Antiviral Agents in Development for Zika Virus Infections


Abstract
1. Introduction
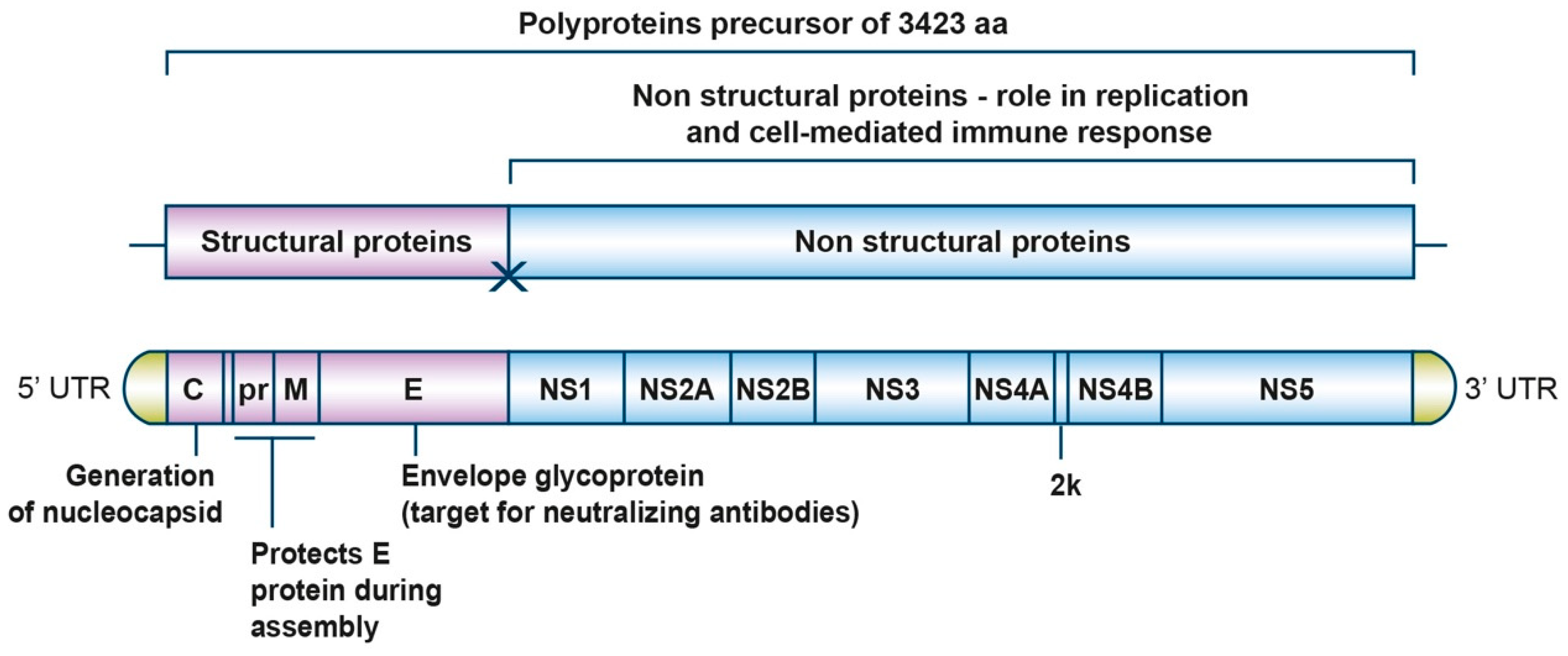
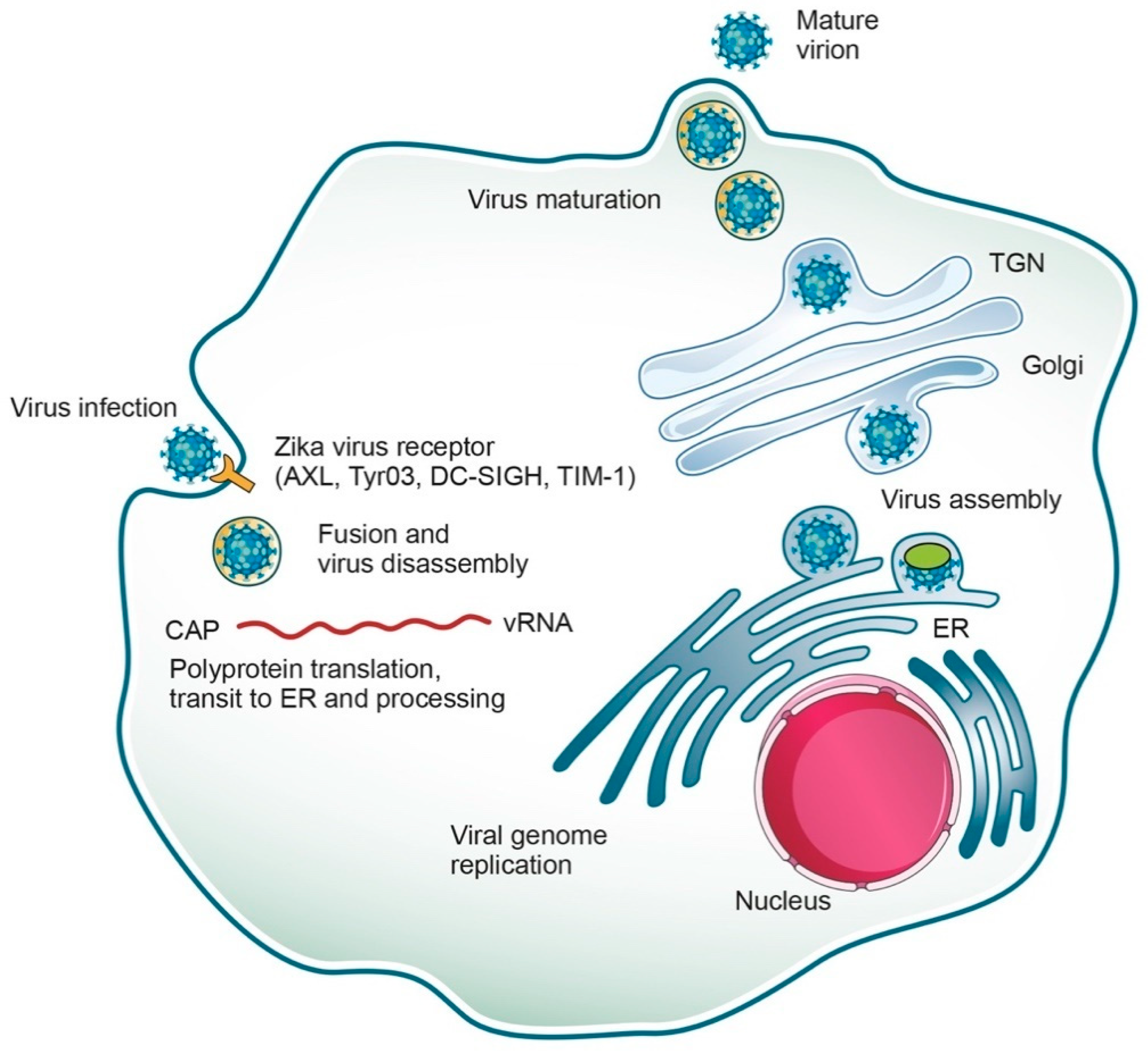
2. Genome and Replicative Cycle
3. Potential Therapeutic Options for the Treatment of ZIKV Infection
3.1. Direct-Acting Antivirals
3.2. Host-Targeting Antivirals
4. Who Benefits from ZIKV Therapies?
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Kuno, G.; Chang, G.J.; Tsuchiya, K.R.; Karabatsos, N.; Cropp, C.B. Phylogeny of the genus Flavivirus. J. Virol. 1998, 72, 73–83. [Google Scholar] [PubMed]
- Centers for Disease Control and Prevention (CDC). Oseltamivir-resistant 2009 pandemic influenza A (H1N1) virus infection in two summer campers receiving prophylaxis-North Carolina, 2009. Morb. Mortal. Wkly. Rep. 2009, 58, 969–972. [Google Scholar]
- Dick, G.W. Zika virus. II. Pathogenicity and physical properties. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 521–534. [Google Scholar] [CrossRef]
- Haddow, A.J.; Williams, M.C.; Woodall, J.P.; Simpson, D.I.; Goma, L.K. Twelve Isolations of Zika Virus from Aedes (Stegomyia) Africanus (Theobald) Taken in and above a Uganda Forest. Bull. World Health Org. 1964, 31, 57–69. [Google Scholar] [PubMed]
- Simpson, D.I. Zika Virus Infection in Man. Trans. R. Soc. Trop. Med. Hyg. 1964, 58, 335–338. [Google Scholar] [CrossRef]
- Duffy, M.R.; Chen, T.H.; Hancock, W.T.; Powers, A.M.; Kool, J.L.; Lanciotti, R.S.; Pretrick, M.; Marfel, M.; Holzbauer, S.; Dubray, C.; et al. Zika virus outbreak on Yap Island, Federated States of Micronesia. N. Engl. J. Med. 2009, 360, 2536–2543. [Google Scholar] [CrossRef] [PubMed]
- Zanluca, C.; Melo, V.C.; Mosimann, A.L.; Santos, G.I.; Santos, C.N.; Luz, K. First report of autochthonous transmission of Zika virus in Brazil. Mem. Inst. Oswaldo Cruz 2015, 110, 569–572. [Google Scholar] [CrossRef] [PubMed]
- Hamelin, M.E.; Baz, M.; Abed, Y.; Couture, C.; Joubert, P.; Beaulieu, E.; Bellerose, N.; Plante, M.; Mallett, C.; Schumer, G.; et al. Oseltamivir-resistant pandemic A/H1N1 virus is as virulent as its wild-type counterpart in mice and ferrets. PLoS Pathog. 2010, 6, e1001015. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, S.A.; Jamieson, D.J.; Honein, M.A.; Petersen, L.R. Zika Virus and Birth Defects—Reviewing the Evidence for Causality. N. Engl. J. Med. 2016, 374, 1981–1987. [Google Scholar] [CrossRef]
- Diagne, C.T.; Diallo, D.; Faye, O.; Ba, Y.; Faye, O.; Gaye, A.; Dia, I.; Faye, O.; Weaver, S.C.; Sall, A.A.; et al. Potential of selected Senegalese Aedes spp. mosquitoes (Diptera: Culicidae) to transmit Zika virus. BMC Infect. Dis. 2015, 15, 492. [Google Scholar] [CrossRef]
- Musso, D.; Gubler, D.J. Zika Virus. Clin. Microbiol. Rev. 2016, 29, 487–524. [Google Scholar] [CrossRef] [PubMed]
- Gourinat, A.C.; O’Connor, O.; Calvez, E.; Goarant, C.; Dupont-Rouzeyrol, M. Detection of Zika virus in urine. Emerg. Infect. Dis. 2015, 21, 84–86. [Google Scholar] [CrossRef] [PubMed]
- Coffey, L.L.; Pesavento, P.A.; Keesler, R.I.; Singapuri, A.; Watanabe, J.; Watanabe, R.; Yee, J.; Bliss-Moreau, E.; Cruzen, C.; Christe, K.L.; et al. Zika Virus Tissue and Blood Compartmentalization in Acute Infection of Rhesus Macaques. PLoS ONE 2017, 12, e0171148. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.J.; Diamond, M.S. Zika Virus Pathogenesis and Tissue Tropism. Cell Host Microbe 2017, 21, 134–142. [Google Scholar] [CrossRef] [PubMed]
- Govero, J.; Esakky, P.; Scheaffer, S.M.; Fernandez, E.; Drury, A.; Platt, D.J.; Gorman, M.J.; Richner, J.M.; Caine, E.A.; Salazar, V.; et al. Zika virus infection damages the testes in mice. Nature 2016, 540, 438–442. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, A.J.; Smith, J.L.; Haese, N.N.; Broeckel, R.M.; Parkins, C.J.; Kreklywich, C.; DeFilippis, V.R.; Denton, M.; Smith, P.P.; Messer, W.B.; et al. Zika Virus infection of rhesus macaques leads to viral persistence in multiple tissues. PLoS Pathog. 2017, 13, e1006219. [Google Scholar] [CrossRef] [PubMed]
- Besnard, M.; Lastere, S.; Teissier, A.; Cao-Lormeau, V.; Musso, D. Evidence of perinatal transmission of Zika virus, French Polynesia, December 2013 and February 2014. Eurosurveillance 2014, 19. [Google Scholar] [CrossRef]
- Imperato, P.J. The Convergence of a Virus, Mosquitoes, and Human Travel in Globalizing the Zika Epidemic. J. Commun. Health 2016, 41, 674–679. [Google Scholar] [CrossRef]
- Foy, B.D.; Kobylinski, K.C.; Chilson Foy, J.L.; Blitvich, B.J.; Travassos da Rosa, A.; Haddow, A.D.; Lanciotti, R.S.; Tesh, R.B. Probable non-vector-borne transmission of Zika virus, Colorado, USA. Emerg. Infect. Dis. 2011, 17, 880–882. [Google Scholar] [CrossRef]
- Dupont-Rouzeyrol, M.; Biron, A.; O’Connor, O.; Huguon, E.; Descloux, E. Infectious Zika viral particles in breastmilk. Lancet 2016, 387, 1051. [Google Scholar] [CrossRef]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonca, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fluid of fetuses with microcephaly in Brazil: A case study. Lancet Infect. Dis. 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Musso, D.; Nhan, T.; Robin, E.; Roche, C.; Bierlaire, D.; Zisou, K.; Shan Yan, A.; Cao-Lormeau, V.M.; Broult, J. Potential for Zika virus transmission through blood transfusion demonstrated during an outbreak in French Polynesia, November 2013 to February 2014. Eurosurveillance 2014, 19. [Google Scholar] [CrossRef] [PubMed]
- Marano, G.; Pupella, S.; Vaglio, S.; Liumbruno, G.M.; Grazzini, G. Zika virus and the never-ending story of emerging pathogens and transfusion medicine. Blood Transfus. 2016, 14, 95–100. [Google Scholar] [PubMed]
- BioCryst Pharmaceuticals Inc. BioCryst’s Partner Shinogi Receives Approval for Pediatric Use of Peramivir in Japan. Available online: http://investor.shareholder.com/biocryst/releasedetail.cfm?releaseid=523371 (accessed on 2 November 2011).
- Chambers, T.J.; Weir, R.C.; Grakoui, A.; McCourt, D.W.; Bazan, J.F.; Fletterick, R.J.; Rice, C.M. Evidence that the N-terminal domain of nonstructural protein NS3 from yellow fever virus is a serine protease responsible for site-specific cleavages in the viral polyprotein. Proc. Natl. Acad. Sci. USA 1990, 87, 8898–8902. [Google Scholar] [CrossRef] [PubMed]
- Wengler, G.; Czaya, G.; Farber, P.M.; Hegemann, J.H. In vitro synthesis of West Nile virus proteins indicates that the amino-terminal segment of the NS3 protein contains the active centre of the protease which cleaves the viral polyprotein after multiple basic amino acids. J. Gen. Virol. 1991, 72, 851–858. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Clum, S.; You, S.; Ebner, K.E.; Padmanabhan, R. The serine protease and RNA-stimulated nucleoside triphosphatase and RNA helicase functional domains of dengue virus type 2 NS3 converge within a region of 20 amino acids. J. Virol. 1999, 73, 3108–3116. [Google Scholar] [PubMed]
- Takegami, T.; Sakamuro, D.; Furukawa, T. Japanese encephalitis virus nonstructural protein NS3 has RNA binding and ATPase activities. Virus Genes 1995, 9, 105–112. [Google Scholar] [CrossRef]
- Bartelma, G.; Padmanabhan, R. Expression, purification, and characterization of the RNA 5’-triphosphatase activity of dengue virus type 2 nonstructural protein 3. Virology 2002, 299, 122–132. [Google Scholar] [CrossRef]
- Guyatt, K.J.; Westaway, E.G.; Khromykh, A.A. Expression and purification of enzymatically active recombinant RNA-dependent RNA polymerase (NS5) of the flavivirus Kunjin. J. Virol. Methods 2001, 92, 37–44. [Google Scholar] [CrossRef]
- Xie, X.; Gayen, S.; Kang, C.; Yuan, Z.; Shi, P.Y. Membrane topology and function of dengue virus NS2A protein. J. Virol. 2013, 87, 4609–4622. [Google Scholar] [CrossRef]
- Erbel, P.; Schiering, N.; D’Arcy, A.; Renatus, M.; Kroemer, M.; Lim, S.P.; Yin, Z.; Keller, T.H.; Vasudevan, S.G.; Hommel, U. Structural basis for the activation of flaviviral NS3 proteases from dengue and West Nile virus. Nat. Struct. Mol. Biol. 2006, 13, 372–373. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Jordan, J.L.; Sanchez-Burgos, G.G.; Laurent-Rolle, M.; Garcia-Sastre, A. Inhibition of interferon signaling by dengue virus. Proc. Natl. Acad. Sci. USA 2003, 100, 14333–14338. [Google Scholar] [CrossRef] [PubMed]
- Roosendaal, J.; Westaway, E.G.; Khromykh, A.; Mackenzie, J.M. Regulated cleavages at the West Nile virus NS4A-2K-NS4B junctions play a major role in rearranging cytoplasmic membranes and Golgi trafficking of the NS4A protein. J. Virol. 2006, 80, 4623–4632. [Google Scholar] [CrossRef] [PubMed]
- Mackenzie, J.M.; Jones, M.K.; Young, P.R. Immunolocalization of the dengue virus nonstructural glycoprotein NS1 suggests a role in viral RNA replication. Virology 1996, 220, 232–240. [Google Scholar] [CrossRef] [PubMed]
- Chambers, T.J.; Hahn, C.S.; Galler, R.; Rice, C.M. Flavivirus genome organization, expression, and replication. Annu. Rev. Microbiol. 1990, 44, 649–688. [Google Scholar] [CrossRef] [PubMed]
- Hamel, R.; Dejarnac, O.; Wichit, S.; Ekchariyawat, P.; Neyret, A.; Luplertlop, N.; Perera-Lecoin, M.; Surasombatpattana, P.; Talignani, L.; Thomas, F.; et al. Biology of Zika Virus Infection in Human Skin Cells. J. Virol. 2015, 89, 8880–8896. [Google Scholar] [CrossRef] [PubMed]
- Van Hemert, F.; Berkhout, B. Nucleotide composition of the Zika virus RNA genome and its codon usage. Virol. J. 2016, 13, 95. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Rice, C.M. Molecular biology of flaviviruses. Adv. Virus Res. 2003, 59, 23–61. [Google Scholar]
- Batista, M.N.; Braga, A.C.S.; Campos, G.R.F.; Souza, M.M.; Matos, R.P.A.; Lopes, T.Z.; Candido, N.M.; Lima, M.L.D.; Machado, F.C.; Andrade, S.T.Q.; et al. Natural Products Isolated from Oriental Medicinal Herbs Inactivate Zika Virus. Viruses 2019, 11, 49. [Google Scholar] [CrossRef]
- Haddad, J.G.; Koishi, A.C.; Gaudry, A.; Nunes Duarte Dos Santos, C.; Viranaicken, W.; Despres, P.; El Kalamouni, C. Doratoxylon apetalum, an Indigenous Medicinal Plant from Mascarene Islands, Is a Potent Inhibitor of Zika and Dengue Virus Infection in Human Cells. Int. J. Mol. Sci. 2019, 20, 2382. [Google Scholar] [CrossRef]
- Tychan Pte Ltd. Safety and Tolerability of an Antibody Against Zika Virus (Tyzivumab) in ZIKV Infected Patients. ClinicalTrials.gov. NCT number: NCT03776695. 2018 Dec. NCT03776695. Available online: https://clinicaltrials.gov/ct2/show/NCT03776695 (accessed on 30 May 2019).
- Niu, X.; Zhao, L.; Qu, L.; Yao, Z.; Zhang, F.; Yan, Q.; Zhang, S.; Liang, R.; Chen, P.; Luo, J.; et al. Convalescent patient-derived monoclonal antibodies targeting different epitopes of E protein confer protection against Zika virus in a neonatal mouse model. Emerg. Microbes Infect. 2019, 8, 749–759. [Google Scholar] [CrossRef] [PubMed]
- De Clercq, E.; Neyts, J. Antiviral agents acting as DNA or RNA chain terminators. Handb. Exp. Pharmacol. 2009, 53–84. [Google Scholar] [CrossRef]
- Eyer, L.; Nencka, R.; Huvarova, I.; Palus, M.; Joao Alves, M.; Gould, E.A.; De Clercq, E.; Ruzek, D. Nucleoside Inhibitors of Zika Virus. J. Infect. Dis. 2016, 214, 707–711. [Google Scholar] [CrossRef] [PubMed]
- Hercik, K.; Kozak, J.; Sala, M.; Dejmek, M.; Hrebabecky, H.; Zbornikova, E.; Smola, M.; Ruzek, D.; Nencka, R.; Boura, E. Adenosine triphosphate analogs can efficiently inhibit the Zika virus RNA-dependent RNA polymerase. Antivir. Res. 2017, 137, 131–133. [Google Scholar] [CrossRef] [PubMed]
- Zmurko, J.; Marques, R.E.; Schols, D.; Verbeken, E.; Kaptein, S.J.; Neyts, J. The Viral Polymerase Inhibitor 7-Deaza-2’-C-Methyladenosine Is a Potent Inhibitor of In Vitro Zika Virus Replication and Delays Disease Progression in a Robust Mouse Infection Model. PLoS Negl. Trop. Dis. 2016, 10, e0004695. [Google Scholar] [CrossRef] [PubMed]
- Arnold, J.J.; Sharma, S.D.; Feng, J.Y.; Ray, A.S.; Smidansky, E.D.; Kireeva, M.L.; Cho, A.; Perry, J.; Vela, J.E.; Park, Y.; et al. Sensitivity of mitochondrial transcription and resistance of RNA polymerase II dependent nuclear transcription to antiviral ribonucleosides. PLoS Pathog. 2012, 8, e1003030. [Google Scholar] [CrossRef]
- Chen, Y.L.; Yin, Z.; Lakshminarayana, S.B.; Qing, M.; Schul, W.; Duraiswamy, J.; Kondreddi, R.R.; Goh, A.; Xu, H.Y.; Yip, A.; et al. Inhibition of dengue virus by an ester prodrug of an adenosine analog. Antimicrob. Agents Chemother. 2010, 54, 3255–3261. [Google Scholar] [CrossRef]
- Furuta, Y.; Takahashi, K.; Shiraki, K.; Sakamoto, K.; Smee, D.F.; Barnard, D.L.; Gowen, B.B.; Julander, J.G.; Morrey, J.D. T-705 (favipiravir) and related compounds: Novel broad-spectrum inhibitors of RNA viral infections. Antivir. Res. 2009, 82, 95–102. [Google Scholar] [CrossRef]
- Baz, M.; Goyette, N.; Griffin, B.D.; Kobinger, G.P.; Boivin, G. In vitro susceptibility of geographically and temporally distinct Zika viruses to favipiravir and ribavirin. Antivir. Ther. 2017, 22, 613–618. [Google Scholar] [CrossRef]
- Cai, L.; Sun, Y.; Song, Y.; Xu, L.; Bei, Z.; Zhang, D.; Dou, Y.; Wang, H. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication. Arch. Virol. 2017, 162, 2847–2853. [Google Scholar] [CrossRef]
- Delang, L.; Abdelnabi, R.; Neyts, J. Favipiravir as a potential countermeasure against neglected and emerging RNA viruses. Antivir. Res. 2018, 153, 85–94. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.Q.; Zhang, N.N.; Li, C.F.; Tian, M.; Hao, J.N.; Xie, X.P.; Shi, P.Y.; Qin, C.F. Adenosine Analog NITD008 Is a Potent Inhibitor of Zika Virus. Open Forum Infect. Dis. 2016, 3, ofw175. [Google Scholar] [CrossRef] [PubMed]
- Munjal, A.; Khandia, R.; Dhama, K.; Sachan, S.; Karthik, K.; Tiwari, R.; Malik, Y.S.; Kumar, D.; Singh, R.K.; Iqbal, H.M.N.; et al. Advances in Developing Therapies to Combat Zika Virus: Current Knowledge and Future Perspectives. Front. Microbiol. 2017, 8, 1469. [Google Scholar] [CrossRef] [PubMed]
- Reznik, S.E.; Ashby, C.R., Jr. Sofosbuvir: An antiviral drug with potential efficacy against Zika infection. Int. J. Infect. Dis. 2017, 55, 29–30. [Google Scholar] [CrossRef] [PubMed]
- Bullard-Feibelman, K.M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M.S.; Geiss, B.J. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antivir. Res. 2017, 137, 134–140. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; de Melo, G.R.; de Freitas, C.S.; Rocha, N.; Hoelz, L.V.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Domingo, E. Antiviral Strategies Based on Lethal Mutagenesis and Error Threshold. Curr. Top. Microbiol. Immunol. 2016, 392, 323–339. [Google Scholar] [PubMed]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Mangia, A.; Piazzolla, V. Overall efficacy and safety results of sofosbuvir-based therapies in phase II and III studies. Dig. Liver Dis. 2014, 46 (Suppl. 5), S179–S185. [Google Scholar] [CrossRef]
- Eyer, L.; Zouharova, D.; Sirmarova, J.; Fojtikova, M.; Stefanik, M.; Haviernik, J.; Nencka, R.; de Clercq, E.; Ruzek, D. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses. Antivir. Res. 2017, 142, 63–67. [Google Scholar] [CrossRef]
- Warren, T.K.; Wells, J.; Panchal, R.G.; Stuthman, K.S.; Garza, N.L.; Van Tongeren, S.A.; Dong, L.; Retterer, C.J.; Eaton, B.P.; Pegoraro, G.; et al. Protection against filovirus diseases by a novel broad-spectrum nucleoside analogue BCX4430. Nature 2014, 508, 402–405. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G.; Siddharthan, V.; Evans, J.; Taylor, R.; Tolbert, K.; Apuli, C.; Stewart, J.; Collins, P.; Gebre, M.; Neilson, S.; et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antivir. Res. 2017, 137, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Stephen, P.; Baz, M.; Boivin, G.; Lin, S.X. Structural Insight into NS5 of Zika Virus Leading to the Discovery of MTase Inhibitors. J. Am. Chem. Soc. 2016, 138, 16212–16215. [Google Scholar] [CrossRef] [PubMed]
- Hamil, R.L.; Hoehn, M.M. A9145, a new adenine-containing antifungal antibiotic. I. Discovery and isolation. J. Antibiot. (Tokyo) 1973, 26, 463–465. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zheng, Y.G. SAM/SAH Analogs as Versatile Tools for SAM-Dependent Methyltransferases. ACS Chem. Biol. 2016, 11, 583–597. [Google Scholar] [CrossRef] [PubMed]
- Hercik, K.; Brynda, J.; Nencka, R.; Boura, E. Structural basis of Zika virus methyltransferase inhibition by sinefungin. Arch. Virol. 2017, 162, 2091–2096. [Google Scholar] [CrossRef] [PubMed]
- Robert-Gero, M.; Lawrence, F.; Lederer, E. Potential Clinical Use of Sinefungin: Reduction of Toxicity and Enhancement of Activity. In Leishmaniasis; NATO ASI Series (Series A: Life Sciences); Hart, D.T., Ed.; Springer: Boston, MA, USA, 1989; Volume 171. [Google Scholar]
- Lee, H.; Ren, J.; Nocadello, S.; Rice, A.J.; Ojeda, I.; Light, S.; Minasov, G.; Vargas, J.; Nagarathnam, D.; Anderson, W.F.; et al. Identification of novel small molecule inhibitors against NS2B/NS3 serine protease from Zika virus. Antivir. Res. 2017, 139, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Bahramsoltani, R.; Sodagari, H.R.; Farzaei, M.H.; Abdolghaffari, A.H.; Gooshe, M.; Rezaei, N. The preventive and therapeutic potential of natural polyphenols on influenza. Expert Rev. Anti Infect. Ther. 2016, 14, 57–80. [Google Scholar] [CrossRef]
- Vazquez-Calvo, A.; Jimenez de Oya, N.; Martin-Acebes, M.A.; Garcia-Moruno, E.; Saiz, J.C. Antiviral Properties of the Natural Polyphenols Delphinidin and Epigallocatechin Gallate against the Flaviviruses West Nile Virus, Zika Virus, and Dengue Virus. Front. Microbiol. 2017, 8, 1314. [Google Scholar] [CrossRef]
- Wu, Y.H. Naturally derived anti-hepatitis B virus agents and their mechanism of action. World J. Gastroenterol. 2016, 22, 188–204. [Google Scholar] [CrossRef]
- Lim, H.J.; Nguyen, T.T.; Kim, N.M.; Park, J.S.; Jang, T.S.; Kim, D. Inhibitory effect of flavonoids against NS2B-NS3 protease of ZIKA virus and their structure activity relationship. Biotechnol. Lett. 2017, 39, 415–421. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Lim, L.; Srivastava, S.; Lu, Y.; Song, J. Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PLoS ONE 2017, 12, e0180632. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Brecher, M.; Deng, Y.Q.; Zhang, J.; Sakamuru, S.; Liu, B.; Huang, R.; Koetzner, C.A.; Allen, C.A.; Jones, S.A.; et al. Existing drugs as broad-spectrum and potent inhibitors for Zika virus by targeting NS2B-NS3 interaction. Cell Res. 2017, 27, 1046–1064. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Chan, J.F.; den-Haan, H.; Chik, K.K.; Zhang, A.J.; Chan, C.C.; Poon, V.K.; Yip, C.C.; Mak, W.W.; Zhu, Z.; et al. Structure-based discovery of clinically approved drugs as Zika virus NS2B-NS3 protease inhibitors that potently inhibit Zika virus infection in vitro and in vivo. Antivir. Res. 2017, 145, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Aguilar, J.S.; Rice, M.; Wagner, E.K. The polysulfonated compound suramin blocks adsorption and lateral difusion of herpes simplex virus type-1 in vero cells. Virology 1999, 258, 141–151. [Google Scholar] [CrossRef] [PubMed]
- Baba, M.; Konno, K.; Shigeta, S.; Wickramasinghe, A.; Mohan, P. Selective inhibition of human cytomegalovirus replication by naphthalenedisulfonic acid derivatives. Antivir. Res. 1993, 20, 223–233. [Google Scholar] [CrossRef]
- Ellenbecker, M.; Lanchy, J.M.; Lodmell, J.S. Inhibition of Rift Valley fever virus replication and perturbation of nucleocapsid-RNA interactions by suramin. Antimicrob. Agents Chemother. 2014, 58, 7405–7415. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Qing, J.; Sun, Y.; Rao, Z. Suramin inhibits EV71 infection. Antivir. Res. 2014, 103, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.C.; Wang, Y.M.; Ho, Y.J.; Chang, T.Y.; Lai, Z.Z.; Tsui, P.Y.; Wu, T.Y.; Lin, C.C. Suramin treatment reduces chikungunya pathogenesis in mice. Antivir. Res. 2016, 134, 89–96. [Google Scholar] [CrossRef] [PubMed]
- Ren, P.; Zou, G.; Bailly, B.; Xu, S.; Zeng, M.; Chen, X.; Shen, L.; Zhang, Y.; Guillon, P.; Arenzana-Seisdedos, F.; et al. The approved pediatric drug suramin identified as a clinical candidate for the treatment of EV71 infection-suramin inhibits EV71 infection in vitro and in vivo. Emerg. Microbes Infect. 2014, 3, e62. [Google Scholar] [CrossRef] [PubMed]
- Basavannacharya, C.; Vasudevan, S.G. Suramin inhibits helicase activity of NS3 protein of dengue virus in a fluorescence-based high throughput assay format. Biochem. Biophys. Res. Commun. 2014, 453, 539–544. [Google Scholar] [CrossRef] [PubMed]
- Albulescu, I.C.; Kovacikova, K.; Tas, A.; Snijder, E.J.; van Hemert, M.J. Suramin inhibits Zika virus replication by interfering with virus attachment and release of infectious particles. Antivir. Res. 2017, 143, 230–236. [Google Scholar] [CrossRef] [PubMed]
- Bekerman, E.; Einav, S. Infectious disease. Combating emerging viral threats. Science 2015, 348, 282–283. [Google Scholar] [CrossRef] [PubMed]
- Graci, J.D.; Cameron, C.E. Mechanisms of action of ribavirin against distinct viruses. Rev. Med. Virol. 2006, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Sidwell, R.W.; Huffman, J.H.; Khare, G.P.; Allen, L.B.; Witkowski, J.T.; Robins, R.K. Broad-spectrum antiviral activity of Virazole: 1-beta-D-ribofuranosyl-1,2,4-triazole-3-carboxamide. Science 1972, 177, 705–706. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, N.; Soma, R.; Hidano, S.; Watanabe, K.; Umekita, H.; Fukuda, C.; Noguchi, K.; Gendo, Y.; Ozaki, T.; Sonoda, A.; et al. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antivir. Res. 2017, 146, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; Smith, J.; Bukreyeva, N.; Koma, T.; Manning, J.T.; Kalkeri, R.; Kwong, A.D.; Paessler, S. Merimepodib, an IMPDH inhibitor, suppresses replication of Zika virus and other emerging viral pathogens. Antivir. Res. 2018, 149, 34–40. [Google Scholar] [CrossRef] [PubMed]
- Rausch, K.; Hackett, B.A.; Weinbren, N.L.; Reeder, S.M.; Sadovsky, Y.; Hunter, C.A.; Schultz, D.C.; Coyne, C.B.; Cherry, S. Screening Bioactives Reveals Nanchangmycin as a Broad Spectrum Antiviral Active against Zika Virus. Cell Rep. 2017, 18, 804–815. [Google Scholar] [CrossRef] [PubMed]
- Barrows, N.J.; Campos, R.K.; Powell, S.T.; Prasanth, K.R.; Schott-Lerner, G.; Soto-Acosta, R.; Galarza-Munoz, G.; McGrath, E.L.; Urrabaz-Garza, R.; Gao, J.; et al. A Screen of FDA-Approved Drugs for Inhibitors of Zika Virus Infection. Cell Host Microbe 2016, 20, 259–270. [Google Scholar] [CrossRef]
- Pascoalino, B.S.; Courtemanche, G.; Cordeiro, M.T.; Gil, L.H.; Freitas-Junior, L. Zika antiviral chemotherapy: Identification of drugs and promising starting points for drug discovery from an FDA-approved library. F1000Research 2016, 5, 2523. [Google Scholar] [CrossRef] [PubMed]
- Ikeda, M.; Abe, K.; Yamada, M.; Dansako, H.; Naka, K.; Kato, N. Different anti-HCV profiles of statins and their potential for combination therapy with interferon. Hepatology 2006, 44, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Rothwell, C.; Lebreton, A.; Young Ng, C.; Lim, J.Y.; Liu, W.; Vasudevan, S.; Labow, M.; Gu, F.; Gaither, L.A. Cholesterol biosynthesis modulation regulates dengue viral replication. Virology 2009, 389, 8–19. [Google Scholar] [CrossRef] [PubMed]
- Sarkey, J.P.; Richards, M.P.; Stubbs, E.B., Jr. Lovastatin attenuates nerve injury in an animal model of Guillain-Barre syndrome. J. Neurochem. 2007, 100, 1265–1277. [Google Scholar] [CrossRef] [PubMed]
- Retallack, H.; Di Lullo, E.; Arias, C.; Knopp, K.A.; Laurie, M.T.; Sandoval-Espinosa, C.; Mancia Leon, W.R.; Krencik, R.; Ullian, E.M.; Spatazza, J.; et al. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.J.; Mitchell, A.A.; Yau, W.P.; Louik, C.; Hernandez-Diaz, S. Safety of macrolides during pregnancy. Am. J. Obstet Gynecol. 2013, 208, 221.e1–221.e8. [Google Scholar] [CrossRef] [PubMed]
- Al-Bari, M.A.A. Targeting endosomal acidification by chloroquine analogs as a promising strategy for the treatment of emerging viral diseases. Pharmacol. Res. Perspect. 2017, 5, e00293. [Google Scholar] [CrossRef]
- Levy, M.; Buskila, D.; Gladman, D.D.; Urowitz, M.B.; Koren, G. Pregnancy outcome following first trimester exposure to chloroquine. Am. J. Perinatol. 1991, 8, 174–178. [Google Scholar] [CrossRef]
- Delvecchio, R.; Higa, L.M.; Pezzuto, P.; Valadao, A.L.; Garcez, P.P.; Monteiro, F.L.; Loiola, E.C.; Dias, A.A.; Silva, F.J.; Aliota, M.T.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef]
- Miner, J.J.; Cao, B.; Govero, J.; Smith, A.M.; Fernandez, E.; Cabrera, O.H.; Garber, C.; Noll, M.; Klein, R.S.; Noguchi, K.K.; et al. Zika Virus Infection during Pregnancy in Mice Causes Placental Damage and Fetal Demise. Cell 2016, 165, 1081–1091. [Google Scholar] [CrossRef]
- Kuivanen, S.; Bespalov, M.M.; Nandania, J.; Ianevski, A.; Velagapudi, V.; De Brabander, J.K.; Kainov, D.E.; Vapalahti, O. Obatoclax, saliphenylhalamide and gemcitabine inhibit Zika virus infection in vitro and differentially affect cellular signaling, transcription and metabolism. Antivir. Res. 2017, 139, 117–128. [Google Scholar] [CrossRef]
- Jurgeit, A.; McDowell, R.; Moese, S.; Meldrum, E.; Schwendener, R.; Greber, U.F. Niclosamide is a proton carrier and targets acidic endosomes with broad antiviral effects. PLoS Pathog. 2012, 8, e1002976. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.K.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.V.; Del Sarto, J.L.; Rocha, R.F.; Silva, F.R.; Doria, J.G.; Olmo, I.G.; Marques, R.E.; Queiroz-Junior, C.M.; Foureaux, G.; Araujo, J.M.S.; et al. N-Methyl-d-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection. mBio 2017, 8. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Name | Amino Acid Residues | Function |
|---|---|---|
| C | 122 | Generation of nucleocapsid (encapsulates genomic RNA) |
| prM | 168 | Protects E protein during assembly |
| E | 500 | Envelope glycoprotein (membrane binding and fusion) |
| NS1 | 352 | Replication and immune response regulation |
| NS2A | 226 | Replication and capsid assembly |
| NS2B | 130 | NS3 cofactor |
| NS3 | 617 | Serine protease NTPase, RNA helicase |
| NS4A | 127 | Viral membrane formation |
| 2K | 23 | Signal peptide |
| NS4B | 251 | Inhibits antiviral state |
| NS5 | 903 | RNA dependent RNA polymerase |
| Direct-Acting Antivirals | |||
| Name | Mode of action | In vitro | In vivo |
| 7-deaza-2-CMA | RdRp inhibitor | √ | √ |
| 2-CMA, 2-CMC, 2-CMG, 2-CMU | RdRp inhibitor | √ | X |
| Favipiravir | RdRp inhibitor | √ | X |
| NITD008 | Pyrimidine synthesis inhibitor | √ | √ |
| Sofosbuvir | RdRp inhibitor | √ | √ |
| BCX4430 * | RdRp inhibitor | √ | √ |
| Sinefungin | Pan-methyltransferase inhibitor | √ | X |
| Myricetin, quercetin, luteolin, isorhamnetin, apigenin, curcumin | NS2B-NS3 protease inhibitor | √ | X |
| Niclosamide, and nitazoxanide | NS2B-NS3 protease inhibitor | √ | X |
| Temoporfin | NS2B-NS3 protease inhibitor | √ | √ |
| Novobiocin | NS2B-NS3 protease inhibitor | √ | √ |
| Suramin | NS3 inhibitor | √ | X |
| Host-Targeting Antivirals | |||
| Name | Mode of action | In vitro | In vivo |
| Ribavirin | Several mechanisms including purine synthesis inhibitor | √ | √ |
| Merimepodib and mycophenolic acid | Inosine monophosphate dehydrogenase (IMPDH) inhibitors | √ | X |
| Azathioprine | Purine synthesis inhibitor | √ | X |
| 6-azauridine, 5-fluorouracil | Pirimidine synthesis inhibitor | √ | X |
| lovastatin | HMG-CoA reductase inhibitor | √ | X |
| Azithromycin | Unknown mechanisms of action against ZIKV | √ | X |
| Chloroquine | Inhibition of pH-dependent steps of viral replication | √ | X |
| Saliphenylhalamide | Viral entry inhibitor | √ | X |
| Obatoclax mesylate (GX15-070) | Bcl-2 protein inhibitor | √ | X |
| PHA-690509 | Cyclin-dependent kinase inhibitor | √ | X |
| MK-801, agmatine, and ifenprodil | Neuronal cell death inhibitor | √ | X |
| Memantine | Neuronal cell death inhibitor | √ | √ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Baz, M.; Boivin, G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals 2019, 12, 101. https://doi.org/10.3390/ph12030101
Baz M, Boivin G. Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals. 2019; 12(3):101. https://doi.org/10.3390/ph12030101
Chicago/Turabian StyleBaz, Mariana, and Guy Boivin. 2019. "Antiviral Agents in Development for Zika Virus Infections" Pharmaceuticals 12, no. 3: 101. https://doi.org/10.3390/ph12030101
APA StyleBaz, M., & Boivin, G. (2019). Antiviral Agents in Development for Zika Virus Infections. Pharmaceuticals, 12(3), 101. https://doi.org/10.3390/ph12030101