Iron Deficiency as a Therapeutic Target in Cardiovascular Disease

{kind=link}
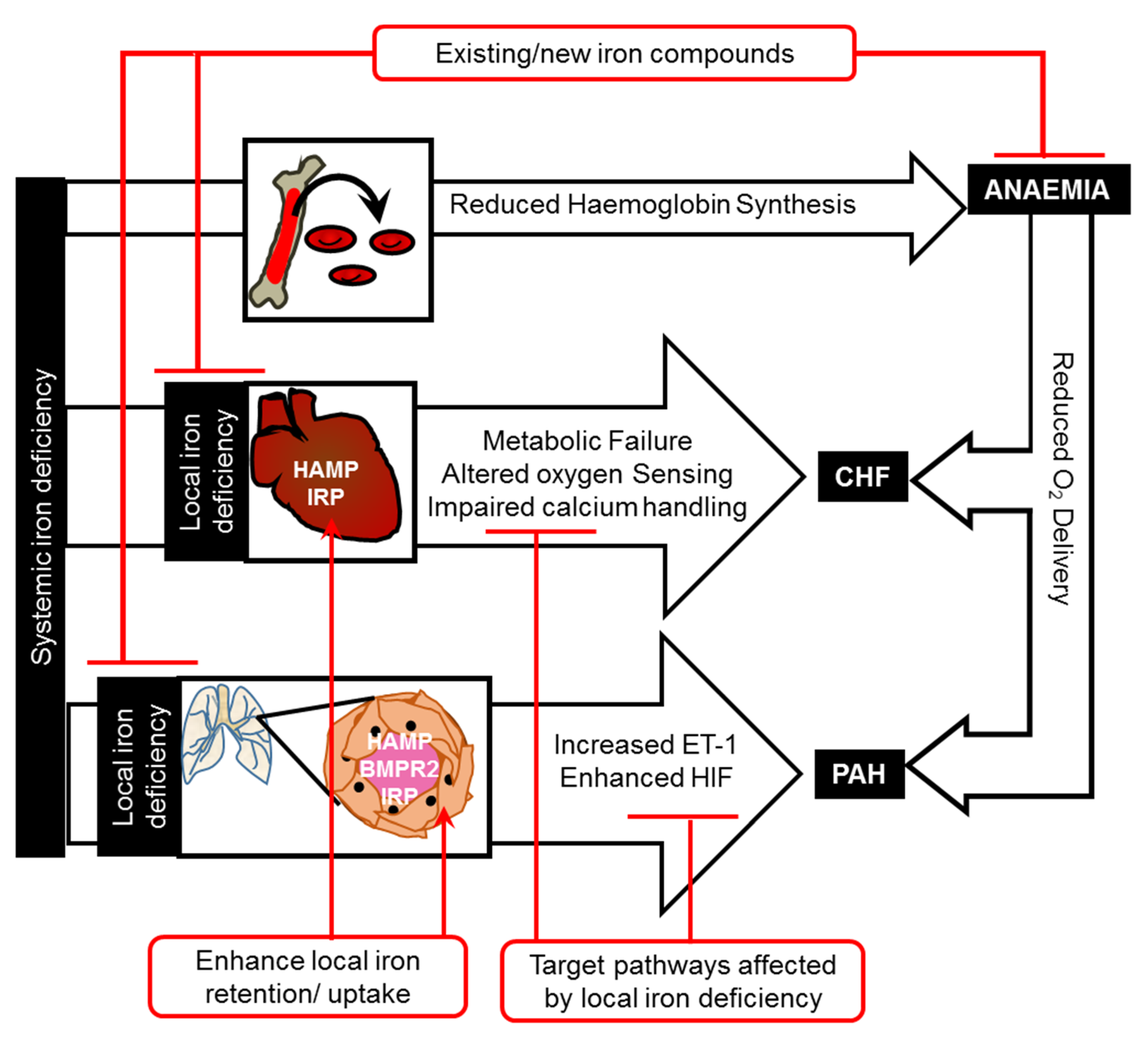
Abstract
1. Iron and Heart Function
1.1. Effects of Iron Deficiency on Heart Function
1.2. Treating Iron Deficiency in Heart Failure—Current Approaches
1.3. Treating Iron Deficiency in Heart Failure—New Approaches
2. Iron and the Pulmonary Vasculature
2.1. Effects of Iron Deficiency on the Pulmonary Vasculature
2.2. Targeting Iron Deficiency in Pulmonary Arterial Hypertension
3. Conclusions
Funding
Conflicts of Interest
References
- Walker, S.P.; Wachs, T.D.; Gardner, J.M.; Lozoff, B.; Wasserman, G.A.; Pollitt, E.; Carter, J.A.; International Child Development Steering Group. Child development: Risk factors for adverse outcomes in developing countries. Lancet 2007, 369, 145–157. [Google Scholar] [CrossRef]
- Buratti, P.; Gammella, E.; Rybinska, I.; Cairo, G.; Recalcati, S. Recent Advances in Iron Metabolism: Relevance for Health, Exercise, and Performance. Med. Sci. Sports Exerc. 2015, 47, 1596–1604. [Google Scholar] [CrossRef]
- Scott, S.P.; Murray-Kolb, L.E. Iron Status Is Associated with Performance on Executive Functioning Tasks in Nonanemic Young Women. J. Nutr. 2016, 146, 30–37. [Google Scholar] [CrossRef]
- Comin-Colet, J.; Enjuanes, C.; Gonzalez, G.; Torrens, A.; Cladellas, M.; Merono, O.; Ribas, N.; Ruiz, S.; Gómez, M.; Verdú, J.M.; et al. Iron deficiency is a key determinant of health-related quality of life in patients with chronic heart failure regardless of anaemia status. Eur. J. Heart Failure 2013, 15, 1164–1172. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Kasztura, M.; Sokolski, M.; Bronisz, M.; Nawrocka, S.; Oleskowska-Florek, W.; Zymliński, R.; Biegus, J.; Siwołowski, P.; Banasiak, W.; et al. Iron deficiency defined as depleted iron stores accompanied by unmet cellular iron requirements identifies patients at the highest risk of death after an episode of acute heart failure. Eur. Heart J. 2014, 35, 2468–2476. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Wharton, J.; Howard, L.; Gibbs, J.S.; Vonk-Noordegraaf, A.; Wilkins, M.R. Iron deficiency in pulmonary arterial hypertension: A potential therapeutic target. Eur. Respir. J. 2011, 38, 1453–1460. [Google Scholar] [CrossRef]
- Jankowska, E.A.; Ponikowski, P. Anaemia (and iron deficiency?) in aortic stenosis—A bystander or a potential therapeutic target? Eur. J. Heart Failure 2015, 17, 994–996. [Google Scholar] [CrossRef]
- Nickol, A.H.; Frise, M.C.; Cheng, H.Y.; McGahey, A.; McFadyen, B.M.; Harris-Wright, T.; Bart, N.K.; Curtis, M.K.; Khandwala, S.; O’Neill, D.P.; et al. A cross-sectional study of the prevalence and associations of iron deficiency in a cohort of patients with chronic obstructive pulmonary disease. BMJ Open 2015, 5, e007911. [Google Scholar] [CrossRef]
- Nemeth, E.; Valore, E.V.; Territo, M.; Schiller, G.; Lichtenstein, A.; Ganz, T. Hepcidin, a putative mediator of anemia of inflammation, is a type II acute-phase protein. Blood 2003, 101, 2461–2463. [Google Scholar] [CrossRef]
- Markousis-Mavrogenis, G.; Tromp, J.; Ouwerkerk, W.; Devalaraja, M.; Anker, S.D.; Cleland, J.G.; Dickstein, K.; Filippatos, G.S.; van der Harst, P.; Lang, C.C.; et al. The clinical significance of interleukin-6 in heart failure: Results from the BIOSTAT-CHF study. Eur. J. Heart Failure 2019, 21, 965–973. [Google Scholar]
- Shuler, T.R.; Pootrakul, P.; Yarnsukon, P.; Nielsen, F.H. Effect of thalassemia/hemoglobin E disease on macro, trace, and ultratrace element concentration in human tissue. J. Trace Elem. Exp. Med. 1990, 3, 31–34. [Google Scholar]
- Chua-anusorna, W.; Tran, K.C.; Webb, J.; Macey, D.J.; St Pierre, T.G. Chemical speciation of iron deposits in thalassemic heart tissue. Inorg. Chim. Acta 2000, 300–302, 932–936. [Google Scholar] [CrossRef]
- Wofford, J.D.; Chakrabarti, M.; Lindahl, P.A. Mossbauer Spectra of Mouse Hearts Reveal Age-dependent Changes in Mitochondrial and Ferritin Iron Levels. J. Biol. Chem. 2017, 292, 5546–5554. [Google Scholar] [CrossRef]
- Beinert, H.; Holm, R.H.; Munck, E. Iron-sulfur clusters: Nature’s modular, multipurpose structures. Science 1997, 277, 653–659. [Google Scholar] [CrossRef]
- Meyer, J. Iron-sulfur protein folds, iron-sulfur chemistry, and evolution. J. Biol. Inorg. Chem. 2008, 13, 157–170. [Google Scholar] [CrossRef]
- Sono, M.; Roach, M.P.; Coulter, E.D.; Dawson, J.H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. [Google Scholar] [CrossRef]
- Solomon, E.I.; Decker, A.; Lehnert, N. Non-heme iron enzymes: Contrasts to heme catalysis. Proc. Natl. Acad. Sci. USA 2003, 100, 3589–3594. [Google Scholar] [CrossRef]
- Glickstein, H.; Ben-El, R.B.; Shvartsman, M.; Cabantchik, Z.I. Intracellular labile iron pools as direct targets of iron chelators: A fluorescence study of chelator action in living cells. Blood 2005, 106, 3242–3250. [Google Scholar] [CrossRef]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef]
- Ratcliffe, P.J.; O’Rourke, J.F.; Maxwell, P.H.; Pugh, C.W. Oxygen sensing, hypoxia-inducible factor-1 and the regulation of mammalian gene expression. J. Exp. Biol. 1998, 201, 1153–1162. [Google Scholar]
- WHO. Available online: http://www.who.int/nutrition/topics/ida/en/ (accessed on 19 July 2019).
- Galan, P.; Yoon, H.C.; Preziosi, P.; Viteri, F.; Valeix, P.; Fieux, B.; Briançon, S.; Malvy, D.; Roussel, A.M.; Favier, A.; et al. Determining factors in the iron status of adult women in the SU.VI.MAX study. SUpplementation en VItamines et Mineraux AntioXydants. Eur. J. Clin. Nutr. 1998, 52, 383–388. [Google Scholar] [CrossRef]
- Sinclair, L.M.; Hinton, P.S. Prevalence of iron deficiency with and without anemia in recreationally active men and women. J. Am. Diet. Assoc. 2005, 105, 975–978. [Google Scholar] [CrossRef]
- Dhur, A.; Galan, P.; Hercberg, S. Effects of different degrees of iron deficiency on cytochrome P450 complex and pentose phosphate pathway dehydrogenases in the rat. J. Nutr. 1989, 119, 40–47. [Google Scholar] [CrossRef]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal Cardiomyopathy in Mice Lacking Transferrin Receptor in the Heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. eLife 2016, 5, e19804. [Google Scholar] [CrossRef]
- Chung, Y.J.; Luo, A.; Park, K.C.; Loonat, A.A.; Lakhal-Littleton, S.; Robbins, P.A.; Swietach, P. Iron-deficiency anemia reduces cardiac contraction by downregulating RyR2 channels and suppressing SERCA pump activity. JCI Insight 2019, 4, 125618. [Google Scholar] [CrossRef]
- Mole, D.R. Iron homeostasis and its interaction with prolyl hydroxylases. Antioxid. Redox Signal. 2010, 12, 445–458. [Google Scholar] [CrossRef]
- Klip, I.T.; Comin-Colet, J.; Voors, A.A.; Ponikowski, P.; Enjuanes, C.; Banasiak, W.; Lok, D.J.; Rosentryt, P.; Torrens, A.; Polonski, L.; et al. Iron deficiency in chronic heart failure: An international pooled analysis. Am. Heart J. 2013, 165, 575–582.e3. [Google Scholar] [CrossRef]
- Okonko, D.O.; Mandal, A.K.; Missouris, C.G.; Poole-Wilson, P.A. Disordered iron homeostasis in chronic heart failure: Prevalence, predictors, and relation to anemia, exercise capacity, and survival. J. Am. Coll. Cardiol. 2011, 58, 1241–1251. [Google Scholar] [CrossRef]
- Mordi, I.R.; Tee, A.; Lang, C.C. Iron Therapy in Heart Failure: Ready for Primetime? Card. Fail. Rev. 2018, 4, 28–32. [Google Scholar] [CrossRef]
- Ponikowski, P.; van Veldhuisen, D.J.; Comin-Colet, J.; Ertl, G.; Komajda, M.; Mareev, V.; McDonagh, T.; Parkhomenko, A.; Tavazzi, L.; Levesque, V.; et al. Beneficial effects of long-term intravenous iron therapy with ferric carboxymaltose in patients with symptomatic heart failure and iron deficiency. Eur. Heart J. 2015, 36, 657–668. [Google Scholar] [CrossRef]
- Anker, S.D.; Comin Colet, J.; Filippatos, G.; Willenheimer, R.; Dickstein, K.; Drexler, H.; Lüscher, T.F.; Bart, B.; Banasiak, W.; Niegowska, J.; et al. Ferric carboxymaltose in patients with heart failure and iron deficiency. N. Engl. J. Med. 2009, 361, 2436–2448. [Google Scholar] [CrossRef]
- Lam, C.S.P.; Doehner, W.; Comin-Colet, J.; IRON CORE Group. Iron deficiency in chronic heart failure: Case-based practical guidance. ESC Heart Fail. 2018, 5, 764–771. [Google Scholar] [CrossRef]
- Florian, A.; Ludwig, A.; Rosch, S.; Yildiz, H.; Klumpp, S.; Sechtem, U.; Yilmaz, A. Positive effect of intravenous iron-oxide administration on left ventricular remodelling in patients with acute ST-elevation myocardial infarction—A cardiovascular magnetic resonance (CMR) study. Int. J. Cardiol. 2014, 173, 184–189. [Google Scholar] [CrossRef]
- Fuernau, G.; Traeder, F.; Lele, S.S.; Rajapurkar, M.M.; Mukhopadhyay, B.; de Waha, S.; Desch, S.; Eitel, I.; Schuler, G.; Adams, V.; et al. Catalytic iron in acute myocardial infarction complicated by cardiogenic shock—A biomarker substudy of the IABP-SHOCK II-trial. Int. J. Cardiol. 2017, 227, 83–88. [Google Scholar] [CrossRef]
- Melenovsky, V.; Petrak, J.; Mracek, T.; Benes, J.; Borlaug, B.A.; Nuskova, H.; Pluhacek, T.; Spatenka, J.; Kovalcikova, J.; Drahota, Z.; et al. Myocardial iron content and mitochondrial function in human heart failure: A direct tissue analysis. Eur. J. Heart Fail. 2017, 19, 522–530. [Google Scholar] [CrossRef]
- Allen, K.J.; Gurrin, L.C.; Constantine, C.C.; Osborne, N.J.; Delatycki, M.B.; Nicoll, A.J.; McLaren, C.E.; Bahlo, M.; Nisselle, A.E.; Vulpe, C.D.; et al. Iron-overload-related disease in HFE hereditary hemochromatosis. N. Engl. J. Med. 2008, 358, 221–230. [Google Scholar] [CrossRef]
- Lekawanvijit, S.; Chattipakorn, N. Iron overload thalassemic cardiomyopathy: Iron status assessment and mechanisms of mechanical and electrical disturbance due to iron toxicity. Can. J. Cardiol. 2009, 25, 213–218. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef]
- Haddad, S.; Wang, Y.; Galy, B.; Korf-Klingebiel, M.; Hirsch, V.; Baru, A.M.; Rostami, F.; Reboll, M.R.; Heineke, J.; Flögel, U.; et al. Iron-regulatory proteins secure iron availability in cardiomyocytes to prevent heart failure. Eur. Heart J. 2017, 38, 362–372. [Google Scholar]
- Smith, T.G.; Talbot, N.P.; Privat, C.; Rivera-Ch, M.; Nickol, A.H.; Ratcliffe, P.J.; Dorrington, K.L.; León-Velarde, F.; Robbins, P.A. Effects of iron supplementation and depletion on hypoxic pulmonary hypertension: Two randomized controlled trials. JAMA 2009, 302, 1444–1450. [Google Scholar] [CrossRef]
- Frise, M.C.; Cheng, H.Y.; Nickol, A.H.; Curtis, M.K.; Pollard, K.A.; Roberts, D.J.; Ratcliffe, P.J.; Dorrington, K.L.; Robbins, P.A. Clinical iron deficiency disturbs normal human responses to hypoxia. J. Clin. Investig. 2016, 126, 2139–2150. [Google Scholar] [CrossRef]
- Bart, N.K.; Curtis, M.K.; Cheng, H.Y.; Hungerford, S.L.; McLaren, R.; Petousi, N.; Dorrington, K.L.; Robbins, P.A. Elevation of iron storage in humans attenuates the pulmonary vascular response to hypoxia. J. Appl. Physiol. 2016, 121, 537–544. [Google Scholar] [CrossRef]
- Smith, T.G.; Balanos, G.M.; Croft, Q.P.; Talbot, N.P.; Dorrington, K.L.; Ratcliffe, P.J.; Robbins, P.A. The increase in pulmonary arterial pressure caused by hypoxia depends on iron status. J. Physiol. 2008, 586, 5999–6005. [Google Scholar] [CrossRef]
- Viethen, T.; Gerhardt, F.; Dumitrescu, D.; Knoop-Busch, S.; ten Freyhaus, H.; Rudolph, T.K.; Baldus, S.; Rosenkranz, S. Ferric carboxymaltose improves exercise capacity and quality of life in patients with pulmonary arterial hypertension and iron deficiency: A pilot study. Int. J. Cardiol. 2014, 175, 233–239. [Google Scholar] [CrossRef]
- Rhodes, C.J.; Howard, L.S.; Busbridge, M.; Ashby, D.; Kondili, E.; Gibbs, J.S.; Wharton, J.; Wilkins, M.R. Iron deficiency and raised hepcidin in idiopathic pulmonary arterial hypertension: Clinical prevalence, outcomes, and mechanistic insights. J. Am. Coll. Cardiol. 2011, 58, 300–309. [Google Scholar] [CrossRef]
- Van Empel, V.P.; Lee, J.; Williams, T.J.; Kaye, D.M. Iron deficiency in patients with idiopathic pulmonary arterial hypertension. Heart Lung Circ. 2014, 23, 287–292. [Google Scholar] [CrossRef]
- Ruiter, G.; Lanser, I.J.; de Man, F.S.; van der Laarse, W.J.; Wharton, J.; Wilkins, M.R.; Howard, L.S.; Vonk-Noordegraaf, A.; Voskuyl, A.E. Iron deficiency in systemic sclerosis patients with and without pulmonary hypertension. Rheumatology 2014, 53, 285–292. [Google Scholar] [CrossRef]
- Plesner, L.L.; Schoos, M.M.; Dalsgaard, M.; Goetze, J.P.; Kjoller, E.; Vestbo, J.; Iversen, K. Iron Deficiency in COPD Associates with Increased Pulmonary Artery Pressure Estimated by Echocardiography. Heart Lung Circ. 2017, 26, 101–104. [Google Scholar] [CrossRef]
- Cotroneo, E.; Ashek, A.; Wang, L.; Wharton, J.; Dubois, O.; Bozorgi, S.; Busbridge, M.; Alavian, K.N.; Wilkins, M.R.; Zhao, L. Iron homeostasis and pulmonary hypertension: Iron deficiency leads to pulmonary vascular remodeling in the rat. Circ. Res. 2015, 116, 1680–1690. [Google Scholar] [CrossRef]
- Lakhal-Littleton, S.; Crosby, A.; Frise, M.C.; Mohammad, G.; Carr, C.A.; Loick, P.A.M.; Robbins, P.A. Intracellular iron deficiency in pulmonary arterial smooth muscle cells induces pulmonary arterial hypertension in mice. Proc. Natl. Acad. Sci. USA 2019, 116, 13122–13130. [Google Scholar] [CrossRef]
- Giaid, A.; Yanagisawa, M.; Langleben, D.; Michel, R.P.; Levy, R.; Shennib, H.; Kimura, S.; Masaki, T.; Duguid, W.P.; Stewart, D.J. Expression of endothelin-1 in the lungs of patients with pulmonary hypertension. N. Engl. J. Med. 1993, 328, 1732–1739. [Google Scholar] [CrossRef]
- Stewart, D.J.; Levy, R.D.; Cernacek, P.; Langleben, D. Increased plasma endothelin-1 in pulmonary hypertension: Marker or mediator of disease? Ann. Intern. Med. 1991, 114, 464–469. [Google Scholar] [CrossRef]
- Channick, R.; Badesch, D.B.; Tapson, V.F.; Simonneau, G.; Robbins, I.; Frost, A.; Roux, S.; Rainisio, M.; Bodin, F.; Rubin, L.J. Effects of the dual endothelin receptor antagonist bosentan in patients with pulmonary hypertension: A placebo-controlled study. J. Heart Lung Transplant. 2001, 20, 262–263. [Google Scholar] [CrossRef]
- Mehmood, M.; Agarwal, R.; Raina, A.; Correa-Jaque, P.; Benza, R.L. Hemodynamic response to treatment of iron deficiency anemia in pulmonary arterial hypertension: Longitudinal insights from an implantable hemodynamic monitor. Pulm. Circ. 2016, 6, 616–618. [Google Scholar] [CrossRef][Green Version]
- Ruiter, G.; Manders, E.; Happe, C.M.; Schalij, I.; Groepenhoff, H.; Howard, L.S.; Wilkins, M.R.; Bogaard, H.J.; Westerhof, N.; van der Laarse, W.J.; et al. Intravenous iron therapy in patients with idiopathic pulmonary arterial hypertension and iron deficiency. Pulm. Circ. 2015, 5, 466–472. [Google Scholar] [CrossRef]
- Ghosh, M.C.; Zhang, D.L.; Jeong, S.Y.; Kovtunovych, G.; Ollivierre-Wilson, H.; Noguchi, A.; Tu, T.; Senecal, T.; Robinson, G.; Crooks, D.R.; et al. Deletion of iron regulatory protein 1 causes polycythemia and pulmonary hypertension in mice through translational derepression of HIF2alpha. Cell Metab. 2013, 17, 271–281. [Google Scholar] [CrossRef]

© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lakhal-Littleton, S. Iron Deficiency as a Therapeutic Target in Cardiovascular Disease. Pharmaceuticals 2019, 12, 125. https://doi.org/10.3390/ph12030125
Lakhal-Littleton S. Iron Deficiency as a Therapeutic Target in Cardiovascular Disease. Pharmaceuticals. 2019; 12(3):125. https://doi.org/10.3390/ph12030125
Chicago/Turabian StyleLakhal-Littleton, Samira. 2019. "Iron Deficiency as a Therapeutic Target in Cardiovascular Disease" Pharmaceuticals 12, no. 3: 125. https://doi.org/10.3390/ph12030125
APA StyleLakhal-Littleton, S. (2019). Iron Deficiency as a Therapeutic Target in Cardiovascular Disease. Pharmaceuticals, 12(3), 125. https://doi.org/10.3390/ph12030125