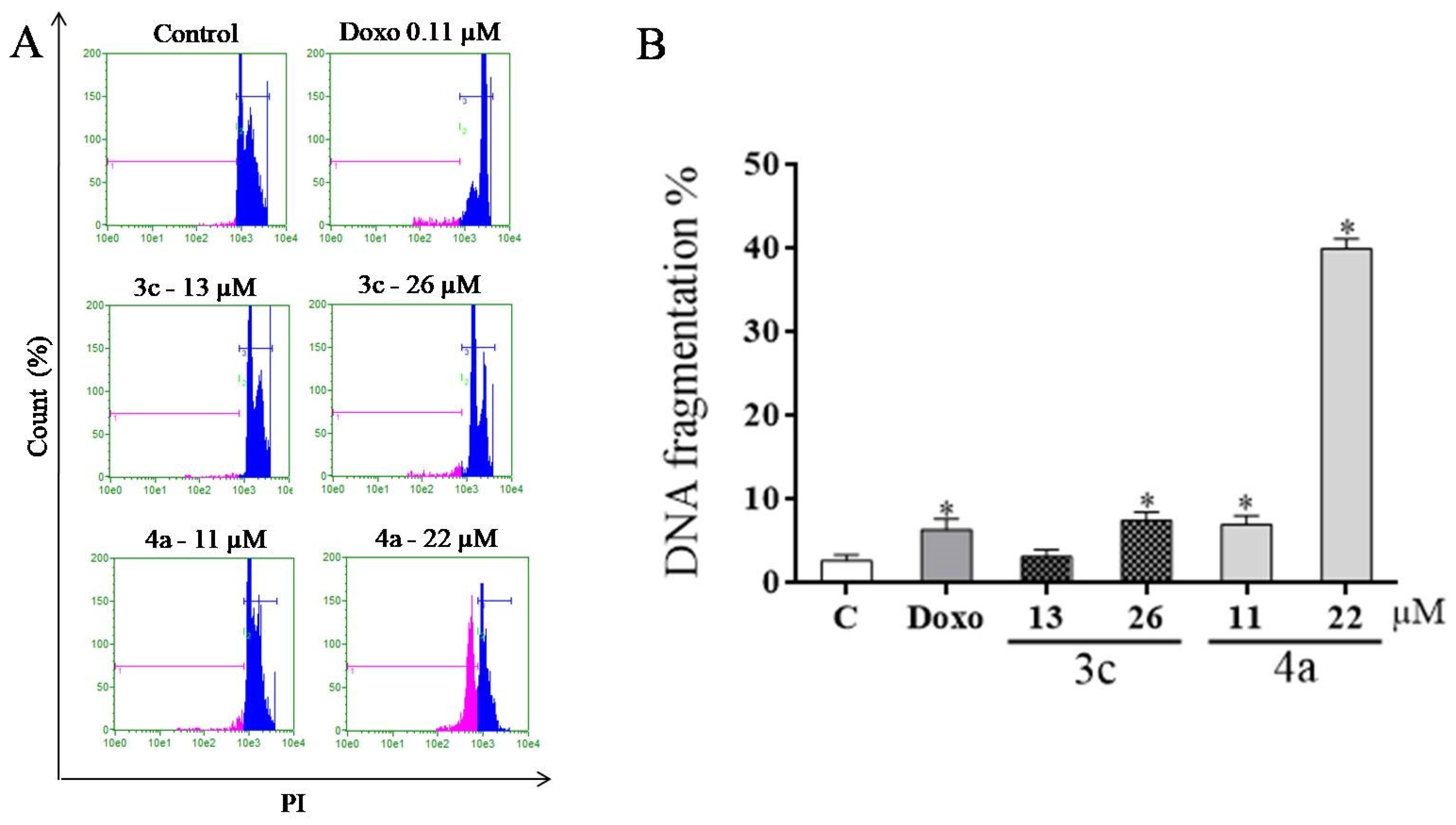
2.8. DNA Fragmentation Assay
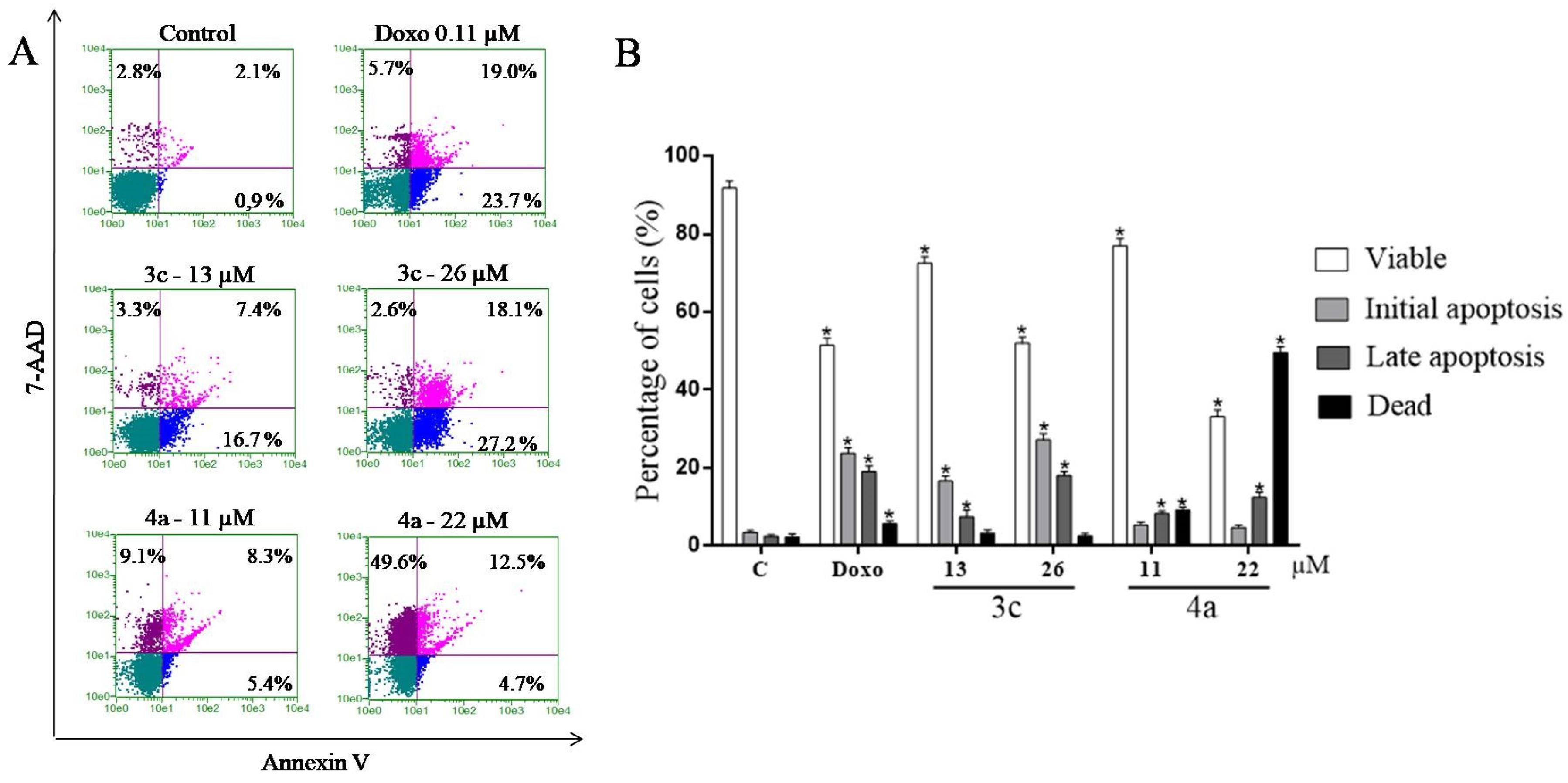
2.9. Study of the Induction of Cell Death by the Annexin V Test
3. Materials and Methods
3.1. Chemistry
3.1.1. General Experimental Procedures
3.1.2. General Procedure for the Preparation of Arylsemicarbazones 3a–3m
4-Methoxybenzaldehyde Semicarbazone (3a)
4-Hydroxy-3-methoxybenzaldehyde Semicarbazone (3b)
2-Hydroxynaphthaldehyde Semicarbazone (3c)
4-Bromobenzaldehyde Semicarbazone (3d)
4-Chlorobenzaldehyde Semicarbazone (3e)
3,4-Dichlorobenzaldehyde Semicarbazone (3f)
4-Methylbenzaldehyde Semicarbazone (3g)
4-Tert-Butylbenzaldehyde Semicarbazone (3h)
4-Pyridinecarboxaldehyde Semicarbazone (3i)
2,6-Pyridinedicarboxaldehyde Semicarbazone (3j)
2-Thiophenecarboxaldehyde Semicarbazone (3k)
2-Furfuraldehyde Semicarbazone (3l)
5-Nitro-2-furfuraldehyde Semicarbazone (3m)
3.1.3. Procedure for the Preparation of N2-alkylated Arylsemicarbazone 4a
5-Nitro-2-(N2-n-butyl)furfuraldehyde Semicarbazone (4a)
3.2. Molecular Modeling
3.3. Biological Activity
3.3.1. Ethical Aspects of the Peripheral Blood Mononuclear Cell Assay
3.3.2. Isolation of Peripheral Blood Mononuclear Cells
3.3.3. Culture of Tumor Cells
3.3.4. Cytotoxicity Assessment by MTT Assay
3.3.5. Kinase Inhibition Assays
3.3.6. Flow Cytometry Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Abdelatef, S.A.; El-Saadi, M.T.; Amin, N.H.; Abdelazeem, A.H.; Omar, H.A.; Abdellatif, K.R.A. Design, synthesis and anticancer evaluation of novel spirobenzo [h] chromene and spirochromane derivatives with dual EGFR and B-RAF inhibitory activities. Eur. J. Med. Chem. 2018, 150, 567–578. [Google Scholar] [CrossRef] [PubMed]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Stewart, B.W.; Wild, C.P. World Cancer Report; International Agency for Research on Cancer: Lyon, France, 2014. [Google Scholar]
- Rottenberg, Y.; Naeim, A.; Uziely, B.; Peretz, T.; Jacobs, J.M. Breast cancer among older women: The influence of age and cancer stage on survival. Arch. Gerontol. Geriatr. 2018, 76, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Dickens, E.; Ahmed, S. Principles of cancer treatment by chemotherapy. Surgery 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Abraham, J.; Staffurth, J. Hormonal therapy for cancer. Medicine 2016, 44, 30–33. [Google Scholar] [CrossRef]
- Walter, H.S.; Ahmed, S. Targeted therapies in cancer. Surgery 2018, 36, 134–138. [Google Scholar] [CrossRef]
- Chen, Y.-L.; Chang, M.-C.; Cheng, W.-F. Metronomic chemotherapy and immunotherapy in cancer treatment. Cancer Lett. 2017, 400, 282–292. [Google Scholar] [CrossRef]
- Giancotti, F.G. Deregulation of cell signaling in cancer. FEBS Lett. 2014, 588, 2558–2570. [Google Scholar] [CrossRef]
- Makin, G. Principles of chemotherapy. Paediatr. Child Health 2018, 28, 183–188. [Google Scholar] [CrossRef]
- Charlton, P.; Spicer, J. Targeted therapy in cancer. Medicine 2016, 44, 34–38. [Google Scholar] [CrossRef]
- Isakov, N. Protein kinase C (PKC) isoforms in cancer, tumor promotion and tumor suppression. Semin. Cancer Biol. 2018, 48, 36–52. [Google Scholar] [CrossRef] [PubMed]
- Czubaty, A.; Piekiełko-Witkowska, A. Protein kinases that phosphorylate splicing factors: Roles in cancer development, progression and possible therapeutic options. Int. J. Biochem. Cell Biol. 2017, 91, 102–115. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.J. Semicarbazone analogs as anticonvulsant agents: A review. Cent. Nerv. Syst. Agents Med. Chem. 2013, 13, 148–158. [Google Scholar] [CrossRef] [PubMed]
- Beraldo, H.; Gambino, D. The wide pharmacological versatility of semicarbazones, thiosemicarbazones and their metal complexes. Mini Rev. Med. Chem. 2004, 4, 31–39. [Google Scholar] [CrossRef]
- Sinha, S.K.; Shrivastava, S.K. Synthesis, evaluation and molecular dynamics study of some new 4-aminopyridine semicarbazones as an antiamnesic and cognition enhancing agents. Bioorg. Med. Chem. 2013, 21, 5451–5460. [Google Scholar] [CrossRef]
- Soares, R.O.A.; Echevarria, A.; Bellieny, M.S.S.; Pinho, R.T.; de Leo, R.M.M.; Seguins, W.S.; Machado, G.M.; Canto-Cavalheiro, M.M.; Leon, L.L. Evaluation of thiosemicarbazones and semicarbazones as potential agents anti-Trypanosoma cruzi. Exp. Parasitol. 2011, 129, 381–387. [Google Scholar] [CrossRef]
- Ho, J.; Lee, W.Y.; Koh, K.J.T.; Lee, P.P.F.; Yan, Y.-K. Rhenium (I) tricarbonyl complexes of salicylaldehyde semicarbazones: Synthesis, crystal structures and cytotoxicity. J. Inorg. Biochem. 2013, 119, 10–20. [Google Scholar] [CrossRef]
- Zhai, X.; Bao, G.; Wang, L.; Cheng, M.; Zhao, M.; Zhao, S.; Zhou, H.; Gong, P. Design, synthesis and biological evaluation of novel 4-phenoxy-6,7-disubstituted quinolines possessing (thio) semicarbazones as c-Met kinase inhibitors. Bioorg. Med. Chem. 2016, 24, 1331–1345. [Google Scholar] [CrossRef]
- Tu, Y.; Wang, C.; Xu, S.; Lan, Z.; Li, W.; Han, J.; Zhou, Y.; Zheng, P.; Zhu, W. Design, synthesis, and docking studies of quinazoline analogues bearing aryl semicarbazone scaffolds as potent EGFR inhibitors. Bioorg. Med. Chem. 2017, 25, 3148–3157. [Google Scholar] [CrossRef]
- Enyedy, É.A.; Bognár, G.M.; Nagy, N.V.; Jakusch, T.; Kiss, T.; Gambino, D. Solution speciation of potential anticancer metal complexes of salicylaldehyde semicarbazone and its bromo derivative. Polyhedron 2014, 67, 242–252. [Google Scholar] [CrossRef]
- Brondani, D.J.; de Magalhaes Moreira, D.R.; de Farias, M.P.A.; da S. Souza, F.R.; Barbosa, F.F.; Leite, A.C.L. A new and efficient N-alkylation procedure for semicarbazides/semicarbazones derivatives. Tetrahedron Lett. 2007, 48, 3919–3923. [Google Scholar] [CrossRef]
- Barros, M.E.S.B.; Freitas, J.C.R.; Oliveira, J.M.; da Cruz, C.H.B.; da Silva, P.B.N.; de Araújo, L.C.C.; Militão, G.C.G.; da Silva, T.G.; Oliveira, R.A.; Menezes, P.H. Synthesis and evaluation of (−)-Massoialactone and analogues as potential anticancer and anti-inflammatory agents. Eur. J. Med. Chem. 2014, 76, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Fortes, M.P.; da Silva, P.B.N.; da Silva, T.G.; Kaufman, T.S.; Militão, G.C.G.; Silveira, C.C. Synthesis and preliminary evaluation of 3-thiocyanato-1H-indoles as potential anticancer agents. Eur. J. Med. Chem. 2016, 118, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Princival, I.M.R.G.; Ferreira, J.G.; Silva, T.G.; Aguiar, J.S.; Princival, J.L. Synthesis and in vitro evaluation of (R), (S) and (R/S)-2-hexyne-1,4-diol, a natural product produced by fungus Clitocybe catinus, and related analogs as potential anticancer agents. Bioorg. Med. Chem. Lett. 2016, 26, 2839–2842. [Google Scholar] [CrossRef] [PubMed]
- Santos, J.A.M.; Santos, C.S.; Almeida, C.L.A.; Silva, T.D.S.; Freitas Filho, J.R.; Militão, G.C.G.; da Silva, T.G.; da Cruz, C.H.B.; Freitas, J.C.R.; Menezes, P.H. Structure-based design, synthesis and antitumoral evaluation of enulosides. Eur. J. Med. Chem. 2017, 128, 192–201. [Google Scholar] [CrossRef]
- De Santana, T.I.; de Oliveira Barbosa, M.; de Moraes Gomes, P.A.T.; da Cruz, A.C.N.; da Silva, T.G.; Leite, A.C.L. Synthesis, anticancer activity and mechanism of action of new thiazole derivatives. Eur. J. Med. Chem. 2018, 144, 874–886. [Google Scholar] [CrossRef]
- Bazin, M.-A.; Rousseau, B.; Marhadour, S.; Tomasoni, C.; Evenou, P.; Piessard, S.; Vaisberg, A.J.; Ruchaud, S.; Bach, S.; Roussakis, C.; et al. Discovery of novel (imidazo[1,2-a]pyrazin-6-yl)ureas as antiproliferative agents targeting P53 in non-small cell lung cancer cell lines. Anticancer Res. 2016, 36, 1621–1630. [Google Scholar]
- Antoine, M.; Gerlach, M.; Günther, E.; Schuster, T.; Czech, M.; Seipelt, I.; Marchand, P. A convenient synthesis of novel 2,8-disubstituted pyrido[3,4-b]pyrazines possessing biological activity. Synthesis 2012, 44, 69–82. [Google Scholar] [CrossRef]
- Marchand, P.; Antoine, M.; Baut, G.L.; Czech, M.; Baasner, S.; Günther, E. Synthesis and structure—Activity relationships of N-aryl(indol-3-yl)glyoxamides as antitumor agents. Bioorg. Med. Chem. 2009, 17, 6715–6727. [Google Scholar] [CrossRef]
- Lima Leite, A.C.; Moreira, D.R.M.; Duarte Coelho, L.C.; Duarte de Menezes, F.; Brondani, D.J. Synthesis of aryl-hydrazones via ultrasound irradiation in aqueous medium. Tetrahedron Lett. 2008, 49, 1538–1541. [Google Scholar] [CrossRef]
- Pontes Espíndola, J.W.; Oliveira da Silva, L.; de Carvalho Silva, D.R.; Victor Hugo Nunes Soares Costa, V.H.; Ferreira de Almeida, A.; Moutinho Lagos de Melo, C.; de Lima Soares, E.C.; de Araújo, J.M.; Alves Pereira, V.R.; Brondani, D.J. Evaluation of antimicrobial activity and cytotoxicity of aryl-semicarbazones derivatives. Rev. Bras. Farm. 2011, 92, 171–175. [Google Scholar]
- Rajak, H.; Agarawal, A.; Parmar, P.; Singh Thakur, B.; Veerasamy, R.; Chander Sharma, P.; Dhar Kharya, M. 2,5-Disubstituted-1,3,4-oxadiazoles/thiadiazole as surface recognition moiety: Design and synthesis of novel hydroxamic acid based histone deacetylase inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 5735–5738. [Google Scholar] [CrossRef] [PubMed]
- Khlebnikov, A.; Schepetkin, I.; Kwon, B.S. Modeling of the anticancer action for radical derivatives of nitroazoles: Quantitative structure-activity relationship (QSAR) study. Cancer Biother. Radiopharm. 2002, 17, 193–203. [Google Scholar] [CrossRef]
- Shinoda, W. Permeability across lipid membranes. Biochim. Biophys. Acta Biomembr. 2016, 1858, 2254–2265. [Google Scholar] [CrossRef]
- Cherukupalli, S.; Chandrasekaran, B.; Kryštof, V.; Aleti, R.R.; Sayyad, N.; Merugu, S.R.; Kushwaha, N.D.; Karpoormath, R. Synthesis, anticancer evaluation, and molecular docking studies of some novel 4,6-disubstituted pyrazolo[3,4-d]pyrimidines as cyclin-dependent kinase 2 (CDK2) inhibitors. Bioorg. Chem. 2018, 79, 46–59. [Google Scholar] [CrossRef]
- Elagawany, M.; Ibrahim, M.A.; Ali Ahmed, H.E.; El-Etrawy, A.S.; Ghiaty, A.; Abdel-Samii, Z.K.; El-Feky, S.A.; Bajorath, J. Design, synthesis, and molecular modelling of pyridazinone and phthalazinone derivatives as protein kinases inhibitors. Bioorg. Med. Chem. Lett. 2013, 23, 2007–2013. [Google Scholar] [CrossRef]
- Rothweiler, U.; Eriksson, J.; Stensen, W.; Leeson, F.; Engh, R.A.; Svendsen, J.S. Luciferin and derivatives as a DYRK selective scaffold for the design of protein kinase inhibitors. Eur. J. Med. Chem. 2015, 94, 140–148. [Google Scholar] [CrossRef]
- Assadieskandar, A.; Yu, C.; Maisonneuve, P.; Liu, X.; Chen, Y.-C.; Prakash, G.K.S.; Kurinov, I.; Sicheri, F.; Zhang, C. Effects of rigidity on the selectivity of protein kinase inhibitors. Eur. J. Med. Chem. 2018, 146, 519–528. [Google Scholar] [CrossRef]
- Loidreau, Y.; Marchand, P.; Dubouilh-Benard, C.; Nourrisson, M.R.; Duflos, M.; Lozach, O.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and biological evaluation of N-arylbenzo[b]thieno[3,2-d]pyrimidin-4-amines and their pyrido and pyrazino analogues as Ser/Thr kinase inhibitors. Eur. J. Med. Chem. 2012, 58, 171–183. [Google Scholar] [CrossRef]
- Loidreau, Y.; Deau, E.; Marchand, P.; Nourrisson, M.-R.; Logé, C.; Coadou, G.; Loaëc, N.; Meijer, L.; Besson, T. Synthesis and molecular modelling studies of 8-arylpyrido[3′,2′:4,5]thieno[3,2-d]pyrimidin-4-amines as multitarget Ser/Thr kinases inhibitors. Eur. J. Med. Chem. 2015, 92, 124–134. [Google Scholar] [CrossRef]
- Reggi, E.; Diviani, D. The role of A-kinase anchoring proteins in cancer development. Cell. Signal. 2017, 40, 143–155. [Google Scholar] [CrossRef] [PubMed]
- Burke, P.J. Mitochondria, Bioenergetics and Apoptosis in Cancer. Trends Cancer 2017, 3, 857–870. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.-J.; Lu, L.; Yang, J.-J.; Wang, X.-X.; Su, G.; Wang, Z.; Chen, G.; Sun, H.; Wang, M.; Yang, Y. Lariciresinol induces apoptosis in HepG2 cells via mitochondrial-mediated apoptosis pathway. Eur. J. Pharmacol. 2018, 821, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, B.Y.; Sarker, M.M.R.; Kamarudin, M.N.A.; Mohan, G. Antiproliferative and apoptosis inducing effects of citral via p53 and ROS-induced mitochondrial-mediated apoptosis in human colorectal HCT116 and HT29 cell lines. Biomed. Pharmacother. 2017, 96, 834–846. [Google Scholar] [CrossRef] [PubMed]
- Hsu, Y.-N.; Shyu, H.-W.; Hu, T.-W.; Yeh, J.-P.; Lin, Y.-W.; Lee, L.-Y.; Yeh, Y.-T.; Dai, H.-Y.; Perng, D.-S.; Su, S.-H.; et al. Anti-proliferative activity of biochanin A in human osteosarcoma cells via mitochondrial-involved apoptosis. Food Chem. Toxicol. 2018, 112, 194–204. [Google Scholar] [CrossRef]
- Brandmaier, A.; Hou, S.Q.; Shen, W.H. Cell Cycle Control by PTEN. J. Mol. Biol. 2017, 429, 2265–2277. [Google Scholar] [CrossRef]
- Tutone, M.; Almerico, A.M. Recent advances on CDK inhibitors: An insight by means of in silico methods. Eur. J. Med. Chem. 2017, 142, 300–315. [Google Scholar] [CrossRef]
- Contreras-Vallejos, E.; Utreras, E.; Gonzalez-Billault, C. Going out of the brain: Non-nervous system physiological and pathological functions of Cdk5. Cell Signal. 2012, 24, 44–52. [Google Scholar] [CrossRef]
- Pozo, K.; Castro-Rivera, E.; Tan, C.; Plattner, F.; Schwach, G.; Siegl, V.D.; Guo, A.; Gundara, J.; Mettlach, G.; Richer, E.; et al. The role of Cdk5 in neuroendocrine thyroid cancer. Cancer Cell 2013, 24, 499–511. [Google Scholar] [CrossRef]
- Huart, A.-S.; MacLaine, N.J.; Meek, D.W.; Hupp, T.R. CK1α plays a central role in mediating MDM2 control of p53 and E2F-1 protein stability. J. Biol. Chem. 2009, 284, 32384–32394. [Google Scholar] [CrossRef]
- Inuzuka, H.; Tseng, A.; Gao, D.; Zhai, B.; Zhang, Q.; Shaik, S.; Wan, L.; Ang, X.L.; Mock, C.; Yin, H.; et al. Phosphorylation by casein kinase I promotes the turnover of the mdm2 oncoprotein via the SCFβ-TRCP ubiquitin ligase. Cancer Cell 2010, 18, 147–159. [Google Scholar] [CrossRef] [PubMed]
- Bibian, M.; Rahaim, R.J.; Choi, J.Y.; Noguchi, Y.; Schürer, S.; Chen, W.; Nakanishi, S.; Licht, K.; Rosenberg, L.H.; Li, L.; et al. Development of highly selective casein kinase 1δ/1ε (CK1δ/ε) inhibitors with potent antiproliferative properties. Bioorg. Med. Chem. Lett. 2013, 23, 4374–4380. [Google Scholar] [CrossRef] [PubMed]
- Monastyrskyi, A.; Nilchan, N.; Quereda, V.; Noguchi, Y.; Ruiz, C.; Grant, W.; Cameron, M.; Duckett, D.; Roush, W. Development of dual casein kinase 1δ/1ε (CK1δ/ε) inhibitors for treatment of breast cancer. Bioorg. Med. Chem. 2018, 26, 590–602. [Google Scholar] [CrossRef] [PubMed]
- Behrend, L.; Milne, D.M.; Stöter, M.; Deppert, W.; Campbell, L.E.; Meek, D.W.; Knippschild, U. IC261, a specific inhibitor of the protein kinases casein kinase 1-delta and -epsilon, triggers the mitotic checkpoint and induces p53-dependent postmitotic effects. Oncogene 2000, 19, 5303–5313. [Google Scholar] [CrossRef] [PubMed]
- Fadeel, B.; Orrenius, S. Apoptosis: A basic biological phenomenon with wide-ranging implications in human disease. J. Intern. Med. 2005, 258, 479–517. [Google Scholar] [CrossRef] [PubMed]
- John, R.; Chand, V.; Chakraborty, S.; Jaiswal, N.; Nag, A. DNA damage induced activation of Cygb stabilizes p53 and mediates G1 arrest. DNA Repair 2014, 24, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Luo, X.-J.; Qin, Q.-P.; Li, Y.-L.; Liu, Y.-C.; Chen, Z.-F.; Liang, H. Three platinum (II) complexes of 2-(methoxyphenyl)imidazo[4,5-f][1,10]phenanthroline: Cell apoptosis induction by sub-G1 phase cell cycle arrest and G-quadruplex binding properties. Inorg. Chem. Commun. 2014, 46, 176–179. [Google Scholar] [CrossRef]
- Gu, W.; Miao, T.-T.; Hua, D.-W.; Jin, X.-Y.; Tao, X.-B.; Huang, C.-B.; Wang, S.-F. Synthesis and in vitro cytotoxic evaluation of new 1H-benzo[d]imidazole derivatives of dehydroabietic acid. Bioorg. Med. Chem. Lett. 2017, 27, 1296–1300. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, F.; Zhang, L.; Wang, H.; Zhang, J.; Zhang, C.; Jiang, Z.; Li, Y.; Liu, Z.; Chen, H. DMXAA-pyranoxanthone hybrids enhance inhibition activities against human cancer cells with multi-target functions. Eur. J. Med. Chem. 2018, 143, 1768–1778. [Google Scholar] [CrossRef]
- Rocha, G.B.; Freire, R.O.; Simas, A.M.; Stewart, J.J. RM1: A reparameterization of AM1 for H, C, N, O, P, S, F, Cl, Br, and I. J. Comput. Chem. 2006, 27, 1101–1111. [Google Scholar] [CrossRef]
- Spartan’08 Tutorial and User’s Guide: Wavefunction. 2008. Available online: http://www. wavefun.com/spartan.html (accessed on 6 September 2019).
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed]
- Durrant, J.D.; McCammon, J.A. BINANA: A novel algorithm for ligand-binding characterization. J. Mol. Graph. Model. 2011, 29, 888–893. [Google Scholar] [CrossRef] [PubMed]
- Buarque, C.D.; Militão, G.C.G.; Lima, D.J.B.; Costa-Lotufo, L.V.; Pessoa, C.; de Moraes, M.O.; Cunha-Junior, E.F.; Torres-Santos, E.C.; Netto, C.D.; Costa, P.R.R. Pterocarpanquinones, aza-pterocarpanquinone and derivatives: Synthesis, antineoplasic activity on human malignant cell lines and antileishmanial activity on Leishmania amazonensis. Bioorg. Med. Chem. 2011, 19, 6885–6891. [Google Scholar] [CrossRef]
- Zegzouti, H.; Zdanovskaia, M.; Hsiao, K.; Goueli, S.A. ADP-Glo: A Bioluminescent and homogeneous ADP monitoring assay for kinases. Assay Drug. Dev. Technol. 2009, 7, 560–572. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| IC50 (μM) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Compound | K562 | HL-60 | MOLT-4 | HEp-2 | NCI-H292 | HT-29 | MCF-7 | PBMC |
| 3a | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3b | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3c | 52.34 ± 1.15 | 13.08 ± 0.62 | 41.00 ± 0.7 | 37.08 ± 1.50 | 56.27 ± 3.05 | 40.57 ± 1.02 | 34.02 ± 1.06 | 82.01 ± 1.63 |
| 3d | 25.19 ± 2.43 | 16.93 ± 1.10 | 37.17 ± 0.84 | 23.13 ± 0.80 | > 100 | > 100 | 33.46 ± 0.75 | 46.26 ± 0.97 |
| 3e | 58.69 ± 0.97 | > 100 | > 100 | 22.26 ± 0.40 | > 100 | > 100 | 33.90 ± 0.80 | > 100 |
| 3f | 26.28 ± 1.86 | 14.22 ± 1.41 | > 100 | 8.61 ± 0.35 | > 100 | > 100 | 23.26 ± 1.55 | > 100 |
| 3g | 33.29 ± 0.71 | 48.53 ± 1.63 | 56.43 ± 0.97 | 24.26 ± 0.97 | > 100 | > 100 | > 100 | > 100 |
| 3h | > 100 | > 100 | 25.53 ± 1.9 | 42.86 ± 0.75 | 48.79 ± 0.97 | 46.97 ± 0.75 | 58.37 ± 1.86 | 49.70 ± 1.06 |
| 3i | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3j | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3k | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3l | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 | > 100 |
| 3m | N.T. | 24.33 ±0.16 | N.T. | N.T. | 31.9±0.07 | 28.42± 8.56 | 8.56 ±0.01 | >100 |
| 4a | 24.21 ± 1.15 | 11.38 ± 0.27 | 19.53 ± 0.62 | 34.37 ± 0.88 | 37.89 ± 1.50 | 36.32 ± 0.97 | 33.59 ± 0.71 | 51.21 ± 0.75 |
| Doxo a | 1.47 ± 0.09 | 0.11 ± 0.01 | 0.73 ± 0.08 | 1.28 ± 0.11 | 0.18 ± 0.02 | 0.73 ± 0.09 | 0.20 ± 0.03 | 5.33 ± 0.62 |
| Compound | Structure | ClogD7.4 | LogS | Solubility (mg/mL) |
|---|---|---|---|---|
| 3a |  | 0.99 | −2.27 | 1.038 |
| 3b |  | 0.70 | −1.87 | 2.822 |
| 3c |  | 1.93 | −3.72 | 0.044 |
| 3d |  | 2.03 | −3.36 | 0.106 |
| 3e |  | 1.76 | −3.01 | 0.193 |
| 3f |  | 2.28 | −3.76 | 0.040 |
| 3g |  | 1.71 | −2.76 | 0.308 |
| 3h |  | 2.87 | −3.87 | 0.030 |
| 3i |  | −0.07 | −1.10 | 13.040 |
| 3j |  | 0.64 | −2.46 | 0.864 |
| 3k |  | 1.06 | −2.24 | 0.974 |
| 3l |  | 0.19 | −1.62 | 3.674 |
| 3m |  | 0.09 | −2.90 | 0.249 |
| 4a |  | 1.54 | −4.24 | 0.015 |
| Protein Kinases | IC50 (μM) | |||||||
|---|---|---|---|---|---|---|---|---|
| 3c | 3d | 3e | 3f | 3g | 3h | 3m | 4a | |
| CDK2/CyclinA | 0.53 | > 10 | > 10 | > 10 | > 10 | > 10 | N.T. | > 10 |
| CDK5/p25 | 0.35 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| CDK9/CyclinT | 0.86 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| HASPIN | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| PIM1 | 0.52 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| CLK1 | 0.29 | > 10 | > 10 | > 10 | > 10 | 1.9 | N.T. | > 10 |
| DYRK1A | 0.073 | > 10 | > 10 | > 10 | > 10 | > 10 | N.T. | > 10 |
| CK1δ/ε | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | N.T. | 0.76 |
| GSK3 α/β | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 | > 10 |
| AURKB | 5.1 | > 10 | > 10 | > 10 | > 10 | > 10 | N.T. | > 10 |
| CDK2 | DYRK1A | CK1δ | |||||
|---|---|---|---|---|---|---|---|
| Residues | Molecules | Residues | Molecules | Residues | Molecule | ||
| 3c | 3g | 3c | 3g | 4a | |||
| ILE-10 | 3.1/3.1 | 2.8 | ILE-165 | 2.9 | 2.9 | ILE-15 | HC |
| VAL-18 | HC | - | ALA-186 | HC | - | LYS-38 | HC |
| ALA-31 | HC | - | LYS-188 | HC | HC | MET-82 | HC |
| LYS-33 | HC | HC | VAL-222 | HC | HC | GLU-83 | 2.9 |
| VAL-64 | HC | HC | PHE-238 | HC | HC | LEU-85 | 2.8 |
| PHE-80 | HC | HC | GLU-239 | 3.0 | - | SER-88 | 2.9 |
| ASP-86 | 2.9 | 2.9/3.0 | LEU-241 | 2.9/HC | HC | LEU-135 | HC |
| LEU-134 | HC | HC | SER-242 | 3.0 | 3.1 | ILE-148 | HC |
| ALA-144 | - | HC | LEU-294 | HC | HC | ||
| ASP-145 | 3.0 | - | VAL-306 | HC | HC | ||
| Docking score | 33.37 | 29.63 | Docking score | 35.00 | 28.64 | Docking score | 23.47 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nascimento da Cruz, A.C.; Brondani, D.J.; I´talo de Santana, T.; Oliveira da Silva, L.; da Oliveira Borba, E.F.; de Faria, A.R.; Ferreira Cavalcanti de Albuquerque, J.; Piessard, S.; Matos Ximenes, R.; Baratte, B.; et al. Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents. Pharmaceuticals 2019, 12, 169. https://doi.org/10.3390/ph12040169
Nascimento da Cruz AC, Brondani DJ, I´talo de Santana T, Oliveira da Silva L, da Oliveira Borba EF, de Faria AR, Ferreira Cavalcanti de Albuquerque J, Piessard S, Matos Ximenes R, Baratte B, et al. Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents. Pharmaceuticals. 2019; 12(4):169. https://doi.org/10.3390/ph12040169
Chicago/Turabian StyleNascimento da Cruz, Anne Cecília, Dalci José Brondani, Temístocles I´talo de Santana, Lucas Oliveira da Silva, Elizabeth Fernanda da Oliveira Borba, Antônio Rodolfo de Faria, Julianna Ferreira Cavalcanti de Albuquerque, Sylvie Piessard, Rafael Matos Ximenes, Blandine Baratte, and et al. 2019. "Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents" Pharmaceuticals 12, no. 4: 169. https://doi.org/10.3390/ph12040169
APA StyleNascimento da Cruz, A. C., Brondani, D. J., I´talo de Santana, T., Oliveira da Silva, L., da Oliveira Borba, E. F., de Faria, A. R., Ferreira Cavalcanti de Albuquerque, J., Piessard, S., Matos Ximenes, R., Baratte, B., Bach, S., Ruchaud, S., Bezerra Mendonça Junior, F. J., Bazin, M.-A., Montenegro Rabello, M., Hernandes, M. Z., Marchand, P., & Gonçalves da Silva, T. (2019). Biological Evaluation of Arylsemicarbazone Derivatives as Potential Anticancer Agents. Pharmaceuticals, 12(4), 169. https://doi.org/10.3390/ph12040169