Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs


 , , and
, , and 
Abstract
1. Introduction
2. Results and Discussion
2.1. Chemical Synthesis
2.2. Biological Activity
3. Materials and Methods
3.1. Generalities
3.2. Synthesis
3.2.1. Synthesis of amines 10
Methyl 4-(2-aminoethyl)benzoate 10a
4-(2-aminoethyl)benzamide 10b
3.2.2. Synthesis of Compounds 4
General Procedure
(R)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-(phenethylamino)ethan-1-ol (4a)
(S)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-(phenethylamino)ethan-1-ol (4b)
(R)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-methylphenethyl)amino)ethan-1-ol (4c)
(S)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-methylphenethyl)amino)ethan-1-ol (4d)
(R)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-methoxyphenethyl)amino)ethan-1-ol (4e)
(S)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-methoxyphenethyl)amino)ethan-1-ol (4f)
(R)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-chlorophenethyl)amino)ethan-1-ol (4g)
(S)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-chlorophenethyl)amino)ethan-1-ol (4h)
(R)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-nitrophenethyl)amino)ethan-1-ol (4i)
(S)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-((4-nitrophenethyl)amino)ethan-1-ol (4j)
Methyl (R)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzoate (4k)
Methyl (S)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzoate (4l)
(R)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzonitrile (4m)
(S)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzonitrile (4n)
(R)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzoic acid (4o)
(S)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzoic acid (4p)
(R)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzamide (4q)
(S)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzamide (4r)
(R)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzene sulfonamide (4s)
(S)-4-(2-((2-(2,8-bis(trifluoromethyl)quinolin-4-yl)-2-hydroxyethyl)amino)ethyl)benzene sulfonamide (4t)
(R)-2-((4-aminophenethyl)amino)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)ethan-1-ol (4u)
(S)-2-((4-aminophenethyl)amino)-1-(2,8-bis(trifluoromethyl)quinolin-4-yl)ethan-1-ol (4v)
3.3. Antibacterial Assays
3.4. Antimycobacterial Tests
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- World Health Organization. The Burden of Health Care-Associated Infection Worldwide; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2018; World Health Organization: Geneva, Switzerland, 2018. [Google Scholar]
- Nahid, P.; Dorman, S.E.; Alipanah, N.; Barry, P.M.; Brozek, J.L.; Cattamanchi, A.; Chaisson, L.H.; Chaisson, R.E.; Daley, C.L.; Grzemska, M.; et al. Official American Thoracic Society/Centers for Disease Control and Prevention/Infectious Diseases Society of America Clinical Practice Guidelines: Treatment of Drug-Susceptible Tuberculosis. Clin. Infect. Dis. 2016, 63, e147–e195. [Google Scholar] [CrossRef] [PubMed]
- Cohn, D.L.; Bustreo, F.; Raviglione, M.C. Drug-Resistant Tuberculosis: Review of the Worldwide Situation and the WHO/IUATLD Global Surveillance Project. Clin. Infect. Dis. 1997, 24, S121–S130. [Google Scholar] [CrossRef] [PubMed]
- Stout, J.E.; Koh, W.-J.; Yew, W.W. Update on pulmonary disease due to non-tuberculous mycobacteria. Int. J. Infect. Dis. 2016, 45, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Haworth, C.S.; Banks, J.; Capstick, T.; Fisher, A.J.; Gorsuch, T.; Laurenson, I.F.; Leitch, A.; Loebinger, M.R.; Milburn, H.J.; Nightingale, M.; et al. British Thoracic Society guidelines for the management of non-tuberculous mycobacterial pulmonary disease (NTM-PD). Thorax 2017, 72, ii1–ii64. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R. Bedaquiline: First FDA-approved tuberculosis drug in 40 years. Int. J. Appl. Basic Med. Res. 2013, 3, 1–2. [Google Scholar] [CrossRef]
- Keri, R.S.; Patil, S.A. Quinoline: A promising antitubercular target. Biomed. Pharmacother. 2014, 68, 1161–1175. [Google Scholar] [CrossRef]
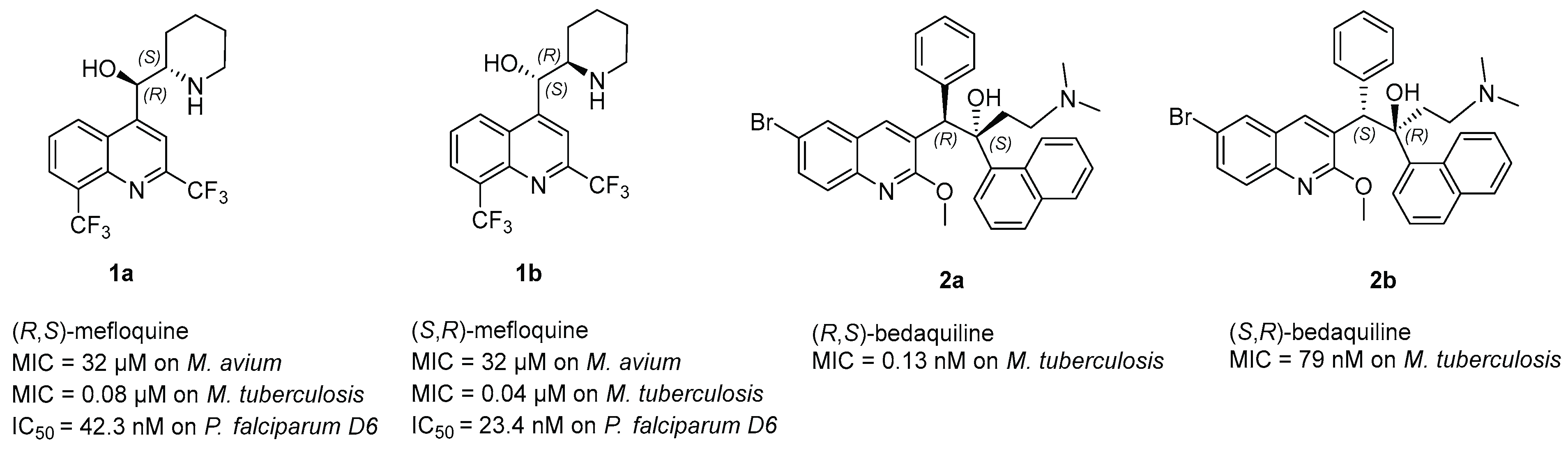
- Kunin, C.M.; Ellis, W.Y. Antimicrobial activities of mefloquine and a series of related compounds. Antimicrob. Agents Chemother. 2000, 44, 848–852. [Google Scholar] [CrossRef]
- World Health Organization. Guidelines for the Treatment of Malaria; World Health Organization: Geneva, Switzerland, 2015; ISBN 978-92-4-154912-7. [Google Scholar]
- Fu, H.-G.; Li, Z.-W.; Hu, X.-X.; Si, S.-Y.; You, X.-F.; Tang, S.; Wang, Y.-X.; Song, D.-Q. Synthesis and biological evaluation of quinoline derivatives as a novel class of broad-spectrum antibacterial agents. Molecules 2019, 24, 548. [Google Scholar] [CrossRef]
- Teng, P.; Li, C.; Peng, Z.; Vanderschouw, A.M.; Nimmagadda, A.; Su, M.; Li, Y.; Sun, X.; Cai, J. Facilely accessible quinoline derivatives as potent antibacterial agents. Bioorg. Med. Chem. 2018, 26, 3573–3579. [Google Scholar] [CrossRef]
- Desai, N.C.; Patel, B.Y.; Dave, B.P. Synthesis and antimicrobial activity of novel quinoline derivatives bearing pyrazoline and pyridine analogues. Med. Chem. Res. 2016, 26, 109–119. [Google Scholar] [CrossRef]
- Brown-Elliott, B.A.; Philley, J.V.; Griffith, D.E.; Thakkar, F.; Wallace, R.J. In Vitro susceptibility testing of Bedaquiline against Mycobacterium avium Complex. Antimicrob. Agents Chemother. 2017, 61, e01798-16. [Google Scholar] [CrossRef] [PubMed]
- Basco, L.K.; Gillotin, C.; Gimenez, F.; Farinotti, R.; Le Bras, J. In vitro activity of the enantiomers of mefloquine, halofantrine and enpiroline against Plasmodium falciparum. Br. J. Clin. Pharmacol. 1992, 33, 517–520. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, L.E.; Inderlied, C.B.; Kolonoski, P.; Chee, C.B.; Aralar, P.; Petrofsky, M.; Parman, T.; Green, C.E.; Lewin, A.H.; Ellis, W.Y.; et al. Identification of (+)-Erythro-Mefloquine as an Active Enantiomer with Greater Efficacy than Mefloquine against Mycobacterium avium Infection in Mice. Antimicrob. Agents Chemother 2012, 56, 4202–4206. [Google Scholar] [CrossRef] [PubMed]
- Bermudez, L.E.; Meek, L. Mefloquine and its enantiomers are active against Mycobacterium tuberculosis in vitro and in macrophages. Tuberc. Res. Treat. 2014, 2014, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Preiss, L.; Langer, J.D.; Yildiz, Ö.; Eckhardt-Strelau, L.; Guillemont, J.E.G.; Koul, A.; Meier, T. Structure of the mycobacterial ATP synthase F1F0 rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015, 1, e1500106. [Google Scholar] [CrossRef]
- De Jonge, M.R.; Koymans, L.H.M.; Guillemont, J.E.G.; Koul, A.; Andries, K. A computational model of the inhibition of Mycobacterium tuberculosis ATPase by a new drug candidate R207910. Proteins Struct. Funct. Bioinform. 2007, 67, 971–980. [Google Scholar] [CrossRef]
- Wong, W.; Bai, X.-C.; Sleebs, B.E.; Triglia, T.; Brown, A.; Thompson, J.K.; Jackson, K.E.; Hanssen, E.; Marapana, D.S.; Fernandez, I.S.; et al. Mefloquine targets the Plasmodium falciparum 80S ribosome to inhibit protein synthesis. Nat. Microbiol. 2017, 2, 17031. [Google Scholar] [CrossRef]
- Martin-Galiano, A.J.; Gorgojo, B.; Kunin, C.M.; de la Campa, A.G. Mefloquine and new related compounds target the F0 Complex of the F0F1 H+-ATPase of Streptococcus pneumoniae. Antimicrob. Agents Chemother. 2002, 46, 1680–1687. [Google Scholar] [CrossRef]
- Mullié, C.; Jonet, A.; Desgrouas, C.; Taudon, N.; Sonnet, P. Differences in anti-malarial activity of 4-aminoalcohol quinoline enantiomers and investigation of the presumed underlying mechanism of action. Malaria J. 2012, 11, 65. [Google Scholar] [CrossRef]
- Dassonville-Klimpt, A.; Cézard, C.; Mullié, C.; Agnamey, P.; Jonet, A.; Da Nascimento, S.; Marchivie, M.; Guillon, J.; Sonnet, P. Absolute configuration and antimalarial activity of erythro-mefloquine enantiomers. ChemPlusChem 2013, 78, 642–646. [Google Scholar] [CrossRef]
- Jonet, A.; Dassonville-Klimpt, A.; Sonnet, P.; Mullié, C. Side chain length is more important than stereochemistry in the antibacterial activity of enantiomerically pure 4-aminoalcohol quinoline derivatives. J. Antibiot. 2013, 66, 683–686. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Craig, P.N. Interdependence between physical parameters and selection of substituent groups for correlation studies. J. Med. Chem. 1971, 14, 680–684. [Google Scholar] [CrossRef] [PubMed]
- Jonet, A.; Dassonville-Klimpt, A.; Da Nascimento, S.; Leger, J.-M.; Guillon, J.; Sonnet, P. First enantioselective synthesis of 4-aminoalcohol quinoline derivatives through a regioselective SN2 epoxide opening mechanism. Tetrahedron: Asymmetry 2011, 22, 138–148. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Methods for Dilution Antimicrobial Susceptibility Tests for Bacteria that Grow Aerobically: Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2015. [Google Scholar]
- Clinical and Laboratory Standards Institute. Susceptibility Testing of Mycobacteria, Nocardiae, and Other Aerobic Actinomycetes: Approved Standard; Clinical and Laboratory Standards Institute: Wayne, PA, USA, 2011. [Google Scholar]
- Jaffe, H.H. A Reëxamination of the Hammett Equation. Chem. Rev. 1953, 53, 191–261. [Google Scholar] [CrossRef]
- Hammett, L.P. Physical Organic Chemistry; McGraw-Hill Book Company, Inc.: New York, NY, USA, 1940; pp. 186–194. [Google Scholar]
- Fujita, T.; Iwasa, J.; Hansch, C. A new substituent constant, π, derived from partition coefficients. J. Am. Chem. Soc. 1964, 86, 5175–5180. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
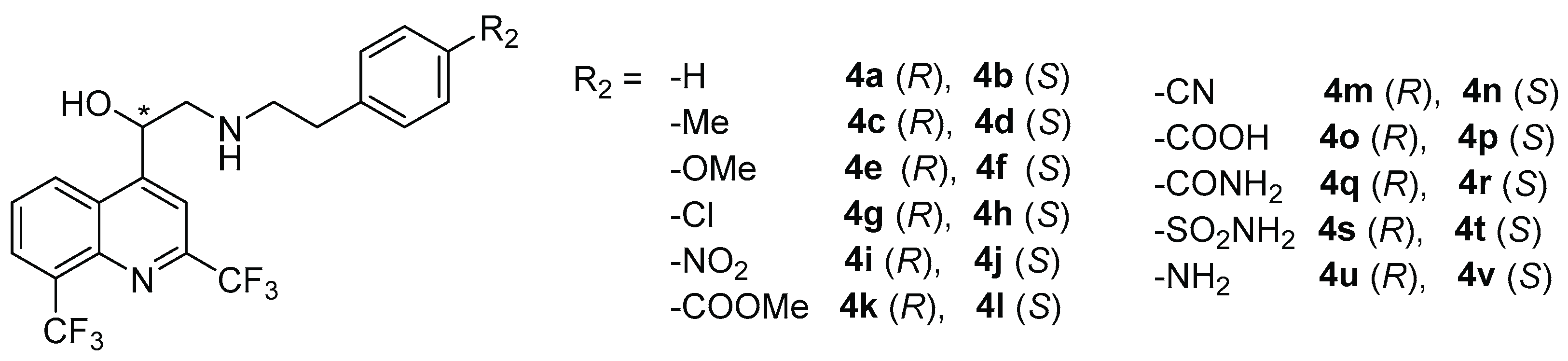{kind=link}
{kind=link}
| Compound | MIC (µg/mL) a | clogP b | |||
|---|---|---|---|---|---|
| S. aureus CIP 103.429 | E. faecalis CIP 103.214 | E. coli DSM 1103 | P. aeruginosa DSM 1117 | ||
| 3a (3b) | 16 (16) | 16 (16) | 32 (64) | >128 (>128) | 4.30 |
| 3c (3d) | 16 (16) | 16 (16) | 16 (32) | >128 (>128) | 4.59 |
| 3e (3f) | 4 (4) | 4 (4) | >128 (16) | >128 (>128) | 4.95 |
| 3g (3h) | 1 (2) | 1 (2) | >128 (>128) | >128 (>128) | 5.31 |
| 3i (3j) | 1 (1) | 1 (1) | >128 (>128) | >128 (>128) | 5.70 |
| 3k (3l) | 16 (>128) | 16 (>128) | >128 (>128) | >128 (>128) | 6.01 |
| 3m (3n) | 8 (8) | 8 (8) | 8 (16) | >128 (>128) | 4.46 |
| mefloquine | 16 | 32 | 64 | >128 | NDc |
| ciprofloxacin | 0.25 | 0.25 | 0.0625 | 0.125 | ND |
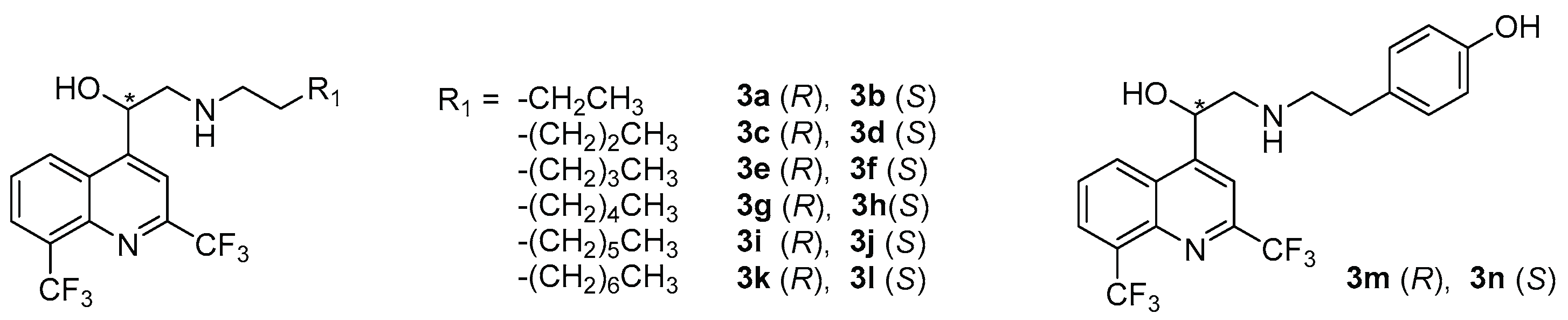
| N° | R2 | AC a | Yield (%) | ee b | c | N° | R2 | AC a | Yield (%) | ee b | c |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 4a | H | R | 73 | 99 | −52.9 | 4l | COOMe | S | 53 | 94 | +55.4 |
| 4b | S | 63 | 94 | +53.0 | |||||||
| 4c | Me | R | 37 | 94 | −52.9 | 4m | CN | R | 86 | 99 | −63.6 |
| 4d | S | 79 | 97.5 | +64.7 | 4n | S | 84 | 90 | +45.1 | ||
| 4e | OMe | R | 80 | 95 | −61.3 | 4o | COOH | R | 72 | ND d | −26.1 |
| 4f | S | 87 | 96 | +64.6 | 4p | S | 68 | ND | +43.6 | ||
| 4g | Cl | R | 68 | 97 | −56.7 | 4q | CONH2 | R | 39 | ND | −37.7 |
| 4h | S | 84 | 94 | +53.4 | 4r | S | 26 | ND | +44.1 | ||
| 4i | NO2 | R | 78 | 98 | −32.6 | 4s | SO2NH2 | R | 76 | ND | −40.0 |
| 4j | S | 61 | 94 | +44.6 | 4t | S | 77 | ND | +43.0 | ||
| 4k | COOMe | R | 77 | 99 | −67.9 | 4u | NH2 | R | 89 | ND | −40.9 |
| 4v | S | 89 | ND | +53.4 |
| N° | R2 | AC a | MIC (µg/mL) b | Physicochemical Constant | |||||
|---|---|---|---|---|---|---|---|---|---|
| S. aureus CIP103.429 | E. faecalis CIP 103214 | E. coli DSM 1103 | P. aeruginosa DSM 1117 | σp c | π f | clogP g | |||
| 4a | H | R | 8 | 8 | >128 | >128 | 0 | 0 | 5.23 |
| 4b | S | 8 | 8 | >128 | >128 | ||||
| 4c | Me | R | 2 | 4 | >128 | >128 | –0.170 | 0.56 | 5.47 |
| 4d | S | 2 | 2 | >128 | >128 | ||||
| 4e | OMe | R | 4 | 8 | >128 | 32 | −0.268 | −0.02 | 5.12 |
| 4f | S | 4 | 8 | >128 | >128 | ||||
| 4g | Cl | R | 1 | 2 | >128 | >128 | 0.227 | 0.71 | 5.75 |
| 4h | S | 1 | 2 | >128 | >128 | ||||
| 4i | NO2 | R | 64 | 128 | >128 | >128 | 0.778d | −0.28 | 4.49 |
| 4j | S | 64 | 128 | >128 | >128 | ||||
| 4k | COOMe | R | 4 | 8 | >128 | >128 | 0.619e | −0.01 | 4.37 |
| 4l | S | 4 | 4 | >128 | >128 | ||||
| 4m | CN | R | 8 | >128 | >128 | >128 | 0.660 | −0.57 | 4.52 |
| 4n | S | 8 | >128 | >128 | >128 | ||||
| 4o | COOH | R | 64 | 64 | >128 | >128 | 0.257 | −0.28 | 2.33 |
| 4p | S | 64 | 64 | >128 | >128 | ||||
| 4q | CONH2 | R | 32 | 32 | 64 | >128 | 0.627 | −1.49 | 3.58 |
| 4r | S | 16 | 32 | 64 | >128 | ||||
| 4s | SO2NH2 | R | 16 | 32 | 64 | >128 | 0.621 | −1.82 | 2.90 |
| 4t | S | 16 | 16 | 64 | >128 | ||||
| 4u | NH2 | R | 8 | 8 | 32 | >128 | −0.660 | −1.23 | 4.12 |
| 4v | S | 8 | 16 | 64 | >128 | ||||
| 3m | OH | R | 8 | 8 | 8 | >128 | −0.357 | −0.67 | 4.46 |
| 3n | S | 8 | 8 | 16 | >128 | ||||
| mefloquine | 16 | 32 | 64 | >128 | |||||
| ciprofloxacin | 0.25 | 0.25 | 0.0625 | 0.125 | |||||
| Compound | MIC (µg/mL) b | clogP e | |||
|---|---|---|---|---|---|
| M. avium ATCC 700898 in CAMBH c | M. avium ATCC 700898 in MB 7H9 d | ||||
 | |||||
| N° | R2 | ACa | |||
| 4a | H | R | >64 | >64 | 5.23 |
| 4b | S | >64 | >64 | ||
| 4c | Me | R | >64 | >64 | 5.47 |
| 4d | S | >64 | >64 | ||
| 4e | OMe | R | >64 | >64 | 5.12 |
| 4f | S | >64 | >64 | ||
| 4g | Cl | R | >64 | >64 | 5.75 |
| 4h | S | >64 | >64 | ||
| 4i | NO2 | R | >64 | >64 | 4.49 |
| 4j | S | >64 | >64 | ||
| 4k | COOMe | R | >64 | >64 | 4.37 |
| 4l | S | >64 | >64 | ||
| 4m | CN | R | >64 | >64 | 4.52 |
| 4n | S | >64 | >64 | ||
| 4o | COOH | R | >64 | >64 | 2.33 |
| 4p | S | >64 | >64 | ||
| 4q | CONH2 | R | 64 | 64 | 3.58 |
| 4r | S | 64 | 64 | ||
| 4s | SO2NH2 | R | 64 | 64 | 2.90 |
| 4t | S | 64 | 64 | ||
| 4u | NH2 | R | 64 | 64 | 4.12 |
| 4v | S | 64 | 64 | ||
| 3m | OH | R | 32 | 32 | 4.46 |
| 3n | S | 32 | 32 | ||
 | |||||
| R1 | ACa | ||||
| 3a | -CH2CH3 | R | 4 | 16 | 4.30 |
| 3b | S | 8 | 32 | ||
| 3c | -(CH2)2CH3 | R | 4 | 16 | 4.59 |
| 3d | S | 8 | 8 | ||
| 3e | -(CH2)3CH3 | R | 8–16 f | 8–16 f | 4.95 |
| 3f | S | 8–16 f | 8–16 f | ||
| 3g | -(CH2)4CH3 | R | 8 | 16 | 5.31 |
| 3h | S | 4 | 4 | ||
| 3i | -(CH2)5CH3 | R | ND g | ND | 5.70 |
| 3j | S | 4 | 4 | ||
| 3k | -(CH2)6CH3 | R | 8–16 f | 8–16 f | 6.01 |
| 3l | S | 2 | 8 | ||
| mefloquine racemic | 4 | 16 | ND | ||
| mefloquine 1a | R,S | 4 | 16 | ||
| mefloquine 1b | S,R | 4 | 8 | ||
| bedaquiline 2a | ≤0.063 | ≤0.063 | ND | ||
| rifampin | 64 | 32–64 | ND | ||
| clarithromycin | 1 | 4–8 | ND | ||
| ethambutol | 8–16 | 8 | ND | ||
| ciprofloxacin | 8 | 4–8 | ND | ||
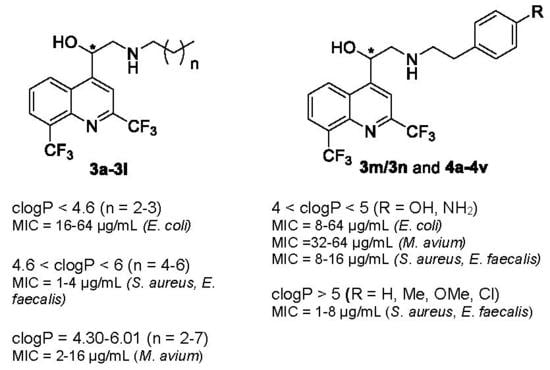
 | clogP < 4.6 | 4.6 < clogP < 6 | clogP > 6 |
| n = 2–3 active against E. coli (MIC = 16–64 µg/mL). | n = 4–6 activity is increased against Gram-positive bacteria (MIC = 1–4 µg/mL). | n = 7 3l is the lead compound against M. avium (MIC = 2 µg/mL). | |
| All compounds were active against M. avium whatever the clogP value | |||
 | clogP < 4 | 4 < clogP < 5 | clogP > 5 |
| R = COOH, CONH2, SO2NH2 activity is decreased against Gram-positive bacteria (MIC = 16–64 µg/mL) and weak on E. coli and M. avium (MIC = 64 µg/mL). | −σ and −π (R = NH2, OH): activity is increased against E. coli (MIC = 8–64 µg/mL) and M. avium (MIC =32–64 µg/mL) and retained against Gram-positive bacteria (MIC = 8–16 µg/mL). +σ and +π (R = NO2): activity is weak against Gram-positive bacteria and no activity against E. coli. | R = H, Me, OMe, Cl activity is increased against Gram-positive bacteria (MIC = 1–8 µg/mL). | |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Laumaillé, P.; Dassonville-Klimpt, A.; Peltier, F.; Mullié, C.; Andréjak, C.; Da-Nascimento, S.; Castelain, S.; Sonnet, P. Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs. Pharmaceuticals 2019, 12, 91. https://doi.org/10.3390/ph12020091
Laumaillé P, Dassonville-Klimpt A, Peltier F, Mullié C, Andréjak C, Da-Nascimento S, Castelain S, Sonnet P. Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs. Pharmaceuticals. 2019; 12(2):91. https://doi.org/10.3390/ph12020091
Chicago/Turabian StyleLaumaillé, Pierre, Alexandra Dassonville-Klimpt, François Peltier, Catherine Mullié, Claire Andréjak, Sophie Da-Nascimento, Sandrine Castelain, and Pascal Sonnet. 2019. "Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs" Pharmaceuticals 12, no. 2: 91. https://doi.org/10.3390/ph12020091
APA StyleLaumaillé, P., Dassonville-Klimpt, A., Peltier, F., Mullié, C., Andréjak, C., Da-Nascimento, S., Castelain, S., & Sonnet, P. (2019). Synthesis and Study of New Quinolineaminoethanols as Anti-Bacterial Drugs. Pharmaceuticals, 12(2), 91. https://doi.org/10.3390/ph12020091