Chain-Branched Polyhydroxylated Octahydro-1H-Indoles as Potential Leads against Lysosomal Storage Diseases



Abstract
:
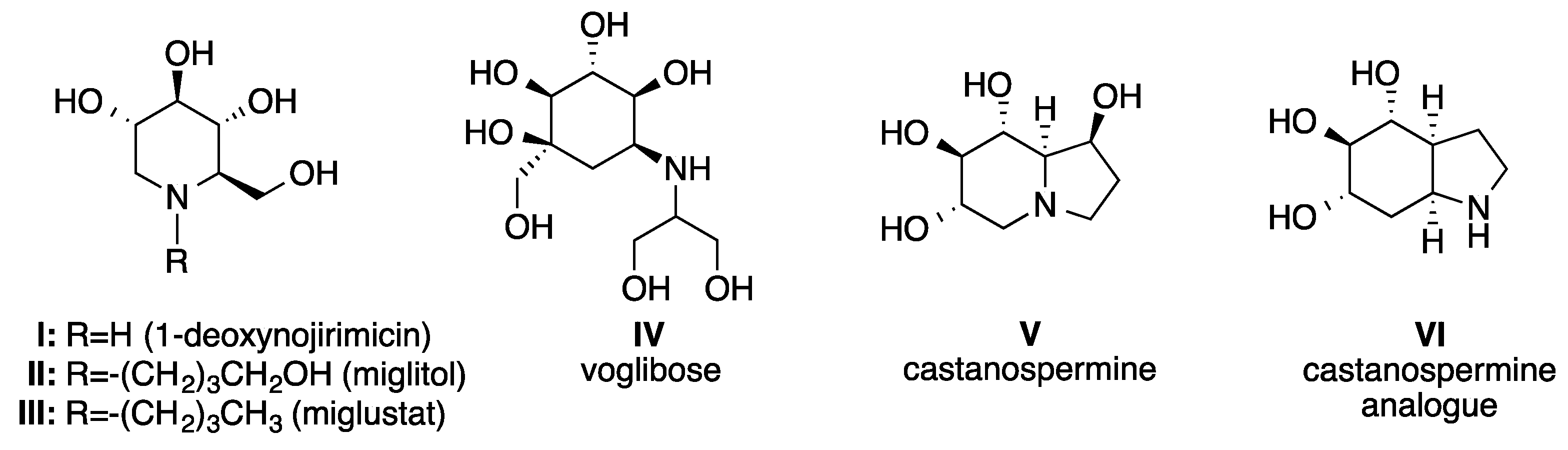
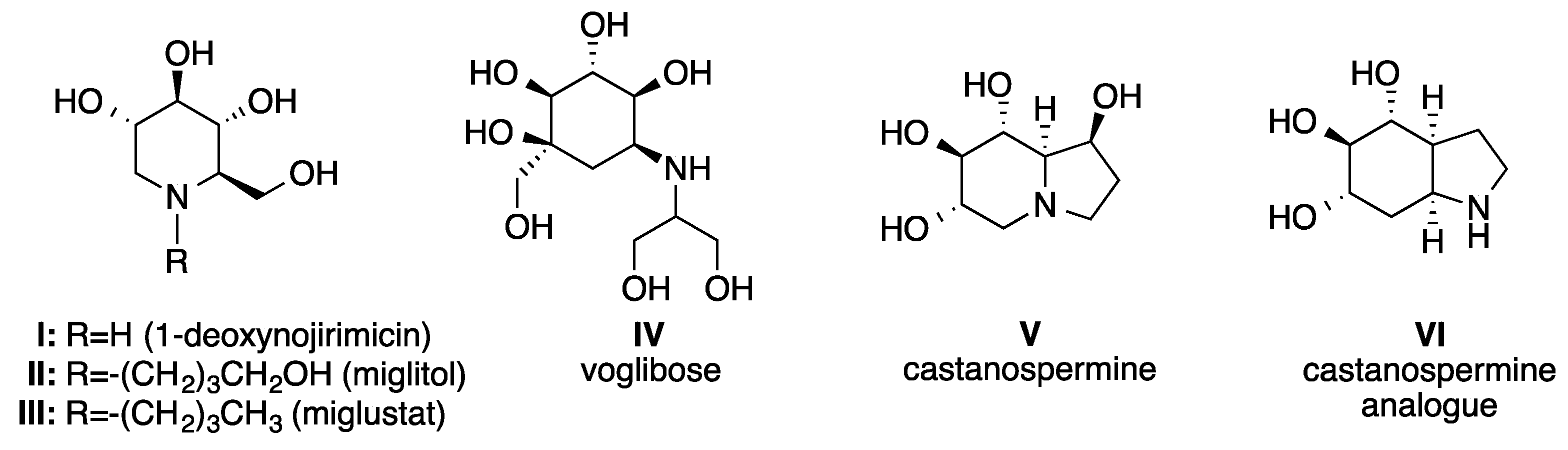
1. Introduction
2. Results
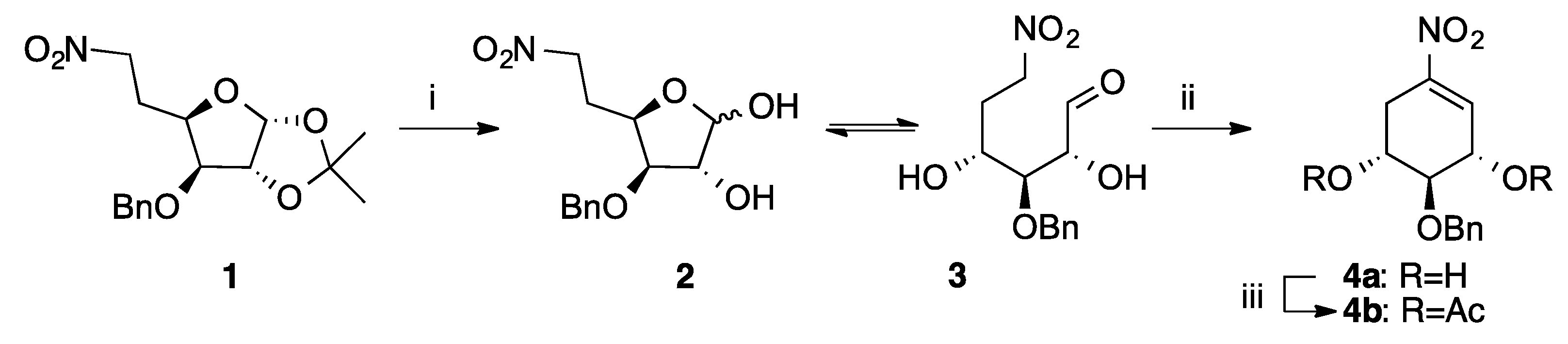
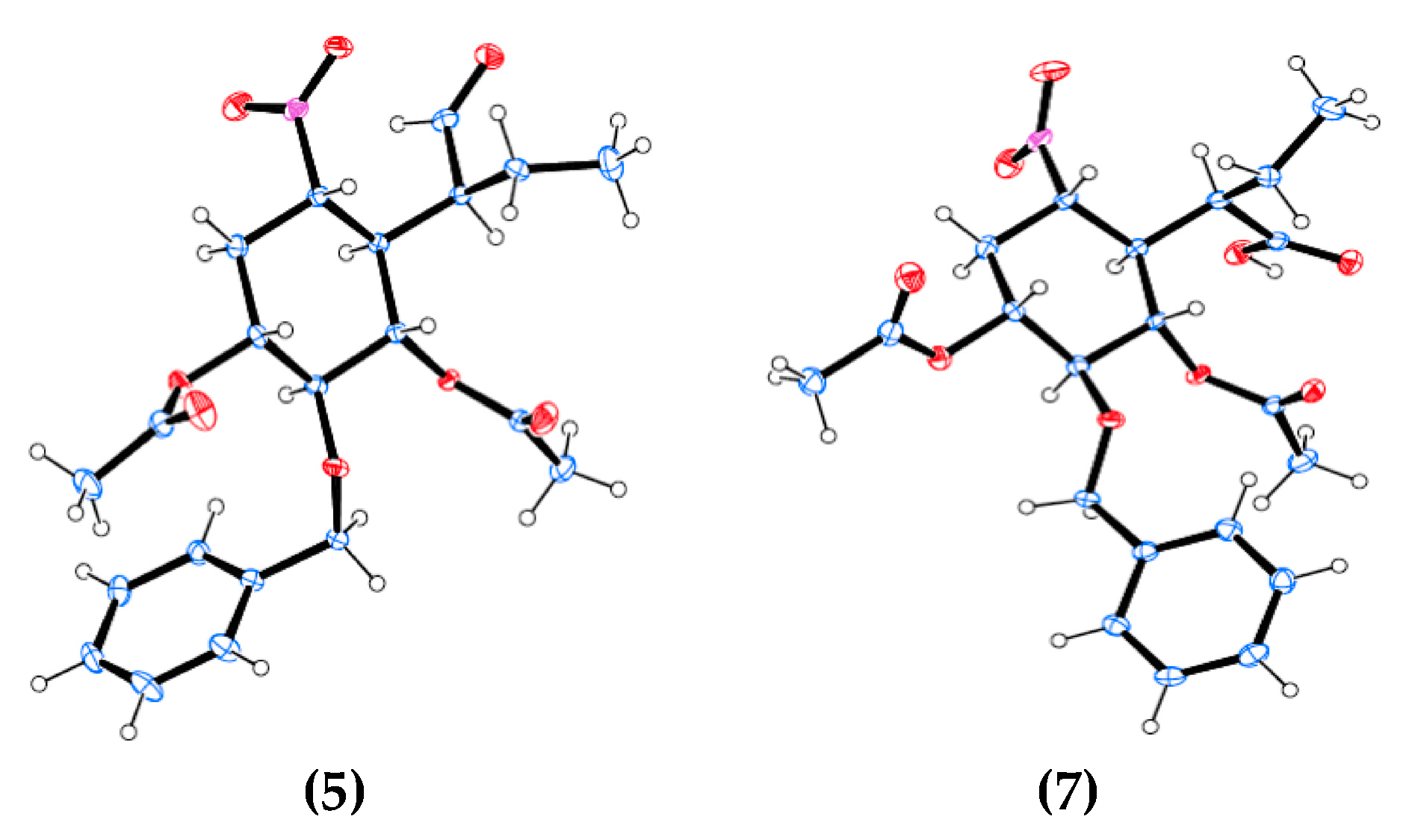
2.1. Chemical Synthesis
2.2. Glycosidase Inhibition Assays
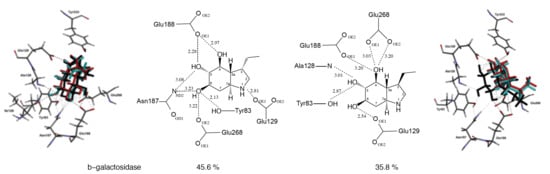
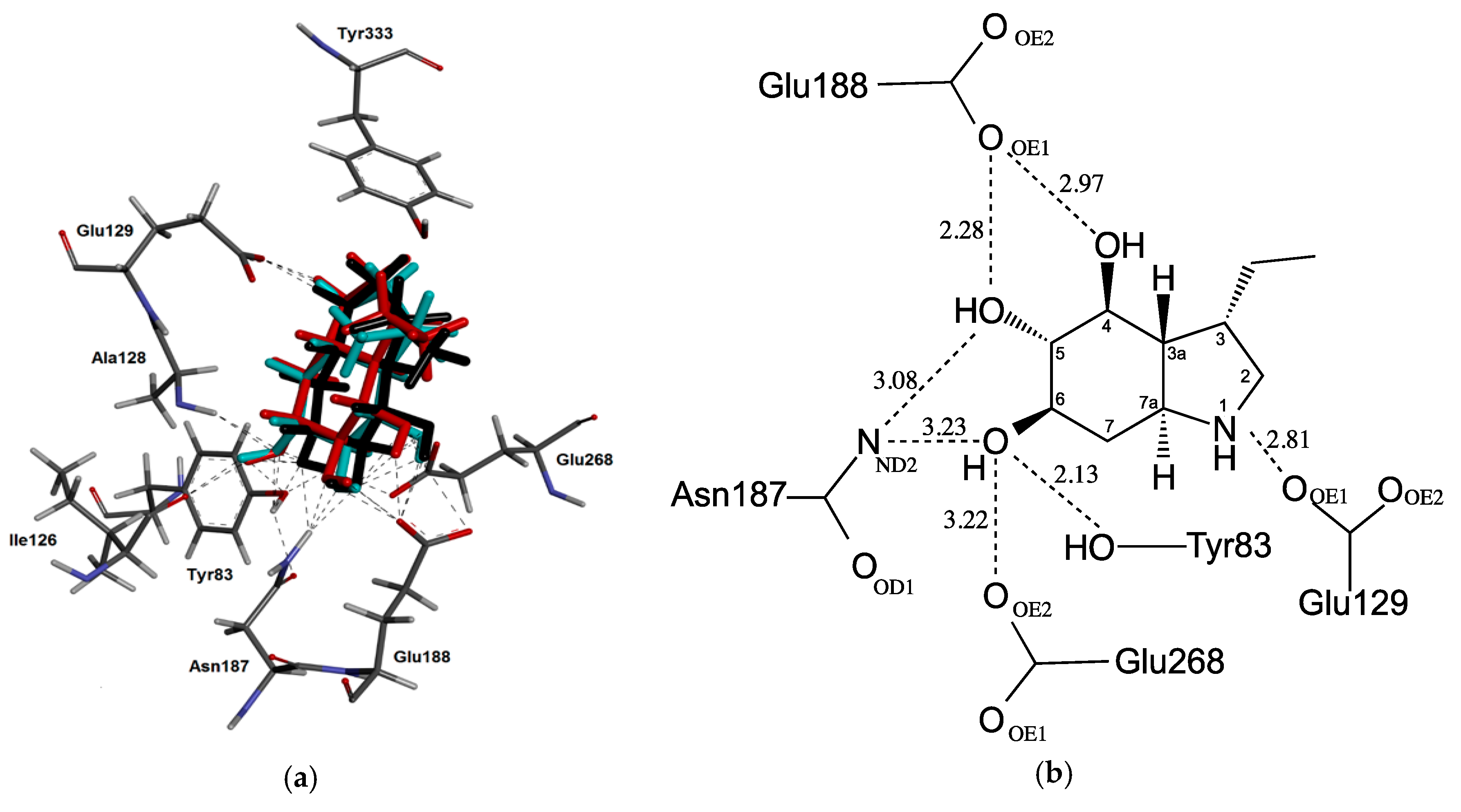
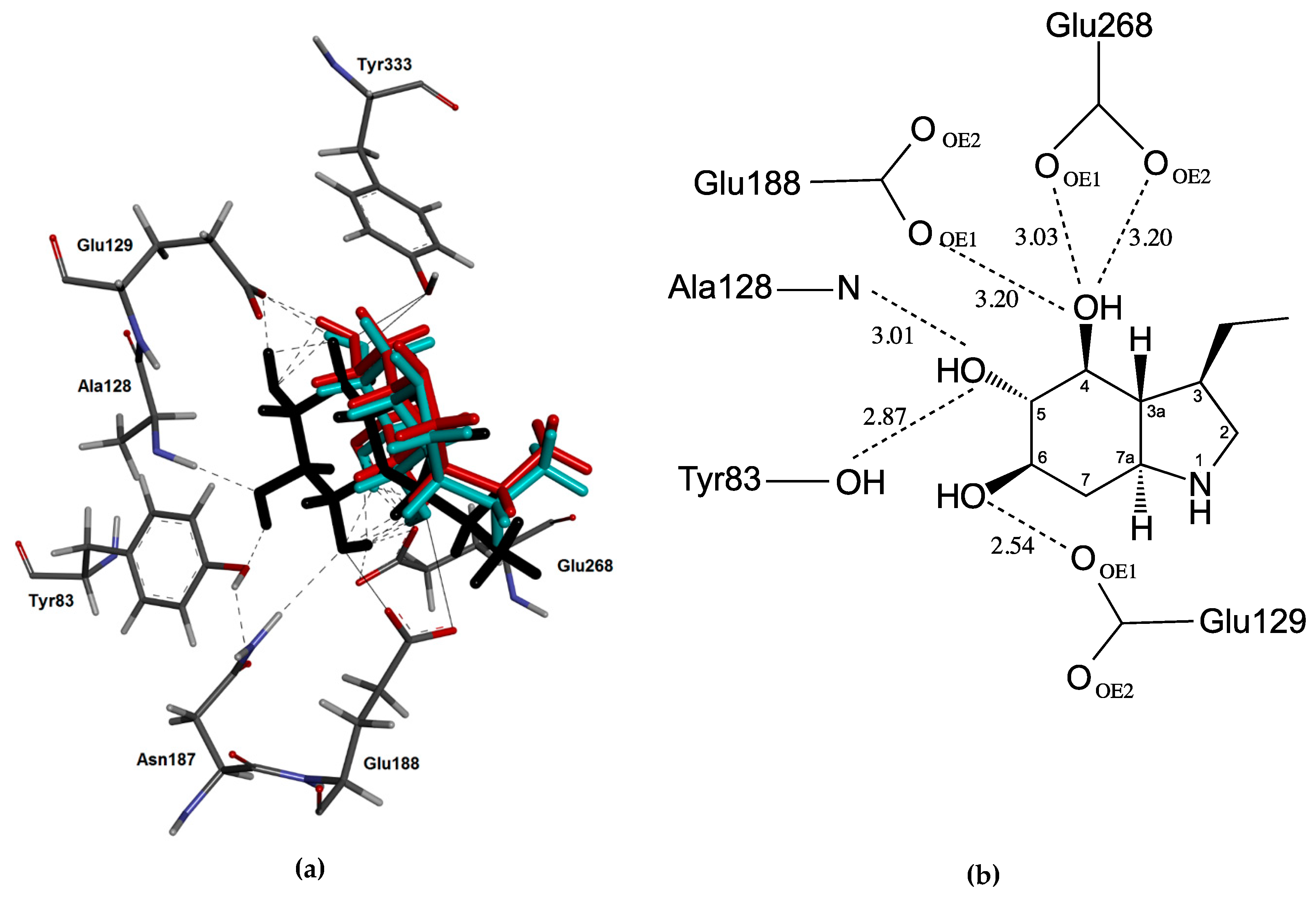
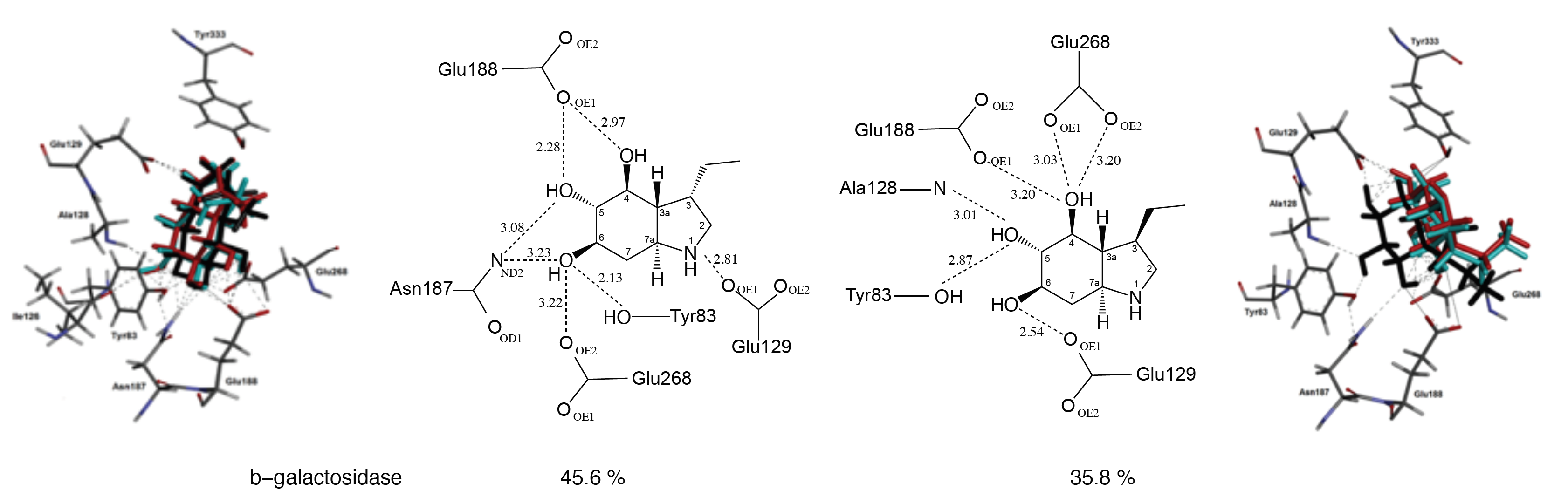
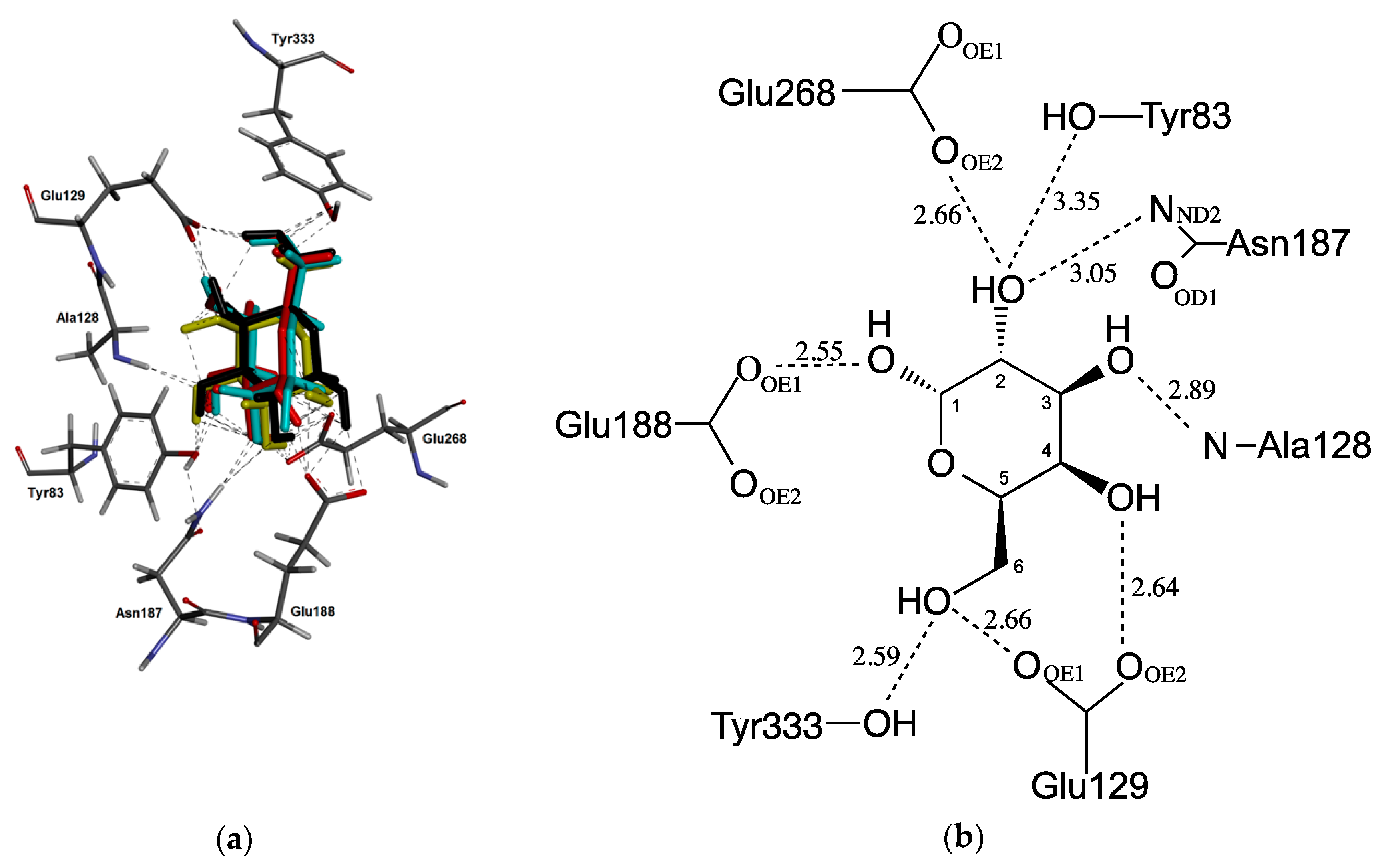
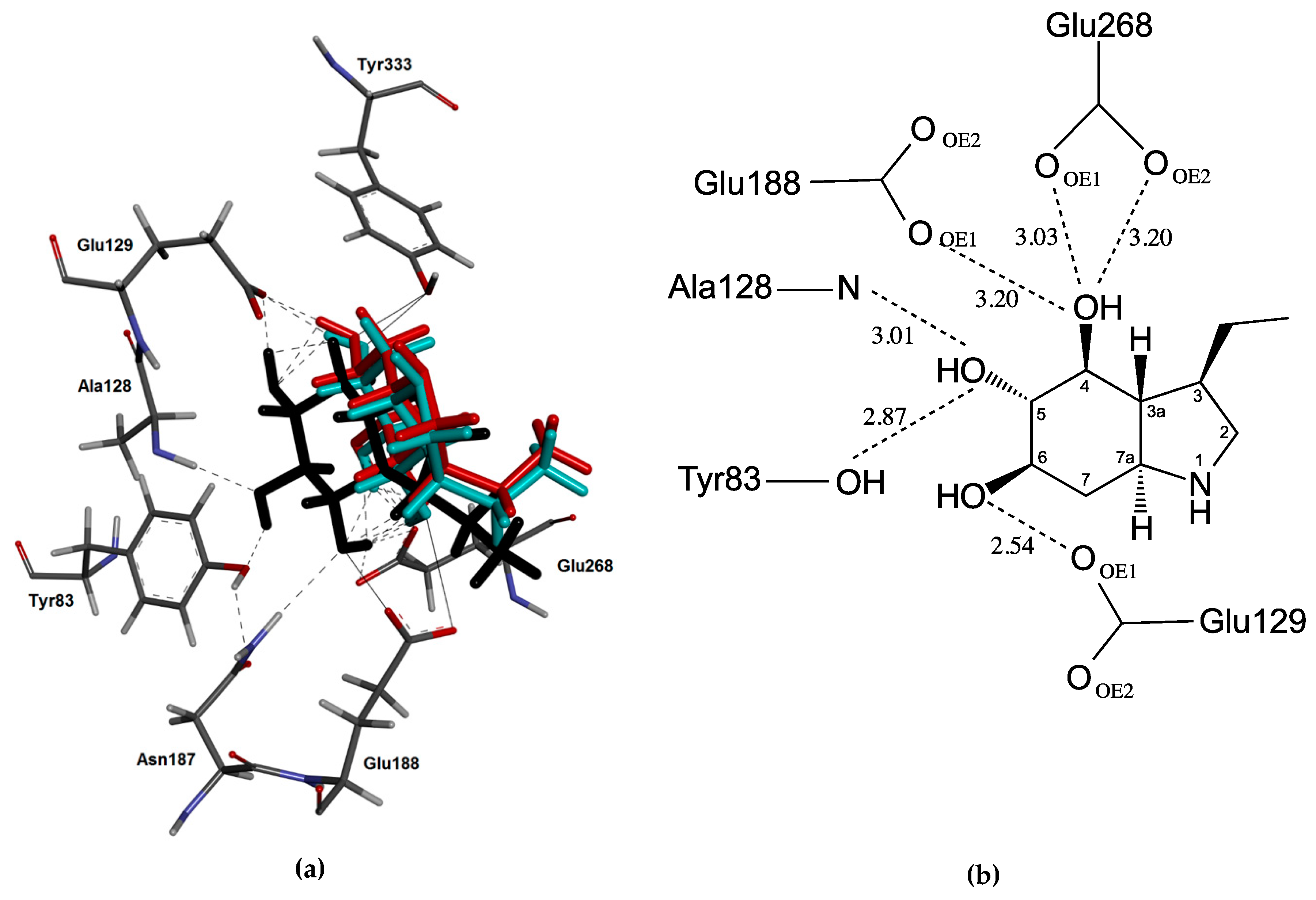
2.3. Docking Studies
3. Discussion
4. Materials and Methods
4.1. Methods for the Glycosidase Inhibition Studies
4.2. Docking Protocol
4.3. Chemical Synthesis Methods
4.3.1. (1R,2S,3S)-2-(Benzyloxy)-5-nitrocyclohex-4-ene-1,3-diyl diacetate (4b)
4.3.2. (1R,2S,3S,4R,5S)-2-(Benzyloxy)-5-nitro-4-((S)-1-oxobutan-2-yl)cyclohexane-1,3-diyl diacetate (5) and (1R,2S,3S,4R,5S)-2-(benzyloxy)-5-nitro-4-((R)-1-oxobutan-2-yl)cyclohexane-1,3-diyl diacetate (6)
4.3.3. (R)-2-((1R,2S,3S,4R,6S)-2,4-Diacetoxy-3-(benzyloxy)-6-nitrocyclohexyl)butanoic acid (7)
4.3.4. (3S,3aR,4S,5S,6R,7aS)-5-(Benzyloxy)-3-ethyloctahydro-1H-indole-4,6-diyl diacetate (8)
4.3.5. (3S,3aR,4S,5S,6R,7aS)-5-(Benzyloxy)-3-ethyloctahydro-1H-indole-4,6-diol (9)
4.3.6. (3S,3aR,4S,5S,6R,7aS)-3-Ethyloctahydro-1H-indole-4,5,6-triol (10)
4.3.7. (3R,3aR,4S,5S,6R,7aS)-5-(Benzyloxy)-3-ethyloctahydro-1H-indole-4,6-diyl diacetate (11)
4.3.8. (3R,3aR,4S,5S,6R,7aS)-5-(Benzyloxy)-3-ethyloctahydro-1H-indole-4,6-diol (12)
4.3.9. (3R,3aR,4S,5S,6R,7aS)-3-ethyloctahydro-1H-indole-4,5,6-triol (13)
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Compain, P.; Martin, O.R. (Eds.) Iminosugars: From Synthesis to Therapeutic Applications; Wiley: Hoboken, NJ, USA, 2007; ISBN 978-0-470-03391-3. [Google Scholar]
- Horne, G.; Wilson, F.X.; Tinsley, J.; Williams, D.H.; Storer, R. Iminosugars past, present and future: Medicines for tomorrow. Drug Discov. Today 2011, 16, 107–118. [Google Scholar] [CrossRef]
- Stütz, A.E. (Ed.) Iminosugars as Glycosidase Inhibitors: Nojirimycin and Beyond; Wiley-VCH: New York, NY, USA, 1999; ISBN 9783527295449. [Google Scholar]
- Harit, V.K.; Ramesh, N.G. Amino-functionalized iminocyclitols: Synthetic glycomimetics of medicinal interest. RSC Adv. 2016, 6, 109528–109607. [Google Scholar] [CrossRef]
- Arjona, O.; Gómez, A.M.; López, J.C.; Plumet, J. Synthesis and conformational and biological aspects of carbasugars. Chem. Rev. 2007, 107, 1919–2036. [Google Scholar] [CrossRef]
- Chen, X.; Fan, Y.; Zheng, Y.; Shen, Y. Properties and production of valienamine and its related analogues. Chem. Rev. 2003, 103, 1955–1977. [Google Scholar] [CrossRef] [PubMed]
- Trapero, A.; Egido-Gabás, M.; Bujons, J.; Llebaria, A. Synthesis and evaluation of hydroxymethylaminocyclitols as glycosidase inhibitors. J. Org. Chem. 2015, 80, 3512–3529. [Google Scholar] [CrossRef]
- Johnston, P.S.; Coniff, R.F.; Hoogwerf, B.J.; Santiago, J.V.; Pi-Sunyer, F.X.; Krol, A. Effects of the carbohydrase inhibitor miglitol in sulfonylurea-treated NIDDM patients. Diabetes Care 1994, 17, 20–29. [Google Scholar] [CrossRef] [PubMed]
- Campbell, L.K.; Baker, D.E.; Campbell, R.K. Miglitol: Assessment of its role in the treatment of patients with diabetes mellitus. Ann. Pharmacother. 2000, 34, 1291–1301. [Google Scholar] [CrossRef] [PubMed]
- Horii, S.; Fukase, H.; Matsuo, T.; Kameda, Y.; Asano, N.; Matsui, K. Synthesis and α-D-glucosidase inhibitory activity of N-substituted valiolamine derivatives as potential oral antidiabetic agents. J. Med. Chem. 1986, 29, 1038–1046. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Yano, M.; Miyake, S.; Ueki, Y.; Yamaguchi, Y.; Akazawa, S.; Tominaga, Y. Effects of voglibose on glycemic excursions, insulin secretion, and insulin sensitivity in non-insulin-ireated NIDDM patients. Diabetes Care 1998, 21, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Krentz, A.J.; Bailey, C.J. Oral antidiabetic agents: Current role in type 2 diabetes mellitus. Drugs 2005, 65, 385–411. [Google Scholar] [CrossRef]
- Joubert, P.H.; Venter, H.L.; Foukaridis, G.N. The effect of miglitol and acarbose after an oral glucose load: A novel hypoglycaemic mechanism? Br. J. Clin. Pharmacol. 1990, 30, 391–396. [Google Scholar] [CrossRef] [PubMed]
- Ficicioglu, C. Review of miglustat for clinical management in Gaucher disease type I. Ther. Clin. Risk Manag. 2008, 4, 425–431. [Google Scholar] [CrossRef] [PubMed]
- Elstein, D.; Hollak, C.; Aerts, J.M.F.G.; van Weely, S.; Maas, M.; Cox, T.M.; Lachmann, R.H.; Hrebicek, M.; Platt, F.M.; Butters, T.D.; et al. Sustained therapeutic effects of oral miglustat (Zavesca, N-butyldeoxynojirimycin, OGT 918) in type I Gaucher disease. J. Inherit. Metab. Dis. 2004, 27, 757–766. [Google Scholar] [CrossRef]
- Molyneux, R.J.; Roitman, J.N.; Dunnheim, G.; Szumilo, T.; Elbein, A.D. 6-Epicastanospermine, a novel indolizidine alkaloid that inhibits α-glucosidase. Arch. Biochem. Biophys. 1986, 251, 450–457. [Google Scholar] [CrossRef]
- Kang, M.S.; Liu, P.S.; Bernotas, R.C.; Harry, B.S.; Sunkara, P.S. Castanospermine analogues: Their inhibition of glycoprotein processing α-glucosidases from porcine kidney and B16F10 cells. Glycobiology 1995, 5, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Michael, J.P. Indolizidine and quinolizidine alkaloids. Nat. Prod. Rep. 1999, 16, 675–696. [Google Scholar] [CrossRef]
- Goss, P.E.; Baker, M.A.; Carver, J.P.; Dennis, J.W. Inhibitors of carbohydrate processing: A new class of anticancer agents. Clin. Cancer Res. 1995, 1, 935–944. [Google Scholar]
- Molyneux, R.J.; Pan, Y.T.; Tropea, J.E.; Benson, M.; Kaushal, G.P.; Elbein, A.D. 6,7-Diepicastanospermine, a tetrahydroxyindolizidine alkaloid inhibitor of amyloglucosidase. Biochemistry 1991, 30, 9981–9987. [Google Scholar] [CrossRef]
- Pastuszak, I.; Molyneux, R.J.; James, L.F.; Elbein, A.D. Lentiginosine, a dihydroxyindolizidine alkaloid that inhibits amyloglucosidase. Biochemistry 1990, 29, 1886–1891. [Google Scholar] [CrossRef]
- Michalik, A.; Hollinshead, J.; Jones, L.; Fleet, G.W.J.; Yu, C.-Y.; Hu, X.-G.; van Well, R.; Horne, G.; Wilson, F.X.; Kato, A.; et al. Steviamine, a new indolizidine alkaloid from Stevia rebaudiana. Phytochem. Lett. 2010, 3, 136–138. [Google Scholar] [CrossRef]
- Gravier-Pelletier, C.; Maton, W.; Le Merrer, Y. A straightforward route to indolizidine and quinolizidine analogs as new potential antidiabetics. Synlett 2003, 333–336. [Google Scholar]
- Gravier-Pelletier, C.; Maton, W.; Bertho, G.; Le Merrer, Y. Synthesis and glycosidase inhibitory activity of enantiopure polyhydroxylated octahydroindoles and decahydroquinolines, analogs to castanospermine. Tetrahedron 2003, 59, 8721–8730. [Google Scholar] [CrossRef]
- González, M.A.; Estévez, A.M.; Campos, M.; Estévez, J.C.; Estévez, R.J. Protocol for the incorporation of γ-amino acids into peptides: Application to (−)-shikimic acid based 2-aminomethyl- cyclohexanecarboxylic acids. J. Org. Chem. 2018, 83, 1543–1550. [Google Scholar] [CrossRef]
- Suzuki, H.; Ohto, U.; Higaki, K.; Mena-Barragán, T.; Aguilar-Moncayo, M.; Ortiz Mellet, C.; Nanba, E.; Garcia-Fernandez, J.M.; Suzuki, Y.; Shimizu, T. Structural basis of pharmacological chaperoning for human β-Galactosidase. J. Biol. Chem. 2014, 289, 14560–14568. [Google Scholar] [CrossRef] [PubMed]
- Ono, N. The Nitro Group in Organic Synthesi; Wiley-VCH: Weinheim, Germany, 2001; ISBN 0-471-22448-0. [Google Scholar]
- Guo, L.; Chi, Y.; Almeida, A.M.; Guzei, I.A.; Parker, B.K.; Gellman, S.H. Stereospecific synthesis of conformationally constrained γ-amino acids: New foldamer building blocks that support helical secondary structure. J. Am. Chem. Soc. 2009, 131, 16018–16020. [Google Scholar] [CrossRef]
- Bhorkade, S.B.; Gavhane, K.B. Multigram synthesis of an advanced nitroalkene intermediate: Application in synthesis of octahydroindol-2-one derivative featuring diastereoselective Michael addition of diethylmalonate. Tetrahedron Lett. 2016, 57, 2575–2578. [Google Scholar] [CrossRef]
- Ballini, R.; Palestini, C. A new, highly efficient synthess of conjugated nitrocycloalkenes. Tetrahedron Lett. 1994, 35, 5731–5734. [Google Scholar] [CrossRef]
- Otero, J.M.; Barcia, J.C.; Salas, C.O.; Thomas, P.; Estévez, J.C.; Estévez, R.J. Studies on the Michael addition of naphthoquinones to sugar nitro olefins: First synthesis of polyhydroxylated hexahydro-11H-benzo[a]carbazole-5,6-diones and hexahydro-11bH-benzo[b]carbazole-6,11-diones. Tetrahedron 2012, 68, 1612–1621. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Jablonska, J.; Pravda, L.; Varekova, R.S.; Thornton, J.M. PDBsum: Structural summaries of PDB entries. Protein Sci. 2018, 27, 129–134. [Google Scholar] [CrossRef]
- Fan, J.-Q. A counterintuitive approach to treat enzyme deficiencies: Use of enzyme inhibitors for restoring mutant enzyme activity. Biol. Chem. 2008, 389, 1–11. [Google Scholar] [CrossRef]
- Ohto, U.; Usui, K.; Ochi, T.; Yuki, K.; Satow, Y.; Shimizu, T. Crystal structure of human β-galactosidase. Structural basis of GM1 gangliosidosis and Morquio B diseases. J. Biol. Chem. 2012, 287, 1801–1812. [Google Scholar] [CrossRef] [PubMed]
- Kessler, M.; Acuto, O.; Storelli, C.; Murer, H.; Müller, M.; Semenza, G. A modified procedure for the rapid preparation of efficiently transporting vesicles from small intestinal brush border membranes. Their use in investigating some properties of D-glucose and choline transport systems. Biochim. Biophys. Acta 1978, 506, 136–154. [Google Scholar] [CrossRef]
- GOLD.; Version 5.1; Cambridge Crystallographic Data Centre: Cambridge, UK, 2011.
- Jones, G.; Willett, P.; Glen, R.C. Molecular recognition of receptor sites using a genetic algorithm with a description of desolvation. J. Mol. Biol. 1995, 245, 43–53. [Google Scholar] [CrossRef]
- Jones, G.; Willett, P.; Glen, R.C.; Leach, A.R.; Taylor, R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baxter, C.A.; Murray, C.W.; Clark, D.E.; Westhead, D.R.; Eldridge, M.D. Flexible docking using Tabu search and an empirical estimate of binding affinity. Proteins 1998, 33, 367–382. [Google Scholar] [CrossRef]
- Eldridge, M.D.; Murray, C.W.; Auton, T.R.; Paolini, G.V.; Mee, R.P. Empirical scoring functions: I. The development of a fast empirical scoring function to estimate the binding affinity of ligands in receptor complexes. J. Comput. Aided Mol. Des. 1997, 11, 425–445. [Google Scholar] [CrossRef] [PubMed]
- Verdonk, M.L.; Cole, J.C.; Hartshorn, M.J.; Murray, C.W.; Taylor, R.D. Improved protein-ligand docking using GOLD. Proteins 2003, 52, 609–623. [Google Scholar] [CrossRef] [Green Version]
- Korb, O.; Stützle, T.; Exner, T.E. Empirical scoring functions for advanced protein-ligand docking with PLANTS. J. Chem. Inf. Model. 2009, 49, 84–96. [Google Scholar] [CrossRef]
- Discovery Studio, Versions 2.1 and 2.5; Acceelrys Inc.: San Diego, CA, USA, 2009.
- Perrin, D.D.; Armarego, W.L.F. Purification of Laboratory Chemicals; Pergamon: New York, NY, USA, 1988. [Google Scholar]
Sample Availability: Samples of the compounds 10 and 13 are available from the authors. |



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Compound 10 | Compound 13 | Miglitol (IC50) |
|---|---|---|---|
| α-Glucosidase | |||
| Yeast | 16% a | 19.5% a | 70 μM |
| Rice | 0% a | 0% a | 0.17 μM |
| Rat intestinal maltase | 4.1% a | 0% a | >1000 μM |
| β-Glucosidase | |||
| Almond | 12% a | 5% a | >1000 μM |
| Bovine liver | 0% a | 21.7% a | >1000 μM |
| α-Galactosidase | |||
| Coffee beans | 15.9% a | 0% a | >1000 μM |
| 0% a | 21.7% a | ||
| β-Galactosidase | |||
| Bovine liver | 45.6% a | 35.8% a | >1000 μM |
| Lactase | 0.6% a | 7.4% a | >1000 μM |
| α-Mannosidase | |||
| Jack bean | 0% a | 0% a | |
| β-Mannosidase | |||
| Snail | 0% a | 2.3% a | >1000 μM |
| α-L-Fucosidase | |||
| Bovine kidney | 7.5% a | 12.9% a | >1000 μM |
| α-L-Rhamnosidase | |||
| Penicillium decumbens | 0% a | 0% a | 803 |
| β-Glucronidase | |||
| E.coli | 18.1% a | 12.3% a | >1000 μM |
| α,α-Trehalase | |||
| Porcine kidney | 0% a | 0% a | 131 |
| Amyloglucosidase | |||
| Aspergillus niger | 0% a | 0% a | >1000 μM |
| Ligand | Protein Residue HB | Fitness & Search Options a | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Early Termination | Diverse Solutions | Side Flexible Search | |||||||||
| CHEM | GOLD | PLP | CHEM | GOLD | PLP | CHEM | GOLD | PLP | PDB | ||
| Galactose | Galactose score | 21.88 | 54.49 | 55.63 | 18.57 | 50.17 | 52.84 | 12.57 | 53.60 | 42.29 | - |
| Tyr83(OH) | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | |||
| Ala128(N) | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | 3-OH | |||||
| Glu129(OE1) | 6-OH | 6-OH | 4,6-OH | 6-OH | 4-OH | 4,6-OH | 4,6-OH | 4-OH | 4-OH | 6-OH | |
| Glu129(OE2) | 4-OH | 4-OH | 4-OH | 4-OH | 4-OH | 4-OH | 6-OH | 6-OH | 4-OH | ||
| Asn187(ND2) | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | |||
| Asn187(OD) | 2-OH | ||||||||||
| Glu188(OE1) | 1-OH | 1-OH | 1-OH | 1-OH | 1-OH | 1-OH | 1-OH | 1-OH | 1-OH | ||
| Glu188(OE2) | 1-OH | 1-OH | 1-OH | 1,2-OH | 1-OH | ||||||
| Glu268(OE1) | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | |||||
| Glu268(OE2) | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | 2-OH | |||
| Tyr333(OH) | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | ||
| 10 | 10 score | 27.58 | 44.31 | 49.82 | 27.19 | 44.25 | 45.78 | 20.47 | 52.90 | 38.17 | |
| Tyr83(OH) | 6-OH | 6-OH | 6-OH | 6-OH | 5-OH | 6-OH | 6-OH | 5-OH | 6-OH | ||
| Ile126(O) | 6-OH | 6-OH | 6-OH | 5-OH | 6-OH | ||||||
| Ala128(N) | 6-OH | 6-OH | 5-OH | 6-OH | |||||||
| Glu129(OE1) | N | N | N | N | 6-OH | N | N | ||||
| Glu129(OE2) | N | 6-OH | N | ||||||||
| Asn187(ND2) | 5-OH | 5,6-OH | 5-OH | 5-OH | 5,6-OH | 4-OH | |||||
| Asn187(OD) | 5,6-OH | 4-OH | 5,6-OH | ||||||||
| Glu188(OE1) | 4,5-OH | 4,5-OH | 4,5-OH | 4,5-OH | 4,5-OH | 4,5-OH | 4,5-OH | ||||
| Glu188(OE2) | 4-OH | 4-OH | |||||||||
| Glu268(OE1) | 4-OH | 4-OH | 4-OH | ||||||||
| Glu268(OE2) | 6-OH | 6-OH | 6-OH | ||||||||
| Tyr333(OH) | N | N | |||||||||
| 13 | 13 score | 25.37 | 46.53 | 43.62 | 25.53 | 45.76 | 43.10 | 17.65 | 56.23 | 39.34 | |
| Tyr83(OH) | 5-OH | 5,6-OH | 5-OH | 6-OH | 6-OH | ||||||
| Ile126(O) | |||||||||||
| Ala128(N) | 5-OH | 6-OH | |||||||||
| Glu129(OE1) | 6-OH | 6-OH | 6-OH | 6-OH | 6-OH | 4-OH | N | ||||
| Glu129(OE2) | N | 5-OH | |||||||||
| Asn187(ND2) | 5-OH | 6-OH | 5-OH | ||||||||
| Asn187(OD) | 6-OH | 5-OH | |||||||||
| Glu188(OE1) | 4-OH | 4-OH | N | ||||||||
| Glu188(OE2) | 4-OH | 4-OH | 4-OH | 5-OH | 4-OH | ||||||
| Glu268(OE1) | 4,5-OH | 4-OH | 4,5-OH | 4-OH | 4,5-OH | 4,5-OH | 6-OH | 5-OH | |||
| Glu268(OE2) | 4-OH | 5-OH | 4,5-OH | ||||||||
| Tyr333(OH) | 6-OH | 6-OH | 6-OH | ||||||||
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Estévez, J.C.; González, M.A.; Villaverde, M.C.; Hirokami, Y.; Kato, A.; Sussman, F.; Reza, D.; Estévez, R.J. Chain-Branched Polyhydroxylated Octahydro-1H-Indoles as Potential Leads against Lysosomal Storage Diseases. Pharmaceuticals 2019, 12, 47. https://doi.org/10.3390/ph12020047
Estévez JC, González MA, Villaverde MC, Hirokami Y, Kato A, Sussman F, Reza D, Estévez RJ. Chain-Branched Polyhydroxylated Octahydro-1H-Indoles as Potential Leads against Lysosomal Storage Diseases. Pharmaceuticals. 2019; 12(2):47. https://doi.org/10.3390/ph12020047
Chicago/Turabian StyleEstévez, Juan C., Marcos A. González, M. Carmen Villaverde, Yuki Hirokami, Atsushi Kato, Fredy Sussman, David Reza, and Ramón J. Estévez. 2019. "Chain-Branched Polyhydroxylated Octahydro-1H-Indoles as Potential Leads against Lysosomal Storage Diseases" Pharmaceuticals 12, no. 2: 47. https://doi.org/10.3390/ph12020047
APA StyleEstévez, J. C., González, M. A., Villaverde, M. C., Hirokami, Y., Kato, A., Sussman, F., Reza, D., & Estévez, R. J. (2019). Chain-Branched Polyhydroxylated Octahydro-1H-Indoles as Potential Leads against Lysosomal Storage Diseases. Pharmaceuticals, 12(2), 47. https://doi.org/10.3390/ph12020047