Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds



Abstract
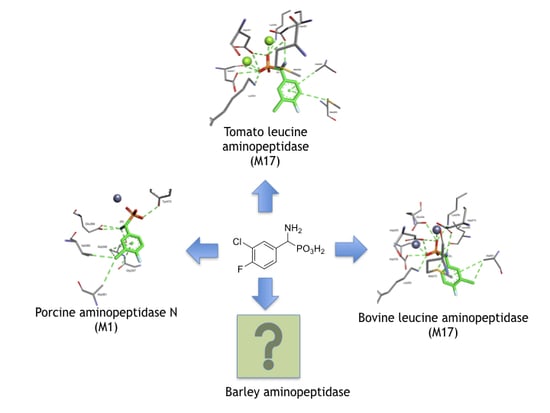
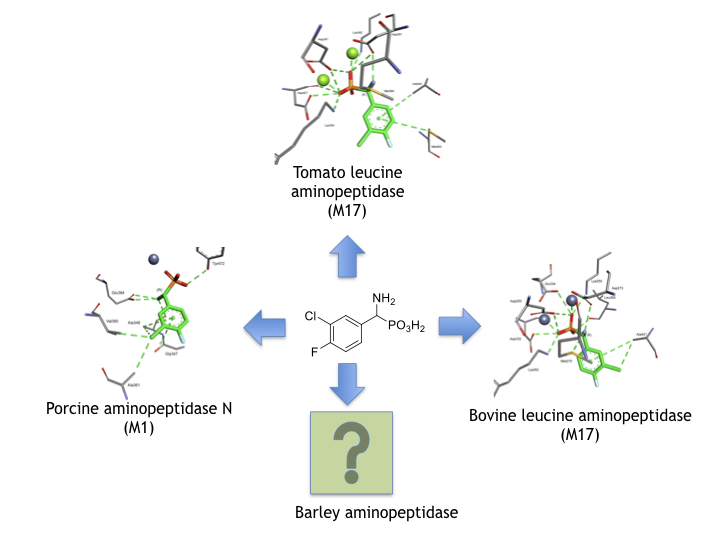
1. Introduction
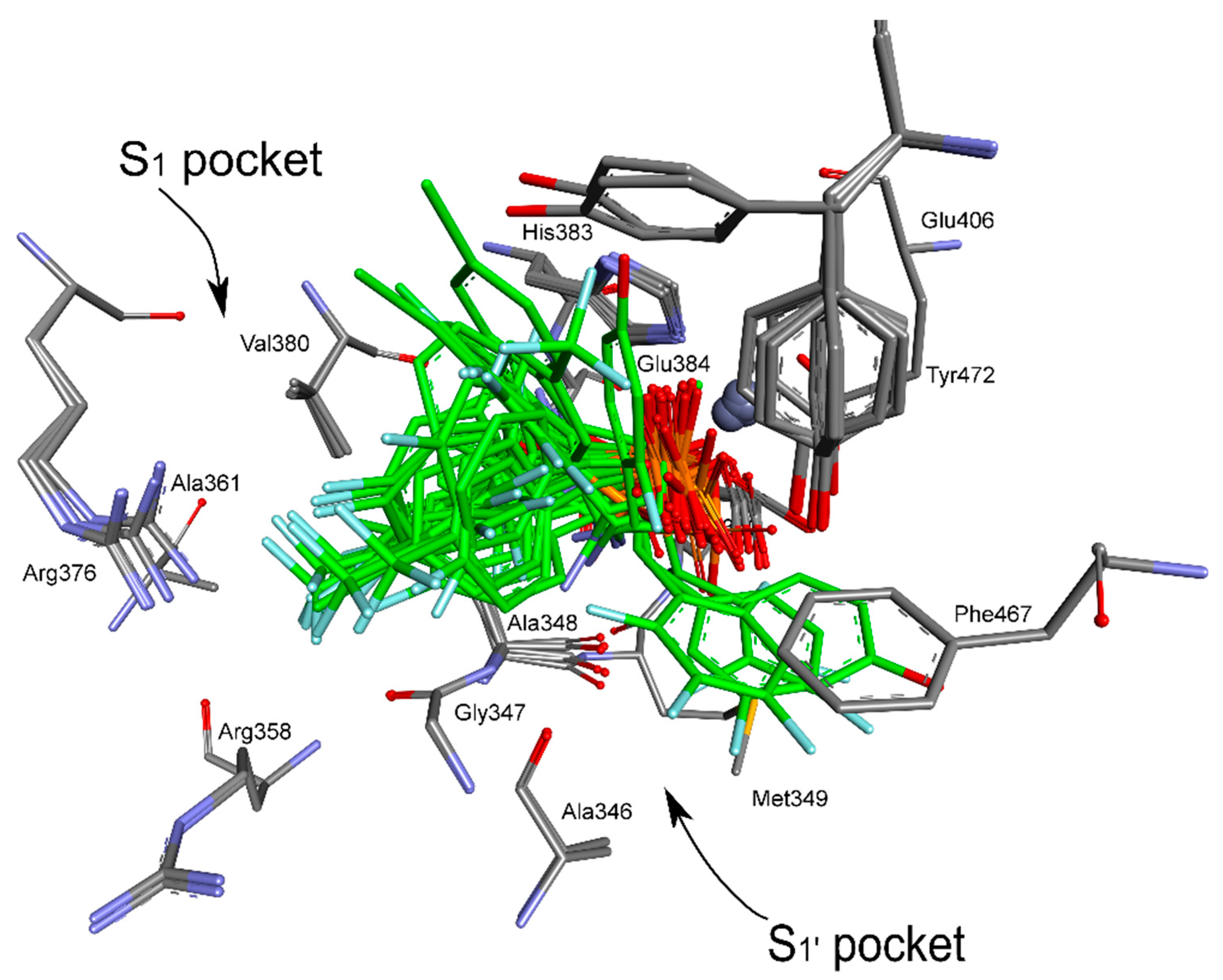
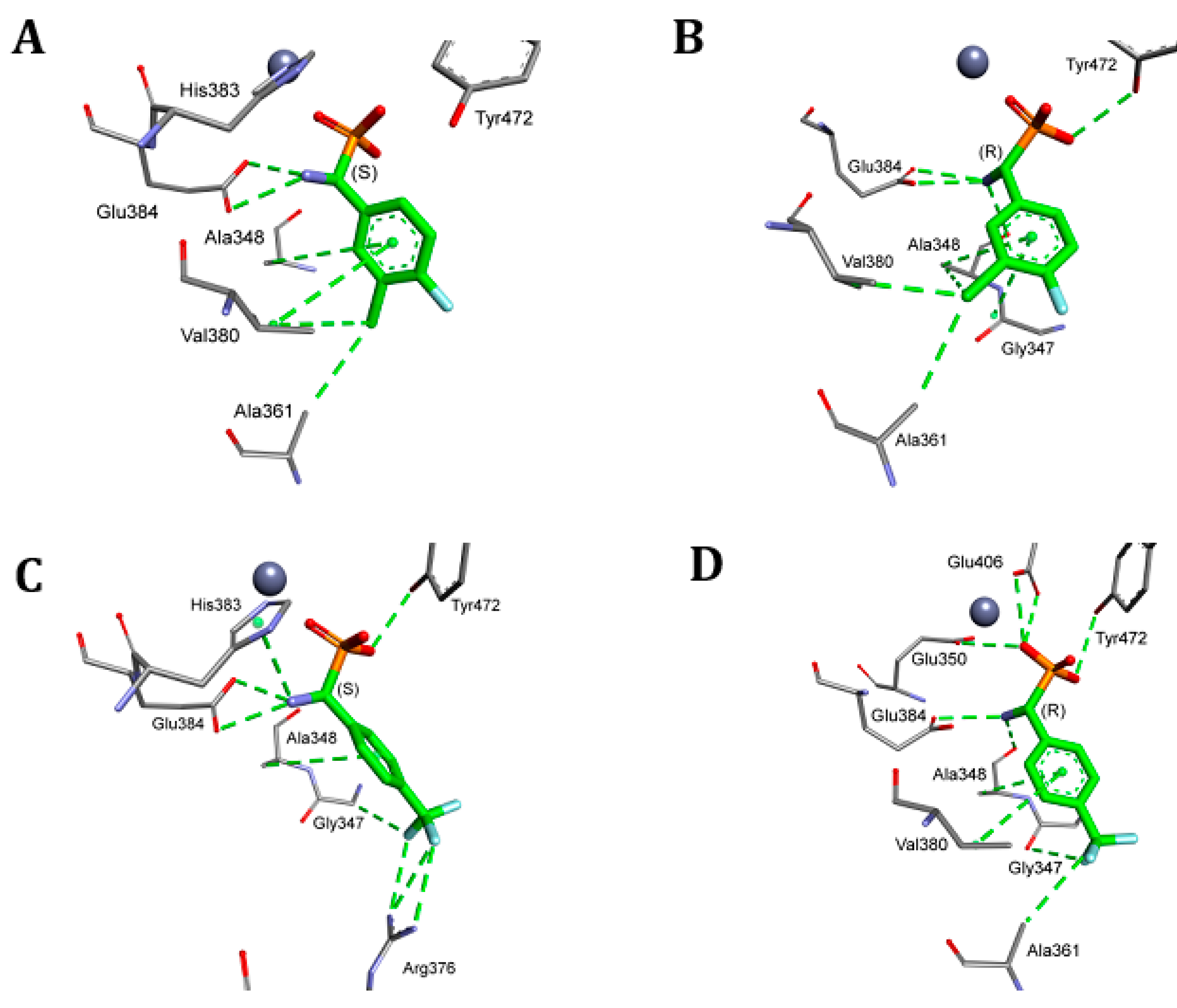
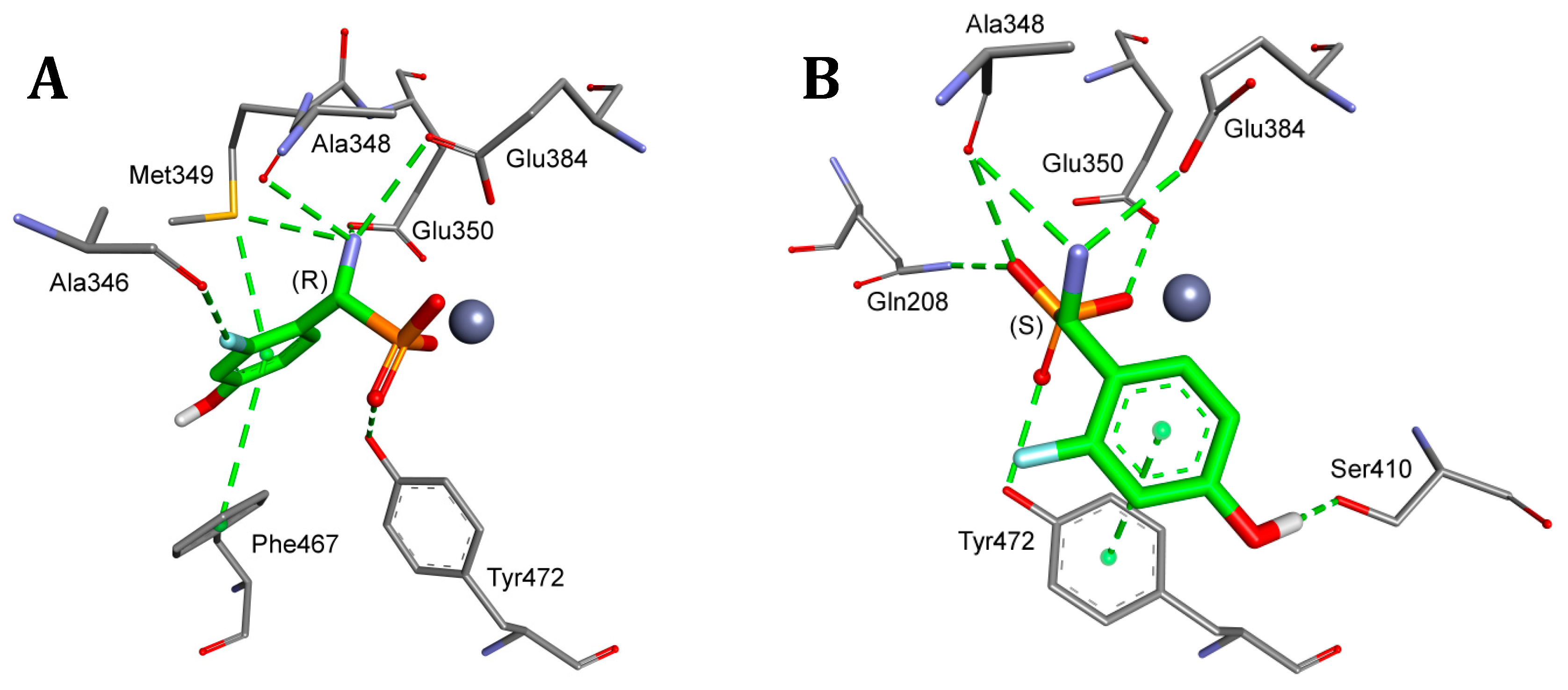
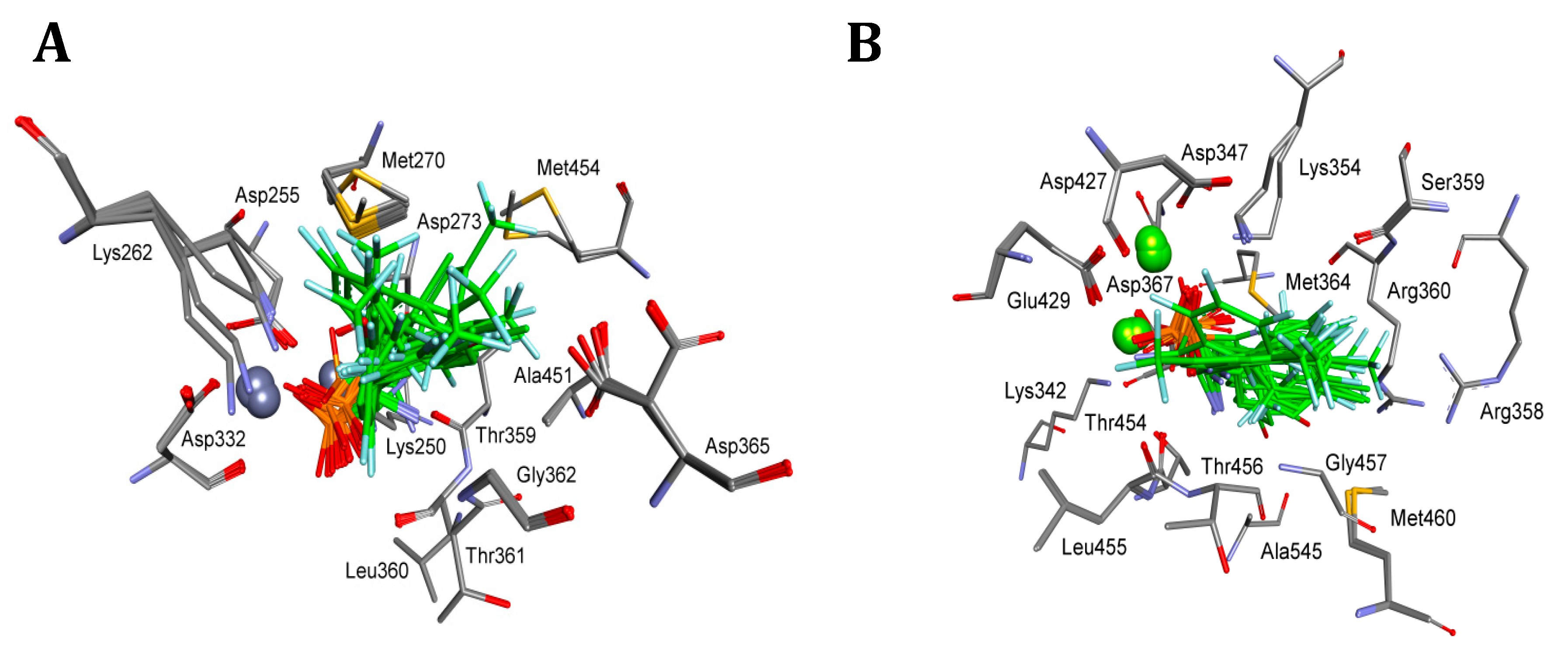
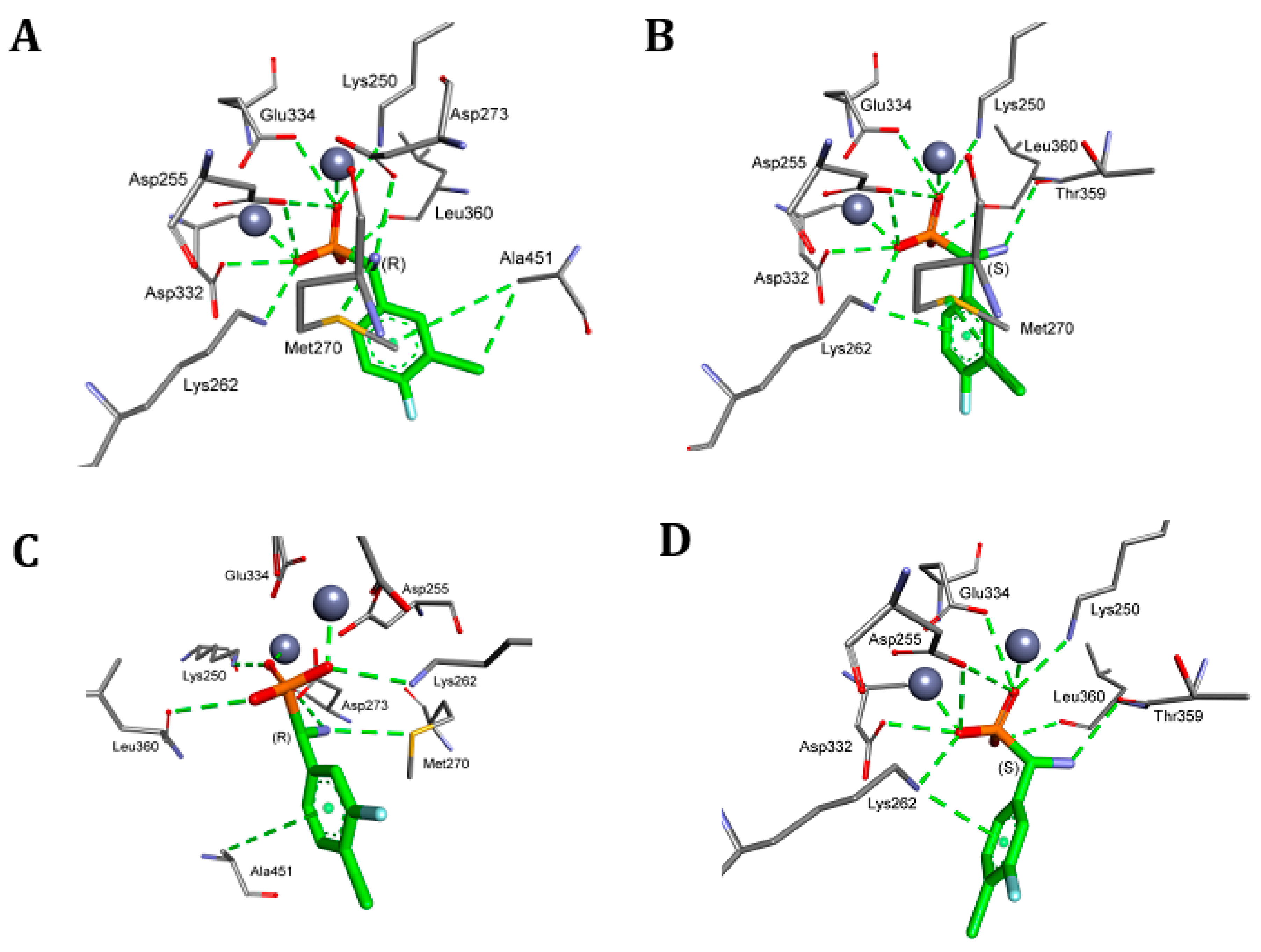
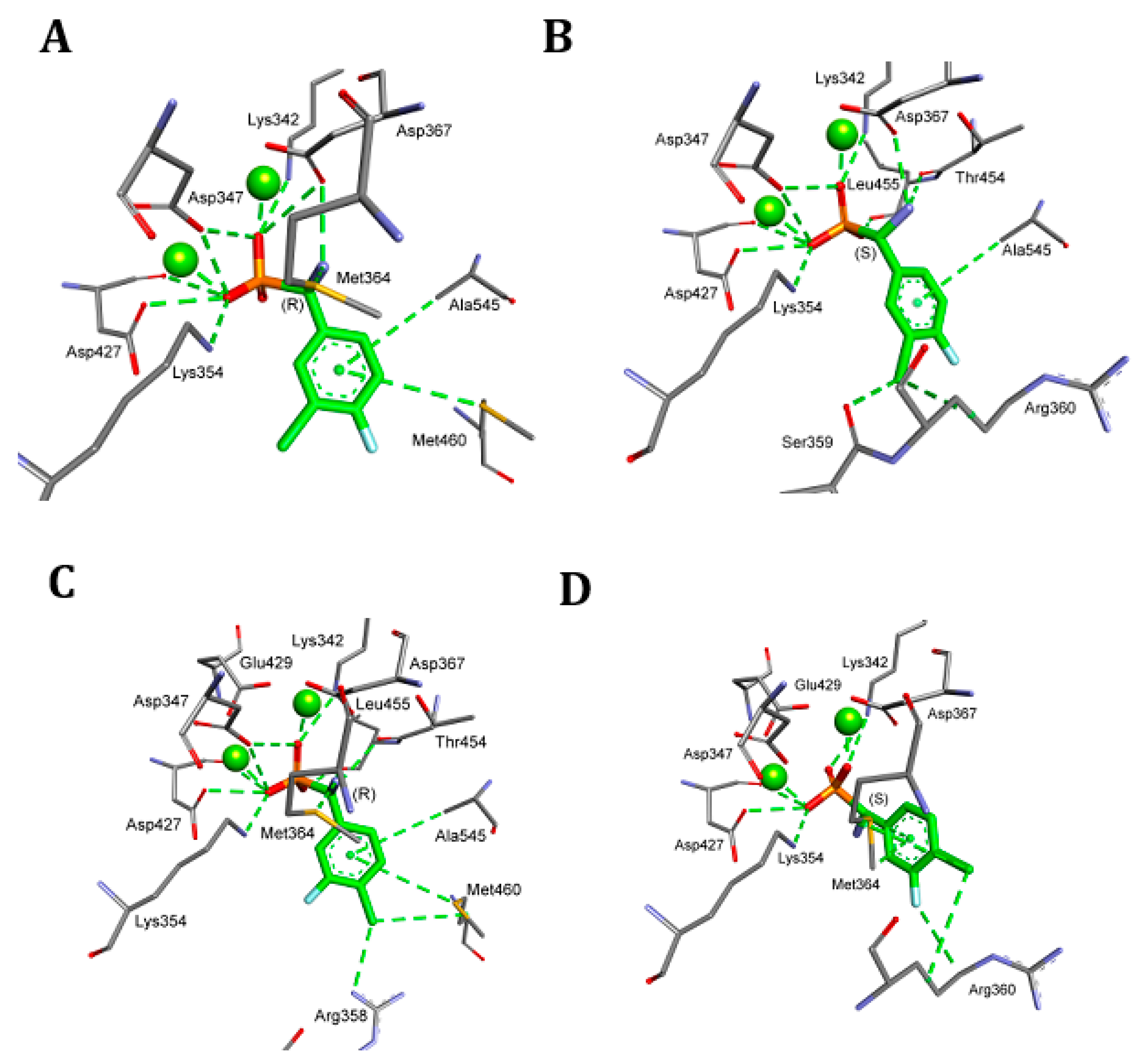
2. Results and Discussion
3. Conclusions
4. Materials and Methods
4.1. Compounds
4.2. Enzyme Preparations
4.3. Kinetic Characterization of Aminopeptidases
4.4. Inhibitory Studies
4.5. Molecular Modeling
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Hooper, N.M.; Lendeckel, U. (Eds.) Aminopeptidases in Biology and Disease; Springer Science+Business Media: New York, NY, USA, 2004; ISBN 978-1-4419-8869-0. [Google Scholar]
- Mucha, A.; Drąg, M.; Dalton, J.P.; Kafarski, P. Metallo-aminopeptidase inhibitors. Biochimie 2010, 92, 1509–1529. [Google Scholar] [CrossRef] [PubMed]
- Matsui, M.; Fowler, J.H.; Walling, L.L. Leucine aminopeptidases: Diversity in structure and function. Biol. Chem. 2006, 152, 1535–1544. [Google Scholar] [CrossRef] [PubMed]
- Amin, S.A.; Adhikari, N.; Jha, T. Design of Aminopeptidase N Inhibitors as Anti-cancer Agents. J. Med. Chem. 2018, 61, 6468–6490. [Google Scholar] [CrossRef] [PubMed]
- Moore, H.E.; Davenport, E.L.; Smith, E.M.; Muralikrishnan, S.; Dunlop, A.S.; Walker, B.A.; Krige, D.; Drummond, A.H.; Hooftman, L.; Morgan, G.J. Aminopeptidase inhibition as a targeted treatment strategy in myeloma. Mol. Cancer Ther. 2009, 8, 762–770. [Google Scholar] [CrossRef] [PubMed]
- Drinkwater, N.; Lee, J.; Yang, W.; Malcolm, T.R.; McGowan, S. M1 aminopeptidases as drug targets: broad applications ortherapeutic niche? FEBS J. 2017, 284, 1473–1488. [Google Scholar] [CrossRef] [PubMed]
- Qiang, W.; Qiao, S.; Lu, H. Novel aminopeptidase N inhibitors with improved antitumor activities. Lett. Drug Des. Discov. 2016, 13, 98–106. [Google Scholar] [CrossRef]
- Vassiliou, S.; Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Mulligan, R.; Joachimiak, A.; Mucha, A. Structure-guided, single-point modifications in the phosphinic dipeptide structure yield highly potent and selective inhibitors of neutral aminopeptidases. J. Med. Chem. 2014, 57, 8140–8151. [Google Scholar] [CrossRef] [PubMed]
- Węglarz-Tomczak, E.; Berlicki, Ł.; Pawełczak, M.; Nocek, B.; Joachimiak, A.; Mucha, A. A structural insight into the P1-S1 binding mode of diaminoethylphosphonic and phosphinic acids, selective inhibitors of alanine aminopeptidases. Eur. J. Med. Chem. 2016, 117, 187–196. [Google Scholar] [CrossRef] [PubMed]
- Grzywa, R.; Oleksyszyn, J.; Salvesen, G.S.; Drąg, M. Identification of very potent inhibitor of human aminopeptidase N (CD13). Bioorg. Med. Chem. Lett. 2010, 20, 2497–2499. [Google Scholar] [CrossRef]
- Wickström, M.; Larsson, R.; Nygren, P.; Gullbo, J. Aminopeptidase N (CD13) as a target for cancer chemotherapy. Cancer Sci. 2010, 102, 501–508. [Google Scholar] [CrossRef]
- Lu, C.; Pi, A.P. (Eds.) Enzyme Inhibition in Drug Discovery and Development. The Good and the Bad; John Wily & Sons Inc.: Hoboken, NJ, USA, 2018; ISBN 978-0-470-28174-1. [Google Scholar]
- Wanat, W.; Talma, M.; Hurek, J.; Pawełczak, M.; Kafarski, P. Substituted phosphonic analogues of phenylglycine as inhibitors of phenylalanine ammonia lyase from potatoes. Biochimie 2018, 151, 119–127. [Google Scholar] [CrossRef] [PubMed]
- Santiago, C.; Mudgal, G.; Reguera, J.; Recacha, R.; Albrecht, S.; Enujanes, L.; Casanovas, J.M. Allosteric inhibition of aminopeptidase N functions related to tumor growth and virus infection. Sci. Rep. 2017, 7, 46045. [Google Scholar] [CrossRef]
- Zhu, X.; Liu, S.; Wang, X.; Luo, Z.; Shi, Y.; Wang, D.; Peng, G.; Chen, H.; Fang, L.; Xiao, S. Contribution of porcine aminopeptidase N to porcine deltacoronavirus infection. Emerg. Microbes Infect. 2018, 7, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Drag, M.; Bogyo, M.; Ellman, J.A.; Salvesen, G.S. Amino peptidase fingerprints, an integrated approach for identification of good substrates and inhibitors. J. Biol. Chem. 2010, 285, 3310–3318. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, C.A.; Smith, B.D. Molecular imaging of aminopeptidase N in cancer and angiogenesis. Contrast Media Mol. Imaging 2018, 2018, 5315172. [Google Scholar] [CrossRef] [PubMed]
- Oszywa, B.; Makowski, M.; Pawełczak, M. Purification and partial characterization of aminopeptidase from barley (Hordeum vulgare L.) seeds. Plant Physiol. Biochem. 2013, 65, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Oszywa, B.; Pawełczak, M.; Kafarski, P. The influence of alpha-aminophosphonic acids on the activity of aminopeptidase from barley seeds—An approach to determine the enzyme specificity. Acta Physiol. Plant. 2015, 37, 44. [Google Scholar] [CrossRef][Green Version]
- Duprez, K.; Scranton, M.A.; Walling, L.L.; Fan, L. Structure of tomato wound-induced leucine aminopeptidase sheds light on substrate specificity. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 1649–1658. [Google Scholar] [CrossRef]
- Mucha, A.; Laemmerhofer, M.; Lindner, W.; Pawełczak, M.; Kafarski, P. Individual stereoisomers of phosphinic dipeptide inhibitor of leucine aminopeptidase. Bioorg. Med. Chem. Lett. 2008, 18, 1550–1554. [Google Scholar] [CrossRef]
- MEROPS. Available online: https://www.ebi.ac.uk/merops/ (accessed on 17 September 2019).
- Grembecka, J.; Mucha, A.; Cierpicki, T.; Kafarski, P. The most potent organophosphorus inhibitors of leucine laminopeptidase. Structure-based design, chemistry, and activity. J. Med. Chem. 2003, 46, 2641–2655. [Google Scholar] [CrossRef]
- Grembecka, J.; Kafarski, P. Leucine aminopeptidase as a target for inhibitor design. Mini Rev. Med. Chem. 2001, 1, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kania, J.; Gillner, D. Characterisation of the aminopeptidase from non-germinated winter rape (Brassica nap-us L.) seeds. Food Chem. 2016, 207, 180–186. [Google Scholar] [CrossRef] [PubMed]
- Fowler, J.H.; Narváez-Vásquez, J.; Aromdee, D.L.; Pautot, V.; Holzer, F.M.; Walling, L. Leucine aminopeptidase regulates defense and wound signaling in tomato downstream of jasmonic acid. Plant Cell 2006, 12, 1239–1251. [Google Scholar] [CrossRef] [PubMed]
- Scranton, M.A.; Yee, A.; Park, S.-Y.; Walling, L. Plant leucine aminopeptidases moonlight as aolecular chaperones to alleviate stress-induced damage. J. Biol. Chem. 2012, 287, 18408–18417. [Google Scholar] [CrossRef] [PubMed]
- Oleksyszyn, J.; Tyka, R.; Mastalerz, P. Direct synthesis of 1-aminoalkanephosphonic and 1-aminoalkanephosphinic acids from phosphorus trichloride or dichlorophosphines. Synthesis 1978, 1978, 479–480. [Google Scholar] [CrossRef]
- Oleksyszyn, J.; Soroka, M.; Rachoń, J. Phosphorus analogs of amino-acids and pepetides. 2. Phosphoanalogs and phosphinanalogs of cycloleucin. Chimia 1978, 32, 253–255. [Google Scholar] [CrossRef]
- Soroka, M. Comments on the synthesis of aminomethylphosphonic acid. Synthesis 1989, 1989, 547–548. [Google Scholar] [CrossRef]
- Lejczak, B.; Kafarski, P.; Zygmunt, J. Inhibition of aminopeptidases by aminophosphonates. Biochemistry 1989, 28, 3549–3555. [Google Scholar] [CrossRef]
- Chen, L.; Lin, Y.L.; Peng, G.; Li, F. Structural basis for multifunctional roles of mammalian aminopeptidase N. Proc. Natl. Acad. Sci. USA 2012, 109, 17966–17971. [Google Scholar] [CrossRef]
- Schrödinger LLC. Schrödinger Release 2018-4: Schrödinger Suite 2018-4 Protein Preparation Wizard; Epik, Schrödinger LLC: New York, NY, USA, 2016; Impact, Schrödinger LLC: New York, NY, USA, 2016; Prime, Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger LLC. Schrödinger Release 2018-4: LigPrep; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger LLC. Schrödinger Release 2018-4: Schrödinger Suite 2018-2 Induced Fit Docking Protocol; Glide; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]
- Schrödinger LLC. Schrödinger Release 2018-4: Prime; Schrödinger LLC: New York, NY, USA, 2018. [Google Scholar]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Com-Pound | Structure | pAPN Ki [μM] + SD 1 | Barley Seed AP Ki [μM] + SD 1 | Com-Pound | Structure | pAPN Ki [μM] + SD 1 | Barley Seed AP Ki [μM] + SD 1 |
|---|---|---|---|---|---|---|---|
| 1 |  | 232 ± 22 | 69 1 ± 68 | 8 |  | 69.1 ± 6.5 | 103 ± 10 |
| 2 |  | 122 ± 10 | 311 ± 27 | 9 |  | 299 ± 11 | 818 ± 43 |
| 3 |  | 123 ± 2.7 | 1105 ± 57 | 10 |  | 56.3 ± 2.2 | 686 ± 70 |
| 4 |  | 258 ± 23 | 1425 ± 83 | 11 |  | 164 ± 7.7 | 1033 ± 73 |
| 5 |  | 101 ± 3.8 | 549 ± 54 | 12 |  | in 2 | in 2 |
| 6 |  | in 2 | in 2 | 13 |  | in 2 | in 2 |
| 7 |  | 45.4 ± 4.5 | 62.1 ± 2.3 | 14 |  | 116 ± 22 | in 2 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wanat, W.; Talma, M.; Pawełczak, M.; Kafarski, P. Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals 2019, 12, 139. https://doi.org/10.3390/ph12030139
Wanat W, Talma M, Pawełczak M, Kafarski P. Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals. 2019; 12(3):139. https://doi.org/10.3390/ph12030139
Chicago/Turabian StyleWanat, Weronika, Michał Talma, Małgorzata Pawełczak, and Paweł Kafarski. 2019. "Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds" Pharmaceuticals 12, no. 3: 139. https://doi.org/10.3390/ph12030139
APA StyleWanat, W., Talma, M., Pawełczak, M., & Kafarski, P. (2019). Phosphonic Acid Analogues of Phenylglycine as Inhibitors of Aminopeptidases: Comparison of Porcine Aminopeptidase N, Bovine Leucine Aminopeptidase, Tomato Acidic Leucine Aminopeptidase and Aminopeptidase from Barley Seeds. Pharmaceuticals, 12(3), 139. https://doi.org/10.3390/ph12030139