Late Neurological Consequences of Zika Virus Infection: Risk Factors and Pharmaceutical Approaches

 ,
,
Abstract
1. Introduction
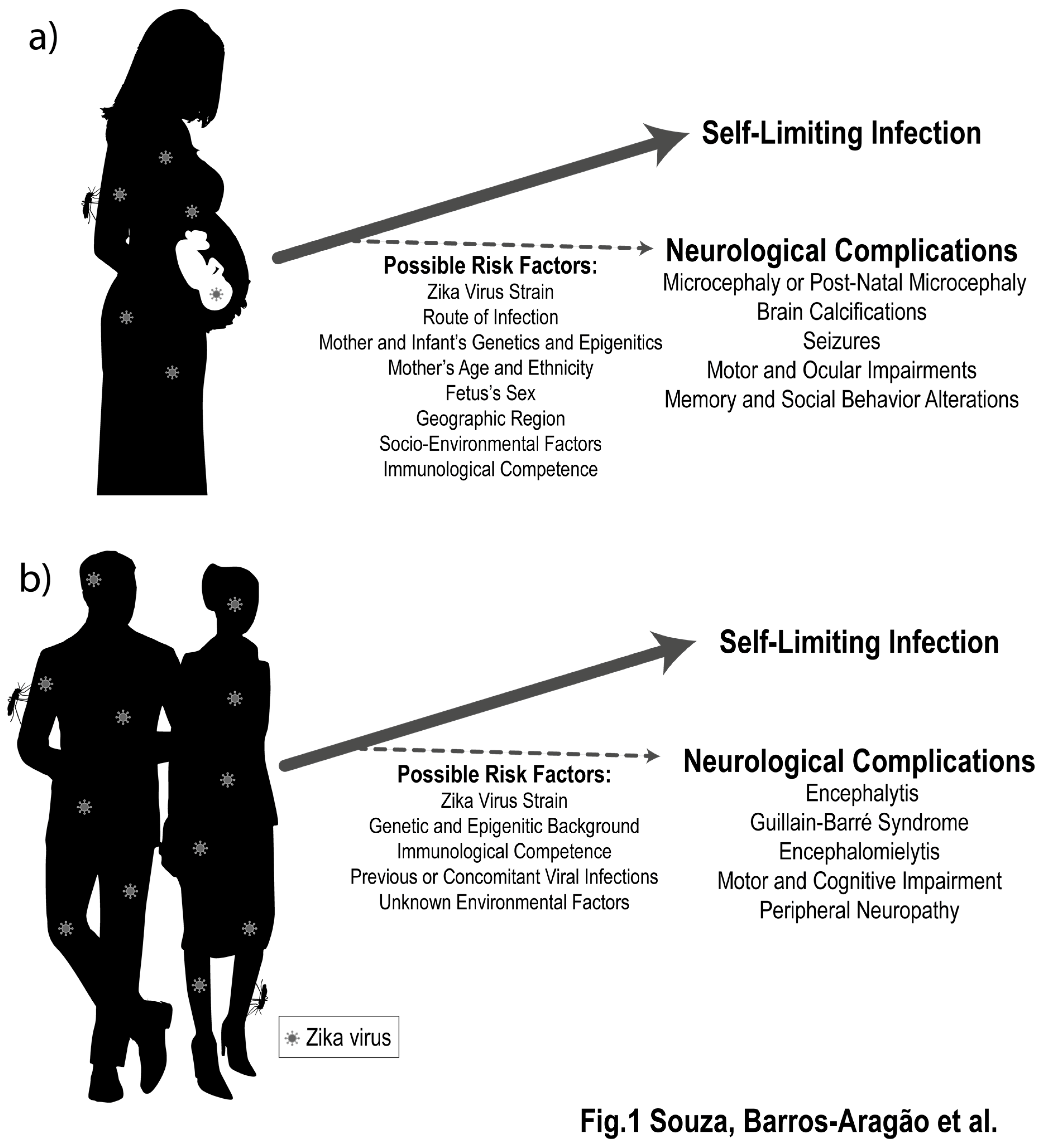
2. ZIKV Infection Is Associated to Late Detrimental Effects to the Developing and Mature Nervous Systems
3. What Genetic, Epigenetic, and Environmental Factors May Increase the Risk of ZIKV-Induced Neurological Damage?
4. Promising Therapeutic Approaches Targeting ZIKV-Associated Symptoms
5. Antiviral Drugs
5.1. Sofosbuvir
5.2. Favipiravir
5.3. Azithromycin
6. Disease-Modifying Drugs
6.1. Memantine
6.2. Infliximab
6.3. PHA-690509
6.4. Emricasan
6.5. Niclosamide
6.6. Chloroquine
6.7. Amodiaquine
6.8. NITD008
6.9. Galidesivir (BCX4430)
6.10. Ribavirin
6.11. Other Reported Candidates
7. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dick, G.W.A.; Kitchen, S.F.; Haddow, A.J. Zika virus. I. Isolations and serological specificity. Trans. R. Soc. Trop. Med. Hyg. 1952, 46, 509–520. [Google Scholar] [CrossRef]
- PAHO/WHO. Zika Cases and Congenital Syndrome Associated with Zika Virus Reported by Countries and Territories in the Americas (Cumulative Cases), 2015–2017; Pan American Health Organization/World Health Organization: Washington, DC, USA, 2018. [Google Scholar]
- Moura da Silva, A.A.; Ganz, J.S.S.; da Sousa, P.S.; Doriqui, M.J.R.; Ribeiro, M.R.C.; dos Branco, M.R.F.C.; de Queiroz, R.C.S.; de Pacheco, M.J.T.; Vieira da Costa, F.R.; Silva, F.D.S.; et al. Early Growth and Neurologic Outcomes of Infants with Probable Congenital Zika Virus Syndrome. Emerg. Infect. Dis. 2016, 22, 1953–1956. [Google Scholar] [CrossRef]
- Carvalho, M.D.C.G.; de Miranda-Filho, D.B.; van der Linden, V.; Sobral, P.F.; Ramos, R.C.F.; Rocha, M.Â.W.; Cordeiro, M.T.; de Alencar, S.P.; Nunes, M.L. Sleep EEG patterns in infants with congenital Zika virus syndrome. Clin. Neurophysiol. 2017, 128, 204–214. [Google Scholar] [CrossRef]
- Chimelli, L.; Pone, S.M.; Avvad-Portari, E.; Vasconcelos, Z.F.M.; Zin, A.A.; Cunha, D.P.; Thompson, N.R.; Moreira, M.E.L.; Wiley, C.A.; da Pone, M.V.S. Persistence of Zika virus after birth: Clinical, virological, neuroimaging, and neuropathological documentation in a 5-month infant with congenital Zika syndrome. J. Neuropathol. Exp. Neurol. 2018, 77, 193–198. [Google Scholar] [CrossRef]
- Cugola, F.R.; Fernandes, I.R.; Russo, F.B.; Freitas, B.C.; Dias, J.L.M.; Guimarães, K.P.; Benazzato, C.; Almeida, N.; Pignatari, G.C.; Romero, S.; et al. The Brazilian Zika virus strain causes birth defects in experimental models. Nature 2016, 534, 267–271. [Google Scholar] [CrossRef]
- van den Pol, A.N.; Mao, G.; Yang, Y.; Ornaghi, S.; Davis, J.N. Zika Virus Targeting in the Developing Brain. J. Neurosci. 2017, 37, 2161–2175. [Google Scholar] [CrossRef] [PubMed]
- Wu, K.-Y.; Zuo, G.-L.; Li, X.-F.; Ye, Q.; Deng, Y.-Q.; Huang, X.-Y.; Cao, W.-C.; Qin, C.-F.; Luo, Z.-G. Vertical transmission of Zika virus targeting the radial glial cells affects cortex development of offspring mice. Cell Res. 2016, 26, 645. [Google Scholar] [CrossRef]
- Rice, M.; Galang, R.; Roth, N.; Ellington, S.; Honein, M. Vital Signs: Zika-Associated Birth Defects and Neurodevelopmental Abnormalities Possibly Associated with Congenital Zika Virus Infection—U.S. Territories and Freely Associated States, 2018. Morb. Mortal. Wkly. Rep. 2018, 67, 858–867. [Google Scholar] [CrossRef]
- The Lancet. The Lancet Zika-associated health and development problems in children. Lancet 2018, 392, 532. [Google Scholar] [CrossRef]
- van der Linden, V.; Pessoa, A.; Dobyns, W.; Barkovich, A.J.; van der Linden, H., Jr.; Filho, E.L.R.; Ribeiro, E.M.; de Leal, M.C.; de Coimbra, P.P.A.; de Aragão, M.F.V.V.; et al. Description of 13 Infants Born During October 2015–January 2016 With Congenital Zika Virus Infection Without Microcephaly at Birth—Brazil. MMWR. Morb. Mortal. Wkly. Rep. 2016, 65, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Prata-Barbosa, A.; Martins, M.M.; Guastavino, A.B.; da Cunha, A.J.L.A. Effects of Zika infection on growth. J. Pediatr (Rio J) 2018. [Google Scholar] [CrossRef]
- Semple, B.D.; Blomgren, K.; Gimlin, K.; Ferriero, D.M.; Noble-Haeusslein, L.J. Brain development in rodents and humans: Identifying benchmarks of maturation and vulnerability to injury across species. Prog. Neurobiol. 2013, 106–107, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Pressler, R.; Auvin, S. Comparison of brain maturation among species: An example in translational research suggesting the possible use of bumetanide in newborn. Front. Neurol. 2013, 4, 36. [Google Scholar] [CrossRef] [PubMed]
- Stagni, F.; Giacomini, A.; Guidi, S.; Ciani, E.; Bartesaghi, R. Timing of therapies for Down syndrome: The sooner, the better. Front. Behav. Neurosci. 2015, 9, 265. [Google Scholar] [CrossRef]
- Ferreira, A.C.; Zaverucha-do-Valle, C.; Reis, P.A.; Barbosa-Lima, G.; Vieira, Y.R.; Mattos, M.; de Silva, P.; Sacramento, C.; de Castro Faria Neto, H.C.; Campanati, L.; et al. Sofosbuvir protects Zika virus-infected mice from mortality, preventing short- and long-term sequelae. Sci. Rep. 2017, 7, 9409. [Google Scholar] [CrossRef]
- Mavigner, M.; Raper, J.; Kovacs-Balint, Z.; Gumber, S.; O’Neal, J.T.; Bhaumik, S.K.; Zhang, X.; Habib, J.; Mattingly, C.; McDonald, C.E.; et al. Postnatal Zika virus infection is associated with persistent abnormalities in brain structure, function, and behavior in infant macaques. Sci. Transl. Med. 2018, 10, eaao6975. [Google Scholar] [CrossRef] [PubMed]
- da Silva, I.R.F.; Frontera, J.A.; de Filippis, A.M.B.; do Nascimento, O.J.M. Neurologic Complications Associated With the Zika Virus in Brazilian Adults. JAMA Neurol. 2017, 74, 1190–1198. [Google Scholar] [CrossRef]
- Muñoz, L.S.; Barreras, P.; Pardo, C.A. Zika Virus-Associated Neurological Disease in the Adult: Guillain-Barré Syndrome, Encephalitis, and Myelitis. Semin. Reprod. Med. 2016, 34, 273–279. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, M.L.B.; de Brito, C.A.A.; Moreira, Á.J.P.; de Morais Machado, M.Í.; Henriques-Souza, A.; Cordeiro, M.T.; de Marques, E.T.; Pena, L.J. Guillain-Barré Syndrome, Acute Disseminated Encephalomyelitis and Encephalitis Associated with Zika Virus Infection in Brazil: Detection of Viral RNA and Isolation of Virus during Late Infection. Am. J. Trop. Med. Hyg. 2017, 97, 1405–1409. [Google Scholar] [CrossRef]
- Sebastián, U.U.; Ricardo, A.V.A.; Alvarez, B.C.; Cubides, A.; Luna, A.F.; Arroyo-Parejo, M.; Acuña, C.E.; Quintero, A.V.; Villareal, O.C.; Pinillos, O.S.; et al. Zika virus-induced neurological critical illness in Latin America: Severe Guillain-Barre Syndrome and encephalitis. J. Crit. Care 2017, 42, 275–281. [Google Scholar] [CrossRef]
- Willison, H.; Jacobs, B.; van Doorn, P. Guillain-Barré Syndrome. Lancet 2016, 23, 1295–1309. [Google Scholar] [CrossRef]
- Parra, B.; Lizarazo, J.; Jiménez-Arango, J.A.; Zea-Vera, A.F.; González-Manrique, G.; Vargas, J.; Angarita, J.A.; Zuñiga, G.; Lopez-Gonzalez, R.; Beltran, C.L.; et al. Guillain–Barré Syndrome Associated with Zika Virus Infection in Colombia. N. Engl. J. Med. 2016, 375, 1513–1523. [Google Scholar] [CrossRef] [PubMed]
- Azevedo, R.S.S.; Araujo, M.T.; Martins Filho, A.J.; Oliveira, C.S.; Nunes, B.T.D.; Cruz, A.C.R.; Nascimento, A.G.P.A.C.; Medeiros, R.C.; Caldas, C.A.M.; Araujo, F.C.; et al. Zika virus epidemic in Brazil. I. Fatal disease in adults: Clinical and laboratorial aspects. J. Clin. Virol. 2016, 85, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.Y.; Lynch, R.; Martins, K.; Encinales, L.; Cadena Bonfanti, A.; Pacheco, N.; Reid, S.P.; Lara Sarabia, O.E.; González Torres, H.J.; Mejia Castillo, S.; et al. Long-term clinical outcomes of Zika-associated Guillain-Barré syndrome. Emerg. Microbes Infect. 2018, 7, 4–7. [Google Scholar] [CrossRef]
- Lanciotti, R.S.; Kosoy, O.L.; Laven, J.J.; Velez, J.O.; Lambert, A.J.; Johnson, A.J.; Stanfield, S.M.; Duffy, M.R. Genetic and serologic properties of Zika virus associated with an epidemic, Yap State, Micronesia, 2007. Emerg. Infect. Dis. 2008, 14, 1232–1239. [Google Scholar] [CrossRef]
- Bhatnagar, J.; Rabeneck, D.B.; Martines, R.B.; Reagan-Steiner, S.; Ermias, Y.; Estetter, L.B.C.; Suzuki, T.; Ritter, J.; Keating, M.K.; Hale, G.; et al. Zika virus RNA replication and persistence in brain and placental tissue. Emerg. Infect. Dis. 2017, 23, 405–414. [Google Scholar] [CrossRef]
- Medina, F.A.; Torres, G.; Acevedo, J.; Fonseca, S.; Casiano, L.; De León-Rodríguez, C.M.; Santiago, G.A.; Doyle, K.; Sharp, T.M.; Alvarado, L.I.; et al. Duration of the Presence of Infectious Zika Virus in Semen and Serum. J. Infect. Dis. 2018, 219, 31–40. [Google Scholar] [CrossRef]
- Oliveira Souto, I.; Alejo-Cancho, I.; Gascón Brustenga, J.; Peiró Mestres, A.; Muñoz Gutiérrez, J.; Martínez Yoldi, M.J. Persistence of Zika virus in semen 93 days after the onset of symptoms. Enferm. Infecc. Microbiol. Clin. 2018, 36, 21–23. [Google Scholar] [CrossRef]
- Aid, M.; Abbink, P.; Larocca, R.A.; Boyd, M.; Nityanandam, R.; Nanayakkara, O.; Martinot, A.J.; Moseley, E.T.; Blass, E.; Borducchi, E.N.; et al. Zika Virus Persistence in the Central Nervous System and Lymph Nodes of Rhesus Monkeys. Cell 2017, 169, 610–620. [Google Scholar] [CrossRef]
- Hirsch, A.J.; Smith, J.L.; Haese, N.N.; Broeckel, R.M.; Parkins, C.J.; Kreklywich, C.; DeFilippis, V.R.; Denton, M.; Smith, P.P.; Messer, W.B.; et al. Zika Virus infection of rhesus macaques leads to viral persistence in multiple tissues. PLoS Pathog. 2017, 13, e1006219. [Google Scholar] [CrossRef]
- Souza, I.N.O.; Frost, P.S.; França, J.V.; Nascimento-Viana, J.B.; Neris, R.L.S.; Freitas, L.; Pinheiro, D.J.L.L.; Nogueira, C.O.; Neves, G.; Chimelli, L.; et al. Acute and chronic neurological consequences of early-life Zika virus infection in mice. Sci. Transl. Med. 2018, 10, eaar2749. [Google Scholar] [CrossRef] [PubMed]
- Simonin, Y.; Loustalot, F.; Desmetz, C.; Foulongne, V.; Constant, O.; Fournier-Wirth, C.; Leon, F.; Molès, J.-P.; Goubaud, A.; Lemaitre, J.-M.; et al. Zika Virus Strains Potentially Display Different Infectious Profiles in Human Neural Cells. EBioMedicine 2016, 12, 161–169. [Google Scholar] [CrossRef] [PubMed]
- Shao, Q.; Herrlinger, S.; Zhu, Y.-N.; Yang, M.; Goodfellow, F.; Stice, S.L.; Qi, X.-P.; Brindley, M.A.; Chen, J.-F. The African Zika virus MR-766 is more virulent and causes more severe brain damage than current Asian lineage and dengue virus. Development 2017, 144, 4114–4124. [Google Scholar] [CrossRef] [PubMed]
- Calvet, G.; Aguiar, R.S.; Melo, A.S.O.; Sampaio, S.A.; de Filippis, I.; Fabri, A.; Araujo, E.S.M.; de Sequeira, P.C.; de Mendonça, M.C.L.; de Oliveira, L.; et al. Detection and sequencing of Zika virus from amniotic fl uid of fetuses with microcephaly in Brazil: A case study. Lancet Infect Dis 2016, 16, 653–660. [Google Scholar] [CrossRef]
- Yuan, L.; Huang, X.-Y.; Liu, Z.-Y.; Zhang, F.; Zhu, X.-L.; Yu, J.-Y.; Ji, X.; Xu, Y.-P.; Li, G.; Li, C.; et al. A single mutation in the prM protein of Zika virus contributes to fetal microcephaly. Science 2017, 358, 933–936. [Google Scholar] [CrossRef]
- Liu, Y.; Liu, J.; Du, S.; Shan, C.; Nie, K.; Zhang, R.; Li, X.-F.; Zhang, R.; Wang, T.; Qin, C.-F.; et al. Evolutionary enhancement of Zika virus infectivity in Aedes aegypti mosquitoes. Nature 2017, 545, 482. [Google Scholar] [CrossRef] [PubMed]
- Xia, H.; Luo, H.; Shan, C.; Muruato, A.E.; Nunes, B.T.D.; Medeiros, D.B.A.; Zou, J.; Xie, X.; Giraldo, M.I.; Vasconcelos, P.F.C.; et al. An evolutionary NS1 mutation enhances Zika virus evasion of host interferon induction. Nat. Commun. 2018, 9, 414. [Google Scholar] [CrossRef] [PubMed]
- Adekolu-John, E.O.; Fagbami, A.H. Arthropod-borne virus antibodies in sera of residents of Kainji Lake Basin, Nigeria 1980. Trans. R. Soc. Trop. Med. Hyg. 1983, 77, 149–151. [Google Scholar] [CrossRef]
- Fagbami, A.H. Zika virus infections in Nigeria: Virological and seroepidemiological investigations in Oyo State. J. Hyg. (Lond) 1979, 83, 213–219. [Google Scholar] [CrossRef]
- Olson, J.G.; Ksiazek, T.G.; Gubler, D.J.; Lubis, S.I.; Simanjuntak, G.; Lee, V.H.; Nalim, S.; Juslis, K.; See, R. A survey for arboviral antibodies in sera of humans and animals in Lombok, Republic of Indonesia. Ann. Trop. Med. Parasitol. 1983, 77, 131–137. [Google Scholar] [CrossRef]
- Olson, J.G.; Ksiazek, T.G.; Suhandiman; Triwibowo. Zika virus, a cause of fever in Central Java, Indonesia. Trans. R. Soc. Trop. Med. Hyg. 1981, 75, 389–393. [Google Scholar] [CrossRef]
- Pond, W.L. Arthropod-borne virus antibodies in sera from residents of South-East Asia. Trans. R. Soc. Trop. Med. Hyg. 1963, 57, 364–371. [Google Scholar] [CrossRef]
- Osuna, C.E.; Lim, S.Y.; Deleage, C.; Griffin, B.D.; Stein, D.; Schroeder, L.T.; Omange, R.; Best, K.; Luo, M.; Hraber, P.T.; et al. Zika viral dynamics and shedding in rhesus and cynomolgus macaques. Nat. Med. 2016, 22, 1448–1455. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Saucedo-Cuevas, L.; Regla-Nava, J.A.; Chai, G.; Sheets, N.; Tang, W.; Terskikh, A.V.; Shresta, S.; Gleeson, J.G. Zika Virus Infects Neural Progenitors in the Adult Mouse Brain and Alters Proliferation. Cell Stem Cell 2016, 19, 593–598. [Google Scholar] [CrossRef]
- Lazear, H.M.; Govero, J.; Smith, A.M.; Platt, D.J.; Fernandez, E.; Miner, J.J.; Diamond, M.S. A Mouse Model of Zika Virus Pathogenesis. Cell Host Microbe 2016, 19, 720–730. [Google Scholar] [CrossRef]
- Smith, D.R.; Hollidge, B.; Daye, S.; Zeng, X.; Blancett, C.; Kuszpit, K.; Bocan, T.; Koehler, J.W.; Coyne, S.; Minogue, T.; et al. Neuropathogenesis of Zika Virus in a Highly Susceptible Immunocompetent Mouse Model after Antibody Blockade of Type I Interferon. PLoS Negl. Trop. Dis. 2017, 11, e0005296. [Google Scholar] [CrossRef] [PubMed]
- Bayer, A.; Lennemann, N.J.; Ouyang, Y.; Bramley, J.C.; Morosky, S.; Marques, E.T.D.A.; Cherry, S.; Sadovsky, Y.; Coyne, C.B.; Coyne, C.B. Type III Interferons Produced by Human Placental Trophoblasts Confer Protection against Zika Virus Infection. Cell Host Microbe 2016, 19, 705–712. [Google Scholar] [CrossRef]
- Yockey, L.J.; Jurado, K.A.; Arora, N.; Millet, A.; Rakib, T.; Milano, K.M.; Hastings, A.K.; Fikrig, E.; Kong, Y.; Horvath, T.L.; et al. Type I interferons instigate fetal demise after Zika virus infection. Sci. Immunol. 2018, 3, eaao1680b. [Google Scholar] [CrossRef]
- Ojha, C.R.; Rodriguez, M.; Karuppan, M.K.M.; Lapierre, J.; Kashanchi, F.; El-Hage, N. Toll-like receptor 3 regulates Zika virus infection and associated host inflammatory response in primary human astrocytes. PLoS ONE 2019, 14, e0208543. [Google Scholar] [CrossRef]
- Wells, M.F.; Salick, M.R.; Wiskow, O.; Ho, D.J.; Worringer, K.A.; Ihry, R.J.; Kommineni, S.; Bilican, B.; Klim, J.R.; Hill, E.J.; et al. Genetic Ablation of AXL Does Not Protect Human Neural Progenitor Cells and Cerebral Organoids from Zika Virus Infection. Cell Stem Cell 2016, 19, 703–708. [Google Scholar] [CrossRef] [PubMed]
- Limonta, D.; Jovel, J.; Kumar, A.; Airo, A.M.; Hou, S.; Saito, L.; Branton, W.; Ka-Shu Wong, G.; Mason, A.; Power, C.; et al. Human Fetal Astrocytes Infected with Zika Virus Exhibit Delayed Apoptosis and Resistance to Interferon: Implications for Persistence. Viruses 2018, 10, 646. [Google Scholar] [CrossRef] [PubMed]
- Aldo, P.; You, Y.; Szigeti, K.; Horvath, T.L.; Lindenbach, B.; Mor, G. HSV-2 enhances ZIKV infection of the placenta and induces apoptosis in first-trimester trophoblast cells. Am. J. Reprod. Immunol. 2016, 76, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Hastings, A.K.; Yockey, L.J.; Jagger, B.W.; Hwang, J.; Uraki, R.; Gaitsch, H.F.; Parnell, L.A.; Cao, B.; Mysorekar, I.U.; Rothlin, C.V.; et al. TAM Receptors Are Not Required for Zika Virus Infection in Mice. Cell Rep. 2017, 19, 558–568. [Google Scholar] [CrossRef]
- Chen, J.; Yang, Y.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.; Cui, Y.; Bai, R.; Liang, Y.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef]
- Liu, Y.; Gordesky-Gold, B.; Leney-Greene, M.; Weinbren, N.L.; Tudor, M.; Cherry, S. Inflammation-Induced, STING-Dependent Autophagy Restricts Zika Virus Infection in the Drosophila Brain. Cell Host Microbe 2018, 24, 57–68. [Google Scholar] [CrossRef]
- Chiramel, A.I.; Best, S.M. Role of autophagy in Zika virus infection and pathogenesis. Virus Res. 2018, 254, 34–40. [Google Scholar] [CrossRef]
- Delgado, F.G.; Torres, K.I.; Castellanos, J.E.; Romero-Sánchez, C.; Simon-Lorière, E.; Sakuntabhai, A.; Roth, C. Improved Immune Responses Against Zika Virus After Sequential Dengue and Zika Virus Infection in Humans. Viruses 2018, 10, 480. [Google Scholar] [CrossRef]
- Wen, J.; Elong Ngono, A.; Regla-Nava, J.A.; Kim, K.; Gorman, M.J.; Diamond, M.S.; Shresta, S. Dengue virus-reactive CD8+ T cells mediate cross-protection against subsequent Zika virus challenge. Nat. Commun. 2017, 8, 1459. [Google Scholar] [CrossRef]
- Brown, J.A.; Singh, G.; Acklin, J.A.; Lee, S.; Duehr, J.E.; Chokola, A.N.; Frere, J.J.; Hoffman, K.W.; Foster, G.A.; Krysztof, D.; et al. Dengue Virus Immunity Increases Zika Virus-Induced Damage during Pregnancy. Immunity 2019. [Google Scholar] [CrossRef]
- Bardina, S.V.; Bunduc, P.; Tripathi, S.; Duehr, J.; Frere, J.J.; Brown, J.A.; Nachbagauer, R.; Foster, G.A.; Krysztof, D.; Tortorella, D.; et al. Enhancement of Zika virus pathogenesis by preexisting antiflavivirus immunity. Science 2017, 356, 175–180. [Google Scholar] [CrossRef]
- Dejnirattisai, W.; Supasa, P.; Wongwiwat, W.; Rouvinski, A.; Barba-Spaeth, G.; Duangchinda, T.; Sakuntabhai, A.; Cao-Lormeau, V.-M.; Malasit, P.; Rey, F.A.; et al. Dengue virus sero-cross-reactivity drives antibody-dependent enhancement of infection with zika virus. Nat. Immunol. 2016, 17, 1102–1108. [Google Scholar] [CrossRef] [PubMed]
- De Araújo, P.S.R.; de Silva Júnior, M.L., Jr.; Tenório, M.; dos Santos, F.G.T. Co-infection ZIKV and HSV-1 associated with meningoencephalitis: Case report and literature review. J. Infect. Public Health 2019, 12, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Raboni, S.M.; Bonfim, C.; Almeida, B.M.; Zanluca, C.; Koishi, A.C.; Rodrigues, P.R.V.P.; Kay, C.K.; Ribeiro, L.L.; Scola, R.H.; dos Santos, C.N.D. Flavivirus cross-reactivity in serological tests and Guillain-Barré syndrome in a hematopoietic stem cell transplant patient: A case report. Transpl. Infect. Dis. 2017, 19, e12700. [Google Scholar] [CrossRef]
- Magalhaes, T.; Robison, A.; Young, M.; Black, W.; Foy, B.; Ebel, G.; Rückert, C.; Magalhaes, T.; Robison, A.; Young, M.C.; et al. Sequential Infection of Aedes aegypti Mosquitoes with Chikungunya Virus and Zika Virus Enhances Early Zika Virus Transmission. Insects 2018, 9, 177. [Google Scholar] [CrossRef] [PubMed]
- Mcgrath, E.L.; Rossi, S.L.; Gao, J.; Widen, S.G.; Grant, A.C.; Dunn, T.J.; Azar, S.R.; Roundy, C.M.; Xiong, Y.; Prusak, D.J.; et al. Differential Responses of Human Fetal Brain Neural Stem Cells to Zika Virus Infection. Stem Cell Rep. 2017, 8, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Caires-Junior, L.C.; Goulart, E.; Melo, U.S.; Araujo, B.H.S.; Alvizi, L.; Soares-Schanoski, A.; de Oliveira, D.F.; Kobayashi, G.S.; Griesi-Oliveira, K.; Musso, C.M.; et al. Discordant congenital Zika syndrome twins show differential in vitro viral susceptibility of neural progenitor cells. Nat. Commun. 2018, 9, 475. [Google Scholar] [CrossRef] [PubMed]
- Quicke, K.M.; Bowen, J.R.; Johnson, E.L.; McDonald, C.E.; Ma, H.; O’Neal, J.T.; Rajakumar, A.; Wrammert, J.; Rimawi, B.H.; Pulendran, B.; et al. Zika Virus Infects Human Placental Macrophages. Cell Host Microbe 2016, 20, 83–90. [Google Scholar] [CrossRef]
- Arumugasaamy, N.; Ettehadieh, L.E.; Kuo, C.-Y.; Paquin-Proulx, D.; Kitchen, S.M.; Santoro, M.; Placone, J.K.; Silveira, P.P.; Aguiar, R.S.; Nixon, D.F.; et al. Biomimetic Placenta-Fetus Model Demonstrating Maternal–Fetal Transmission and Fetal Neural Toxicity of Zika Virus. Ann. Biomed. Eng. 2018, 46, 1963–1974. [Google Scholar] [CrossRef]
- Ribeiro, M.R.; Moreli, J.B.; Marques, R.E.; Papa, M.P.; Meuren, L.M.; Rahal, P.; de Arruda, L.B.; Oliani, A.H.; Oliani, D.C.M.V.; Oliani, S.M.; et al. Zika-virus-infected human full-term placental explants display pro-inflammatory responses and undergo apoptosis. Arch. Virol. 2018, 163, 2687–2699. [Google Scholar] [CrossRef]
- Hermanns, K.; Göhner, C.; Kopp, A.; Schmidt, A.; Merz, W.M.; Markert, U.R.; Junglen, S.; Drosten, C. Zika virus infection in human placental tissue explants is enhanced in the presence of dengue virus antibodies in-vitro. Emerg. Microbes Infect. 2018, 7, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Jurado, K.A.; Simoni, M.K.; Tang, Z.; Uraki, R.; Hwang, J.; Householder, S.; Wu, M.; Lindenbach, B.D.; Abrahams, V.M.; Guller, S.; et al. Zika virus productively infects primary human placenta-specific macrophages. JCI Insight 2016, 1, e88461. [Google Scholar] [CrossRef] [PubMed]
- Duggal, N.K.; Mcdonald, E.M.; Ritter, J.M.; Brault, A.C. Sexual transmission of Zika virus enhances in utero transmission in a mouse model OPEN. Sci. Rep. 2018, 8, 4510. [Google Scholar] [CrossRef] [PubMed]
- Barbeito-Andrés, J.; Schuler-Faccini, L.; Garcez, P.P. Why is congenital Zika syndrome asymmetrically distributed among human populations? PLoS Biol. 2018, 16, e2006592. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, W.K.; de França, G.V.A.; Carmo, E.H.; Duncan, B.B.; de Souza Kuchenbecker, R.; Schmidt, M.I. Infection-related microcephaly after the 2015 and 2016 Zika virus outbreaks in Brazil: A surveillance-based analysis. Lancet 2017, 390, 861–870. [Google Scholar] [CrossRef]
- Souza, A.I.; de Siqueira, M.T.; Ferreira, A.L.C.G.; de Freitas, C.U.; Bezerra, E.V.; Ribeiro, A.G.; Nardocci, A.C. Geography of Microcephaly in the Zika Era: A Study of Newborn Distribution and Socio-environmental Indicators in Recife, Brazil, 2015-2016. Public Health Rep. 2018, 133, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Center for Disease Control and Prevention Zika Virus Treatment. Available online: https://www.cdc.gov/zika/symptoms/treatment.html (accessed on 23 January 2019).
- World Health Organization. Zika Fact Sheet. Available online: https://www.who.int/news-room/fact-sheets/detail/zika-virus (accessed on 23 January 2019).
- Sisk, J.M.; Frieman, M.B. Screening of FDA-Approved Drugs for Treatment of Emerging Pathogens. ACS Infect. Dis. 2016, 1, 401–402. [Google Scholar] [CrossRef]
- Zhou, T.; Tan, L.; Cederquist, G.Y.; Fan, Y.; Hartley, B.J.; Mukherjee, S.; Tomishima, M.; Brennand, K.J.; Zhang, Q.; Schwartz, R.E.; et al. High-Content Screening in hPSC-Neural Progenitors Identifies Drug Candidates that Inhibit Zika Virus Infection in Fetal-like Organoids and Adult Brain. Cell Stem Cell 2017, 21, 274–283.e5. [Google Scholar] [CrossRef]
- Han, Y.; Mesplède, T.; Xu, H.; Quan, Y.; Wainberg, M.A. The antimalarial drug amodiaquine possesses anti-ZIKA virus activities. J. Med. Virol. 2018, 90, 796–802. [Google Scholar] [CrossRef]
- Retallack, H.; Di, E.; Arias, C.; Knopp, K.A.; Laurie, M.T. Zika virus cell tropism in the developing human brain and inhibition by azithromycin. Proc. Natl. Acad. Sci. USA 2016, 113, 14408–14413. [Google Scholar] [CrossRef]
- Wu, Y.H.; Tseng, C.K.; Lin, C.K.; Wei, C.K.; Lee, J.C.; Young, K.C. ICR suckling mouse model of Zika virus infection for disease modeling and drug validation. PLoS Negl. Trop. Dis. 2018, 12, e0006848. [Google Scholar] [CrossRef]
- Delvecchio, R.; Higa, L.; Pezzuto, P.; Valadão, A.; Garcez, P.; Monteiro, F.; Loiola, E.; Dias, A.; Silva, F.; Aliota, M.; et al. Chloroquine, an Endocytosis Blocking Agent, Inhibits Zika Virus Infection in Different Cell Models. Viruses 2016, 8, 322. [Google Scholar] [CrossRef]
- Shiryaev, S.A.; Mesci, P.; Pinto, A.; Fernandes, I.; Sheets, N.; Shresta, S.; Farhy, C.; Huang, C.-T.; Strongin, A.Y.; Muotri, A.R.; et al. Repurposing of the anti-malaria drug chloroquine for Zika Virus treatment and prophylaxis. Sci. Rep. 2017, 7, 15771. [Google Scholar] [CrossRef]
- Li, C.; Zhu, X.; Ji, X.; Quanquin, N.; Deng, Y.-Q.; Tian, M.; Aliyari, R.; Zuo, X.; Yuan, L.; Afridi, S.K.; et al. Chloroquine, a FDA-approved Drug, Prevents Zika Virus Infection and its Associated Congenital Microcephaly in Mice. EBioMedicine 2017, 24, 189–194. [Google Scholar] [CrossRef]
- Xu, M.; Lee, E.M.; Wen, Z.; Cheng, Y.; Huang, W.; Qian, X.; Tcw, J.; Kouznetsova, J.; Ogden, S.C.; Hammack, C.; et al. Identification of small-molecule inhibitors of Zika virus infection and induced neural cell death via a drug repurposing screen. Nat. Med. 2016, 22, 1101–1107. [Google Scholar] [CrossRef]
- Baz, M.; Goyette, N.; Griffin, B.D.; Kobinger, G.P.; Boivin, G. In vitro susceptibility of geographically and temporally distinct Zika viruses to favipiravir and ribavirin. Antivir. Ther. 2017, 22, 613–618. [Google Scholar] [CrossRef]
- Cai, L.; Sun, Y.; Song, Y.; Xu, L.; Bei, Z.; Zhang, D.; Dou, Y.; Wang, H. Viral polymerase inhibitors T-705 and T-1105 are potential inhibitors of Zika virus replication. Arch. Virol. 2017, 162, 2847–2853. [Google Scholar] [CrossRef]
- Lanko, K.; Eggermont, K.; Patel, A.; Kaptein, S.; Delang, L.; Verfaillie, C.M.; Neyts, J. Replication of the Zika virus in different iPSC-derived neuronal cells and implications to assess efficacy of antivirals. Antiviral Res. 2017, 145, 82–86. [Google Scholar] [CrossRef]
- Kim, J.A.; Seong, R.K.; Kumar, M.; Shin, O.S. Favipiravir and ribavirin inhibit replication of Asian and African strains of zika virus in different cell models. Viruses 2018, 10, 72. [Google Scholar] [CrossRef] [PubMed]
- Julander, J.G.; Siddharthan, V.; Evans, J.; Taylor, R.; Tolbert, K.; Apuli, C.; Stewart, J.; Collins, P.; Gebre, M.; Neilson, S.; et al. Efficacy of the broad-spectrum antiviral compound BCX4430 against Zika virus in cell culture and in a mouse model. Antivir. Res. 2017, 137, 14–22. [Google Scholar] [CrossRef]
- Lim, S.-Y.; Osuna, C.; Taylor, R.; Mathis, A.; Kamath, V.; Berger, E.; Babu, Y.; Sheridan, W.; Whitney, J.B. BCX4430, a Broad-Spectrum Adenosine Analog Direct-Acting Antiviral Drug, Abrogates Viremia in Rhesus Macaques Challenged with Zika Virus. Open Forum Infectious Diseases 2016. [Google Scholar] [CrossRef]
- Costa, V.V.; Del Sarto, J.L.; Rocha, R.F.; Silva, F.R.; Doria, J.G.; Olmo, I.G.; Marques, R.E.; Queiroz-Junior, C.M.; Foureaux, G.; Araújo, J.M.S.; et al. N-Methyl-d-Aspartate (NMDA) Receptor Blockade Prevents Neuronal Death Induced by Zika Virus Infection. MBio 2017, 8, e00350-17. [Google Scholar] [CrossRef] [PubMed]
- Cairns, D.M.; Boorgu, D.S.S.K.; Levin, M.; Kaplan, D.L. Niclosamide rescues microcephaly in a humanized in vitro model of Zika infection using human induced neural stem cells. Biol. Open 2018, 7, bio031807. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhang, N.; Li, C.; Tian, M.; Hao, J.; Xie, X.-P.; Shi, P.-Y.; Qin, C.-F. Adenosine Analog NITD008 Is a Potent Inhibitor of Zika Virus. Open Forum Infect. Dis. 2016, 3, ofw175. [Google Scholar] [CrossRef] [PubMed]
- Adcock, R.S.; Chu, Y.K.; Golden, J.E.; Chung, D.H. Evaluation of anti-Zika virus activities of broad-spectrum antivirals and NIH clinical collection compounds using a cell-based, high-throughput screen assay. Antivir. Res. 2017, 138, 47–56. [Google Scholar] [CrossRef]
- Yin, Z.; Chen, Y.-L.; Schul, W.; Wang, Q.-Y.; Gu, F.; Duraiswamy, J.; Kondreddi, R.R.; Niyomrattanakit, P.; Lakshminarayana, S.B.; Goh, A.; et al. An adenosine nucleoside inhibitor of dengue virus. Proc. Natl. Acad. Sci. USA 2009, 106, 20435–20439. [Google Scholar] [CrossRef] [PubMed]
- Kamiyama, N.; Soma, R.; Hidano, S.; Watanabe, K.; Umekita, H.; Fukuda, C.; Noguchi, K.; Gendo, Y.; Ozaki, T.; Sonoda, A.; et al. Ribavirin inhibits Zika virus (ZIKV) replication in vitro and suppresses viremia in ZIKV-infected STAT1-deficient mice. Antiviral Res. 2017, 146, 1–11. [Google Scholar] [CrossRef]
- Elfiky, A.A. Zika viral polymerase inhibition using anti-HCV drugs both in market and under clinical trials. J. Med. Virol. 2016, 88, 2044–2051. [Google Scholar] [CrossRef]
- Onorati, M.; Li, Z.; Liu, F.; Sousa, A.M.M.; Nakagawa, N.; Li, M.; Dell’Anno, M.T.; Gulden, F.O.; Pochareddy, S.; Tebbenkamp, A.T.N.; et al. Zika Virus Disrupts Phospho-TBK1 Localization and Mitosis in Human Neuroepithelial Stem Cells and Radial Glia. Cell Rep. 2016, 16, 2576–2592. [Google Scholar] [CrossRef] [PubMed]
- Bullard-Feibelman, K.M.; Govero, J.; Zhu, Z.; Salazar, V.; Veselinovic, M.; Diamond, M.S.; Geiss, B.J. The FDA-approved drug sofosbuvir inhibits Zika virus infection. Antiviral Res. 2017, 137, 134–140. [Google Scholar] [CrossRef]
- Sacramento, C.Q.; De Melo, G.R.; De Freitas, C.S.; Rocha, N.; Hoelz, L.V.B.; Miranda, M.; Fintelman-Rodrigues, N.; Marttorelli, A.; Ferreira, A.C.; Barbosa-Lima, G.; et al. The clinically approved antiviral drug sofosbuvir inhibits Zika virus replication. Sci. Rep. 2017, 7, 40920. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Deng, Y.Q.; Zou, P.; Wang, Q.; Dai, Y.; Yu, F.; Du, L.; Zhang, N.N.; Tian, M.; Hao, J.N.; et al. A peptide-based viral inactivator inhibits Zika virus infection in pregnant mice and fetuses. Nat. Commun. 2017, 8, 15672. [Google Scholar] [CrossRef]
- Snyder, B.; Goebel, S.; Koide, F.; Ptak, R.; Kalkeri, R. Synergistic antiviral activity of Sofosbuvir and type-I interferons (α and β) against Zika virus. J. Med. Virol. 2018, 90, 8–12. [Google Scholar] [CrossRef]
- Chappell, C.; Gilead Sciences; University of Nebraska. Study of Hepatitis C Treatment During Pregnancy. Available online: https://clinicaltrials.gov/ct2/show/NCT02683005 (accessed on 19 March 2019).
- Wiwanitkit, S.; Wiwanitkit, V. Doubled dosage of sofosbuvir is expected for inhibiting Zika virus infection. Asian Pac. J. Trop. Med. 2017, 10, 612–613. [Google Scholar] [CrossRef]
- Gilead Sciences Inc. Sovaldi Package Insert; Gilead Sciences Inc.: Foster City, CA, USA, 2017; pp. 1–37. [Google Scholar]
- Bassi, M.R.; Sempere, R.N.; Meyn, P.; Polacek, C.; Arias, A. Extinction of zika virus and usutu virus by lethal mutagenesis reveals different patterns of sensitivity to three mutagenic drugs. Antimicrob. Agents Chemother. 2018, 62, e00380-18. [Google Scholar] [CrossRef]
- Pires De Mello, C.P.; Tao, X.; Kim, T.H.; Bulitta, J.B.; Rodriquez, J.L.; Pomeroy, J.J.; Brown, A.N. Zika virus replication is substantially inhibited by novel favipiravir and interferon alpha combination regimens. Antimicrob. Agents Chemother. 2018, 62, e01983-17. [Google Scholar] [CrossRef] [PubMed]
- Best, K.; Guedj, J.; Madelain, V.; de Lamballerie, X.; Lim, S.-Y.; Osuna, C.E.; Whitney, J.B.; Perelson, A.S. Zika plasma viral dynamics in nonhuman primates provides insights into early infection and antiviral strategies. Proc. Natl. Acad. Sci. USA 2017, 114, 8847–8852. [Google Scholar] [CrossRef] [PubMed]
- MDVI LCC. Dose-Finding Study of Favipiravir in the Treatment of Uncomplicated Influenza. Available online: https://clinicaltrials.gov/ct2/show/results/NCT01068912 (accessed on 19 March 2019).
- Food and Drug Administration. ZITHROMAX® (Azithromycin Tablets) and (Azithromycin for Oral Suspension); Food and Drug Administration: Silver Spring, MD, USA, 2012; pp. 2011–2012.
- Ramsey, P.S.; Vaules, M.B.; Vasdev, G.M.; Andrews, W.W.; Ramin, K.D. Maternal and transplacental pharmacokinetics of azithromycin. Am. J. Obstet. Gynecol. 2003, 188, 714–718. [Google Scholar] [CrossRef] [PubMed]
- Jaruratanasirikul, S.; Hortiwakul, R.; Tantisarasart, T.; Phuenpathom, N.; Tussanasunthornwong, S. Distribution of azithromycin into brain tissue, cerebrospinal fluid, and aqueous humor of the eye. Antimicrob. Agents Chemother. 1996, 40, 825–826. [Google Scholar] [CrossRef] [PubMed]
- Lewerenz, J.; Maher, P. Chronic glutamate toxicity in neurodegenerative diseases-What is the evidence? Front. Neurosci. 2015, 9, 469. [Google Scholar] [CrossRef] [PubMed]
- Food and Drug Administration. FDA Label: Namenda (Memantine HCI); Food and Drug Administration: Silver Spring, MD, USA, 2003.
- Janssen Pharmaceutica. Remicade (Package Insert); Janssen Pharmaceutica: Beerse, Belgium, 2015. [Google Scholar]
- Pfizer. IBRANCE (palbociclib) Fact Sheet; Pfizer: New York, NY, USA, 2015. [Google Scholar]
- Conatus Pharmaceuticals Inc. A Trial of IDN-6556 in Post Orthotopic Liver Transplant for Chronic HCV (POLT-HCV-SVR). Available online: https://clinicaltrials.gov/ct2/show/NCT02138253 (accessed on 19 March 2019).
- Ofori-Adjei, D.; Dodoo, A.N.O.; Appiah-Danquah, A.; Couper, M. A review of the safety of niclosamide, pyrantel, triclabendazole and oxamniquine. Int. J. Risk Saf. Med. 2008, 20, 113–122. [Google Scholar]
- Kao, J.C.; HuangFu, W.C.; Tsai, T.T.; Ho, M.R.; Jhan, M.K.; Shen, T.J.; Tseng, P.C.; Wang, Y.T.; Lin, C.F. The antiparasitic drug niclosamide inhibits dengue virus infection by interfering with endosomal acidification independent of mTOR. PLoS Negl. Trop. Dis. 2018, 12, e0006715. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-K.; Bai, M.-Y.; Hu, T.-M.; Wang, Y.-C.; Chao, T.-K.; Weng, S.-J.; Huang, R.-L.; Su, P.-H.; Lai, H.-C. Preclinical evaluation of a nanoformulated antihelminthic, niclosamide, in ovarian cancer. Oncotarget 2016, 7, 8993–9006. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, Y.; Zhang, T.; Zhang, J.; Wu, B. Significantly enhanced bioavailability of niclosamide through submicron lipid emulsions with or without PEG-lipid: A comparative study. J. Microencapsul. 2015, 32, 496–502. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.X.; Ding, K.; Wang, C.Y. Niclosamide, an old antihelminthic agent, demonstrates antitumor activity by blocking multiple signaling pathways of cancer stem cells. Chin. J. Cancer 2012, 31, 178–184. [Google Scholar] [CrossRef]
- Bayer. Yomesan (Package Insert); Bayer: Leverkusen, Germany, 1988. [Google Scholar]
- Vliet, S.M.; Dasgupta, S.; Volz, D.C. Niclosamide Induces Epiboly Delay During Early Zebrafish Embryogenesis. Toxicol. Sci. 2018, 166, 306–317. [Google Scholar] [CrossRef]
- Borges, M.C.; Castro, L.A.; da Fonseca, B.A.L. Chloroquine use improves dengue-related symptoms. Mem. Inst. Oswaldo Cruz 2013, 108, 596–599. [Google Scholar] [CrossRef]
- Savarino, A.; Boelaert, J.R.; Cassone, A.; Majori, G.; Cauda, R. Effects of chloroquine on viral infections: An old drug against today’s diseases. Lancet Infect. Dis. 2003, 3, 722–727. [Google Scholar] [CrossRef]
- Taylor, W.R.J.; White, N.J. Antimalarial Drug Toxicity. Drug Saf. 2004, 27, 25–61. [Google Scholar] [CrossRef]
- De Clercq, E.; Bergstrom, D.E.; John, A.H. Broad-spectrum antiviral activity of adenosine analogues. Antivir. Res. 1984, 4, 119–133. [Google Scholar] [CrossRef]
- Lo, M.K.; Shi, P.-Y.; Chen, Y.-L.; Flint, M.; Spiropoulou, C.F. In vitro antiviral activity of adenosine analog NITD008 against tick-borne flaviviruses. Antivir. Res. 2016, 130, 46–49. [Google Scholar] [CrossRef]
- Eyer, L.; Zouharová, D.; Širmarová, J.; Fojtíková, M.; Štefánik, M.; Haviernik, J.; Nencka, R.; de Clercq, E.; Růžek, D. Antiviral activity of the adenosine analogue BCX4430 against West Nile virus and tick-borne flaviviruses. Antivir. Res. 2017, 142, 63–67. [Google Scholar] [CrossRef] [PubMed]
- BioCryst Pharmaceuticals. A Study to Evaluate the Single Dose Safety, Tolerability and Pharmacokinetics of IV BCX4430. Available online: https://clinicaltrials.gov/ct2/show/NCT03800173 (accessed on 19 March 2019).
- Bernatchez, J.A.; Yang, Z.; Coste, M.; Li, J.; Beck, S.; Liu, Y.; Clark, A.E.; Zhu, Z.; Luna, L.A.; Sohl, C.D.; et al. Development and Validation of a Phenotypic High-Content Imaging Assay for Assessing the Antiviral Activity of Small-Molecule Inhibitors Targeting Zika Virus. Antimicrob. Agents Chemother. 2018, 62, e00725-18. [Google Scholar] [CrossRef] [PubMed]
- FDA. Copegus Packet Insert; FDA: Silver Spring, MD, USA, 2011.
- Leonardi, W.; Zilbermintz, L.; Cheng, L.W.; Zozaya, J.; Tran, S.H.; Elliott, J.H.; Polukhina, K.; Manasherob, R.; Li, A.; Chi, X.; et al. Bithionol blocks pathogenicity of bacterial toxins, ricin, and Zika virus. Sci. Rep. 2016, 6, 34475. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Lim, L.; Srivastava, S.; Lu, Y.; Song, J. Solution conformations of Zika NS2B-NS3pro and its inhibition by natural products from edible plants. PLoS ONE 2017, 12, e0180632. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Chen, J.; Cheng, T.; Gindulyte, A.; He, J.; He, S.; Li, Q.; Shoemaker, B.; Thiessen, P.; Yu, B.; et al. PubChem 2019 update: Improved access to chemical data. Nucleic Acids Res. 2019, 47, D1102–D1109. [Google Scholar] [CrossRef]


{kind=link}
| Drug | Proposed Mechanism of Action | In Vitro Efficacy | In Vitro Efficacy | Food and Drug Administration (FDA) Approved? | FDA Category | Refs. |
|---|---|---|---|---|---|---|
| Amodiaquine | Unknown for ZIKV (antimalarial) | Vero cell line human pluripotent stem cell-derived neural progenitors | 40 mg/kg for 7 days s.c. to adult SCID-beige mice | Discontinued | - | [80,81] |
| Azithromycin | Unknown for ZIKV (antibiotic) | U87 glial cell line | 10 mg/kg i.p. to neonatal ICR mice | Yes | B | [82,83] |
| Chloroquine | Unknown for ZIKV (antimalarial) | human neural progenitor cells human brain microvascular endothelial cells mouse neurospheres | 50 mg/kg for 5 days + 5 mg/kg maintenance p.o. to adult AG129 mice 20 mg/kg for 6 days s.c. to pregnant BALB/c dams | Yes | Not assigned | [84,85,86] |
| Emricasan | pan-caspase inhibitor | human neural progenitor cells human astrocytes | N.D. | No | - | [87] |
| Favipiravir | RNA-dependent RNA polymerase inhibitor | Vero cell line induced-pluripotent neural stem cells human embryonic stem cells | N.D. | No | - | [88,89,90,91] |
| Galidesivir (BCX4430) | RNA-dependent RNA polymerase inhibitor | Vero76 cell line Huh7 cell line RD cell line | 150 mg/kg for 8 days b.i.d. i.m.to adult AG129 mice 100 mg/kg + 25 mg/kg for 9 days b.i.d. i.m. to adult rhesus monkeys | No | - | [92,93] |
| Infliximab | TNF-α neutralizing antibody | N.D. | 20 μg for 13 days i.p. to neonatal Swiss mice | Yes | B | [32] |
| Memantine | NMDA receptor blocker | glial primary cells neuronal primary cells | 30 mg/kg for 4 days p.o. to adult IFN-α/βR−/− mice | Yes | B | [94] |
| Niclosamide | endolysosomal pH neutralizer | human glioblastoma cell line (SNB-19) human astrocytes | 50 mg/kg for 2 days to chick embryos | Yes | B | [87,95] |
| NITD008 | RNA-dependent RNA polymerase inhibitor | Vero cell line | 50 mg/kg for 5 days i.p. to adult A129 mice | No | - | [96,97,98] |
| PHA-690509 | cyclin-dependent kinase inhibitor | human neural progenitor cells induced-pluripotent neural stem cells | N.D. | No | - | [87] |
| Ribavirin | RNA-dependent RNA polymerase inhibitor | Vero cell line human neural progenitor cells | 15 mg for 3 days i.p. to adult STAT-1 deficient mice | Yes | X | [88,91,99] |
| Sofosbuvir | RNA-dependent RNA polymerase inhibitor | human neuroepithelial stem cell placental cell line neuroblastoma cell line human fetal-derived neuronal stem cells induced-pluripotent neural stem cells brain organoids | 33 mg/kg for 7 days p.o. to adult C57BL/6 mice 20 mg/kg for 7 days i.p. to neonatal Swiss mice | Yes | B | [16,100,101,102,103] |
| Z2 | Direct inhibitor | Vero cell line BHK21 cell line | Single dose of 10 mg/kg i.p. to pregnant C57BL/6 mice Single dose of 10 mg/kg i.p. to A129 and AG6 adult mice | No | - | [104] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Souza, I.N.O.; Barros-Aragão, F.G.Q.; Frost, P.S.; Figueiredo, C.P.; Clarke, J.R. Late Neurological Consequences of Zika Virus Infection: Risk Factors and Pharmaceutical Approaches. Pharmaceuticals 2019, 12, 60. https://doi.org/10.3390/ph12020060
Souza INO, Barros-Aragão FGQ, Frost PS, Figueiredo CP, Clarke JR. Late Neurological Consequences of Zika Virus Infection: Risk Factors and Pharmaceutical Approaches. Pharmaceuticals. 2019; 12(2):60. https://doi.org/10.3390/ph12020060
Chicago/Turabian StyleSouza, Isis N. O., Fernanda G. Q. Barros-Aragão, Paula S. Frost, Claudia P. Figueiredo, and Julia R. Clarke. 2019. "Late Neurological Consequences of Zika Virus Infection: Risk Factors and Pharmaceutical Approaches" Pharmaceuticals 12, no. 2: 60. https://doi.org/10.3390/ph12020060
APA StyleSouza, I. N. O., Barros-Aragão, F. G. Q., Frost, P. S., Figueiredo, C. P., & Clarke, J. R. (2019). Late Neurological Consequences of Zika Virus Infection: Risk Factors and Pharmaceutical Approaches. Pharmaceuticals, 12(2), 60. https://doi.org/10.3390/ph12020060