In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations

 , ,
, ,  , ,
, ,  ,
,
Abstract
1. Introduction
2. Results and Discussion
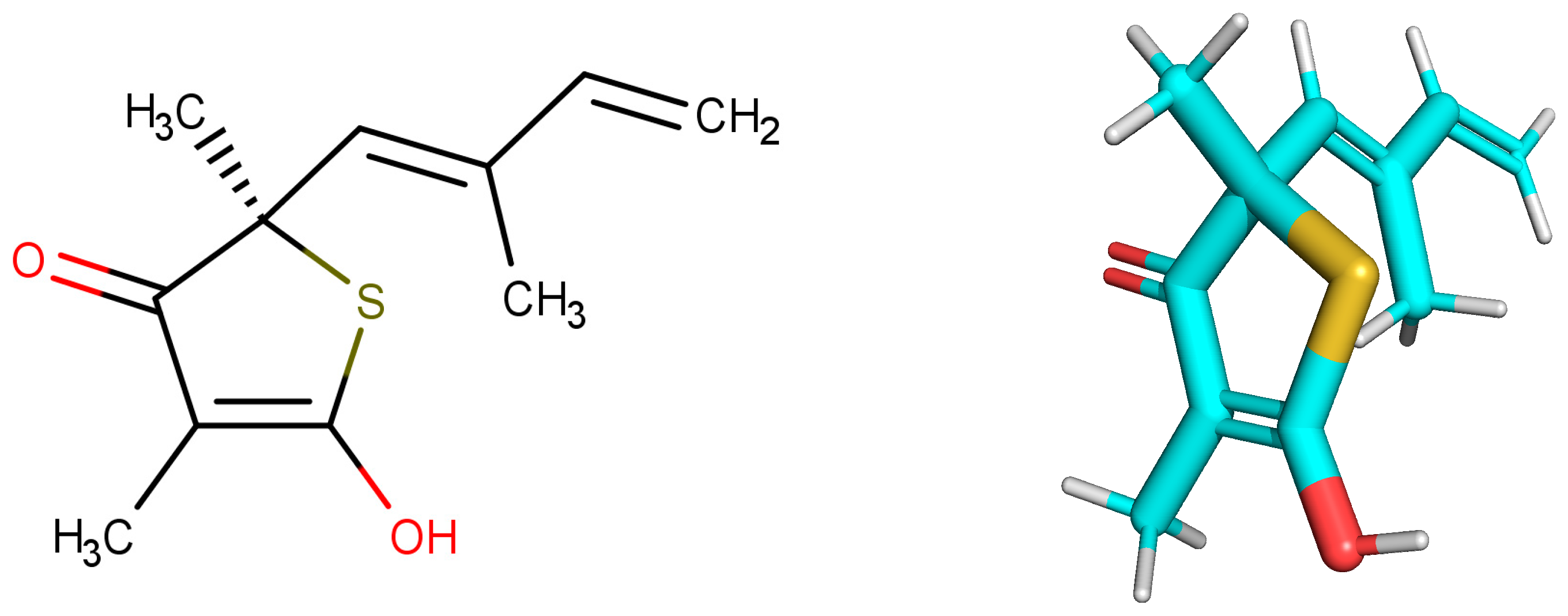
2.1. Selected Compounds that Exhibit Biological Activity with Target
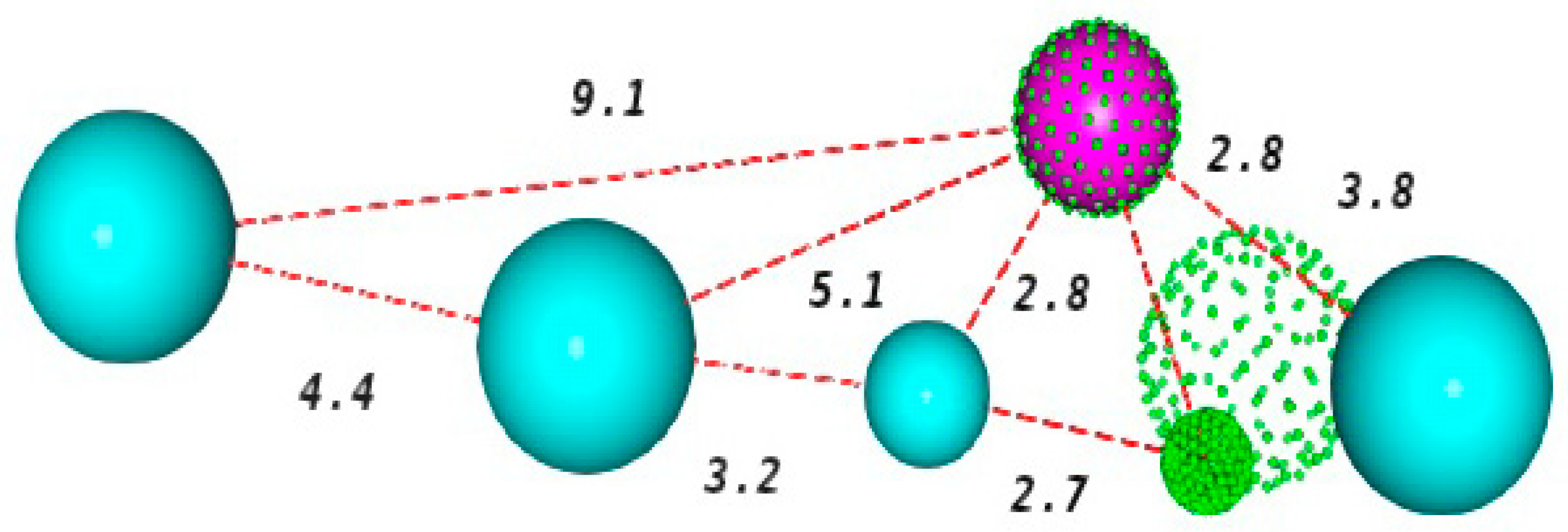
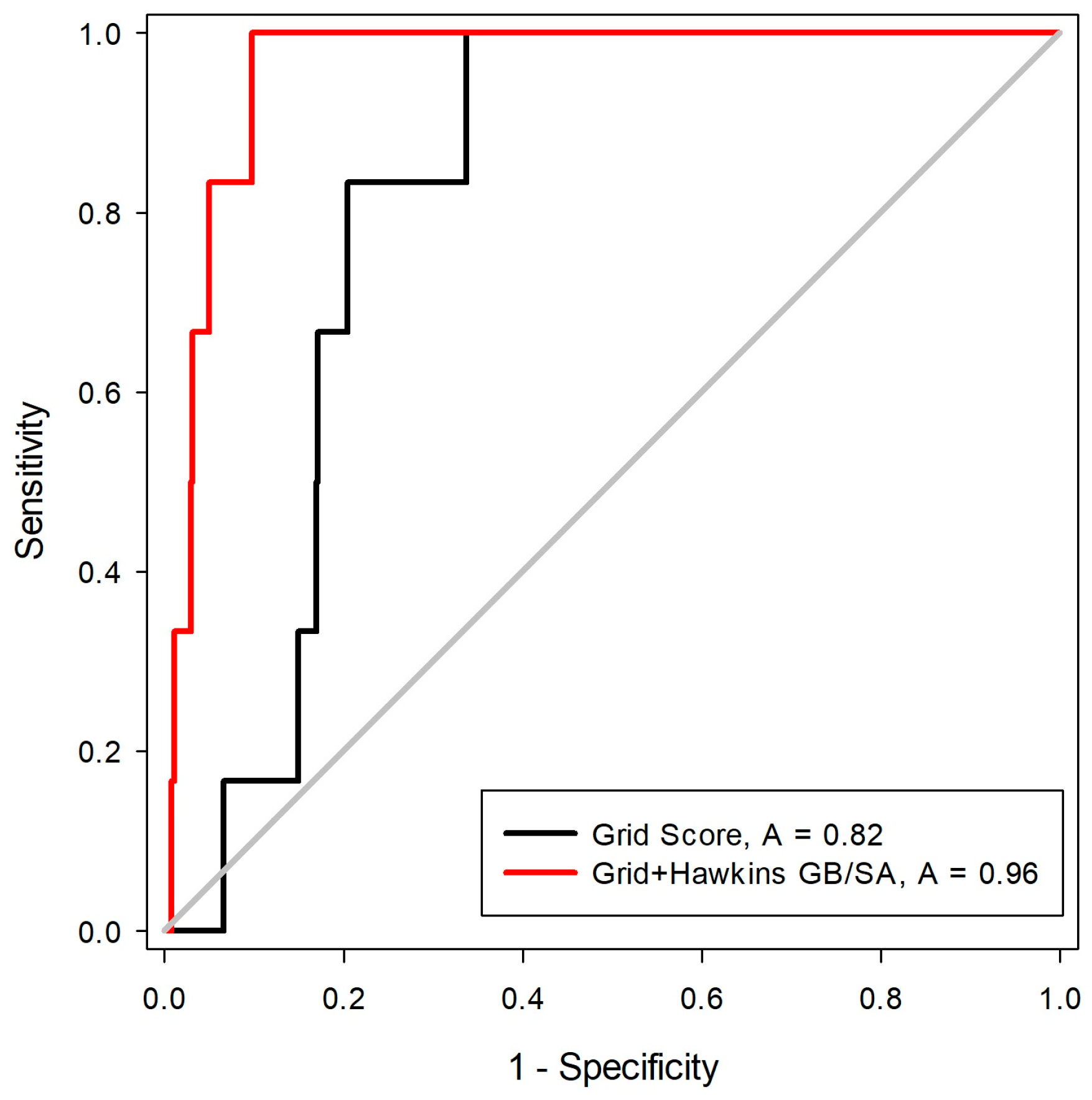
2.2. Construction and Evaluation of Pharmacophore Models
Pharmacophore-Based Virtual Screening
2.3. Docking-Based Virtual Screening
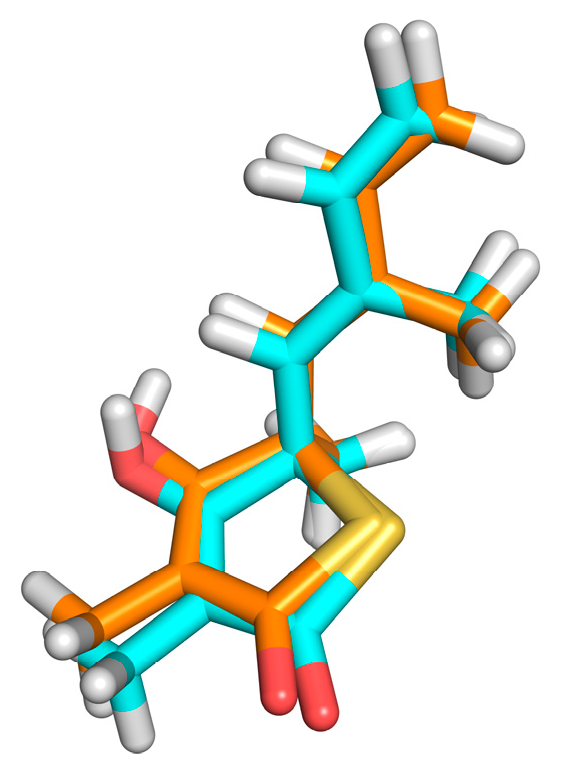
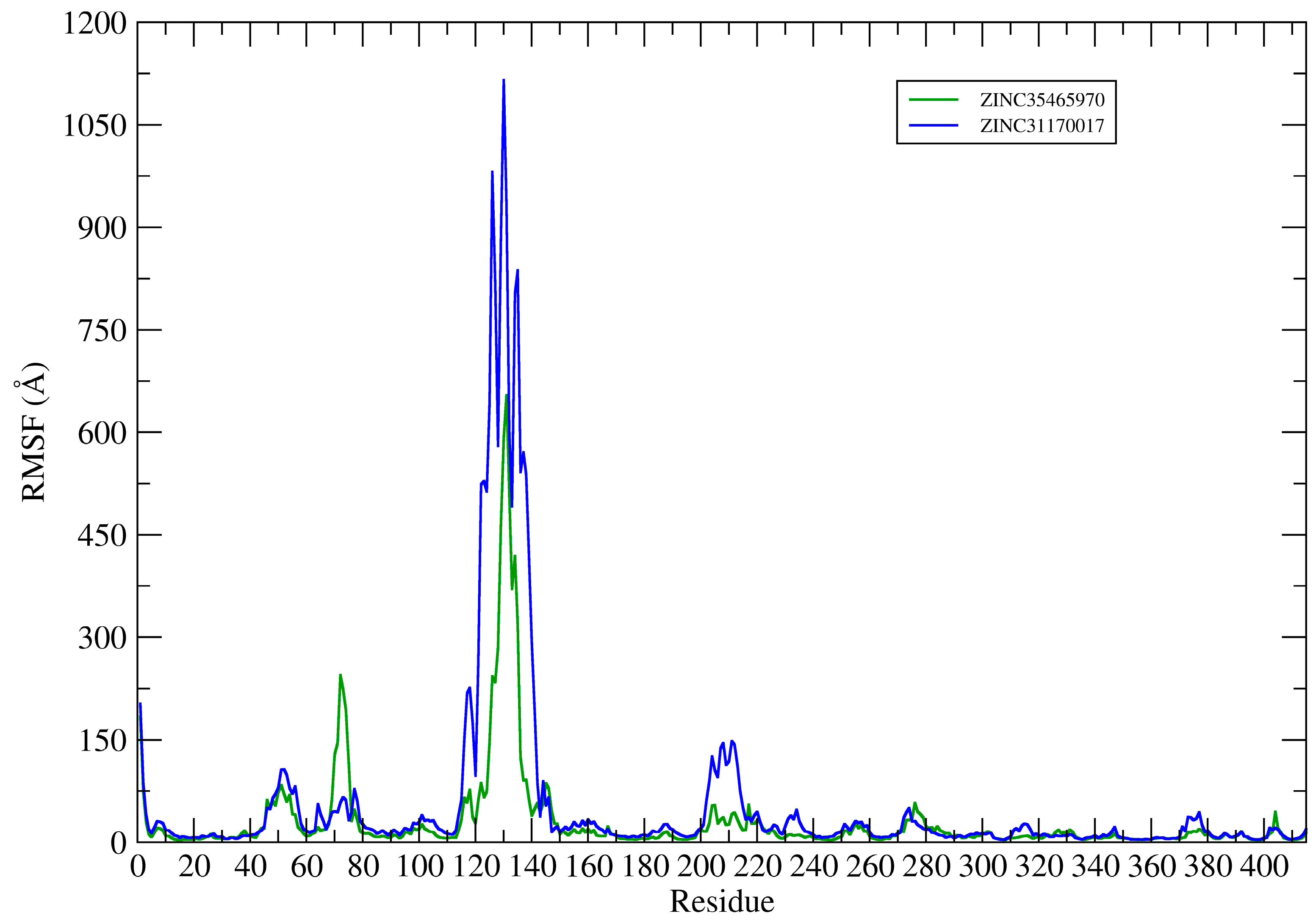
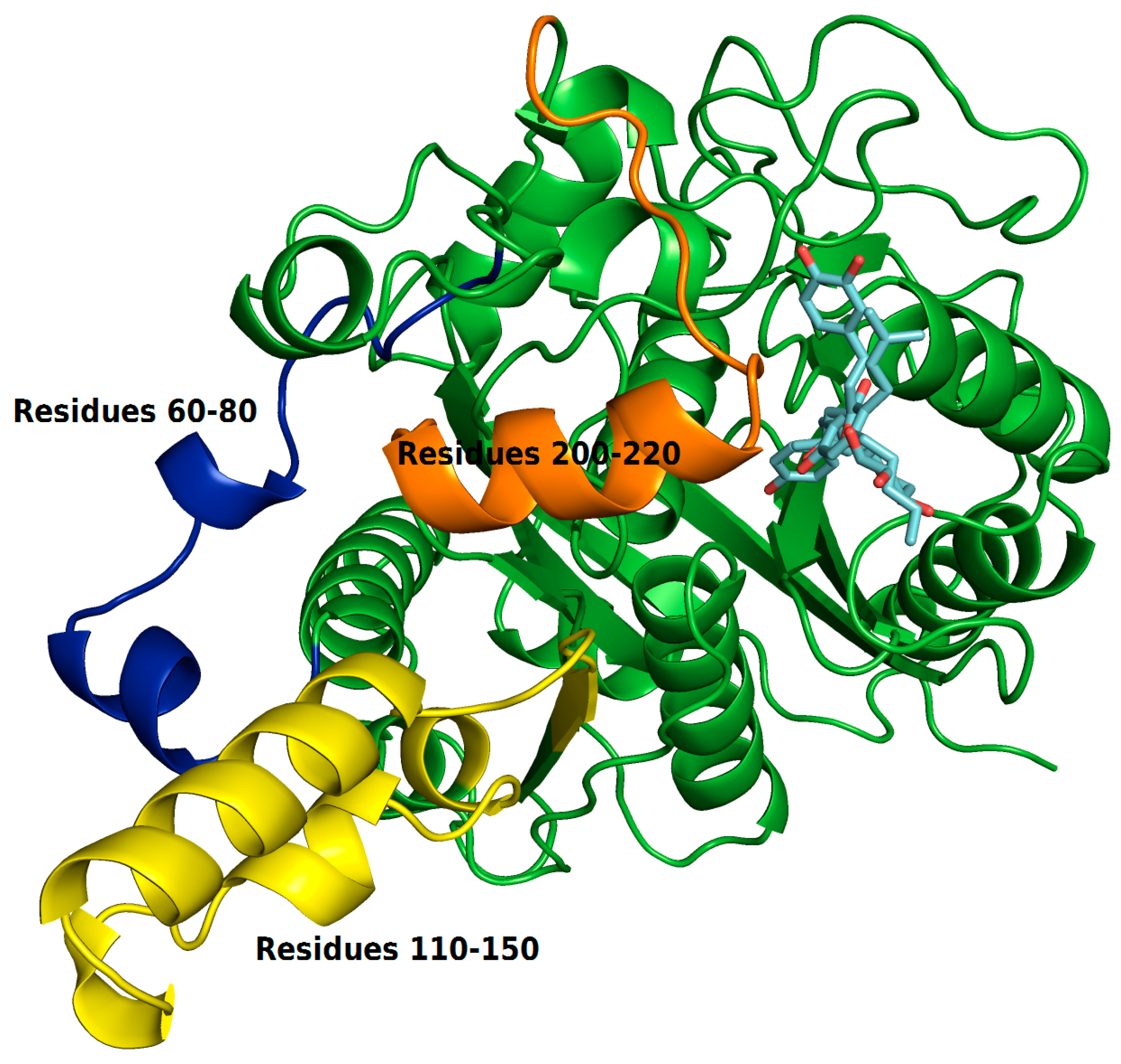
2.4. Structural Analysis of Systems
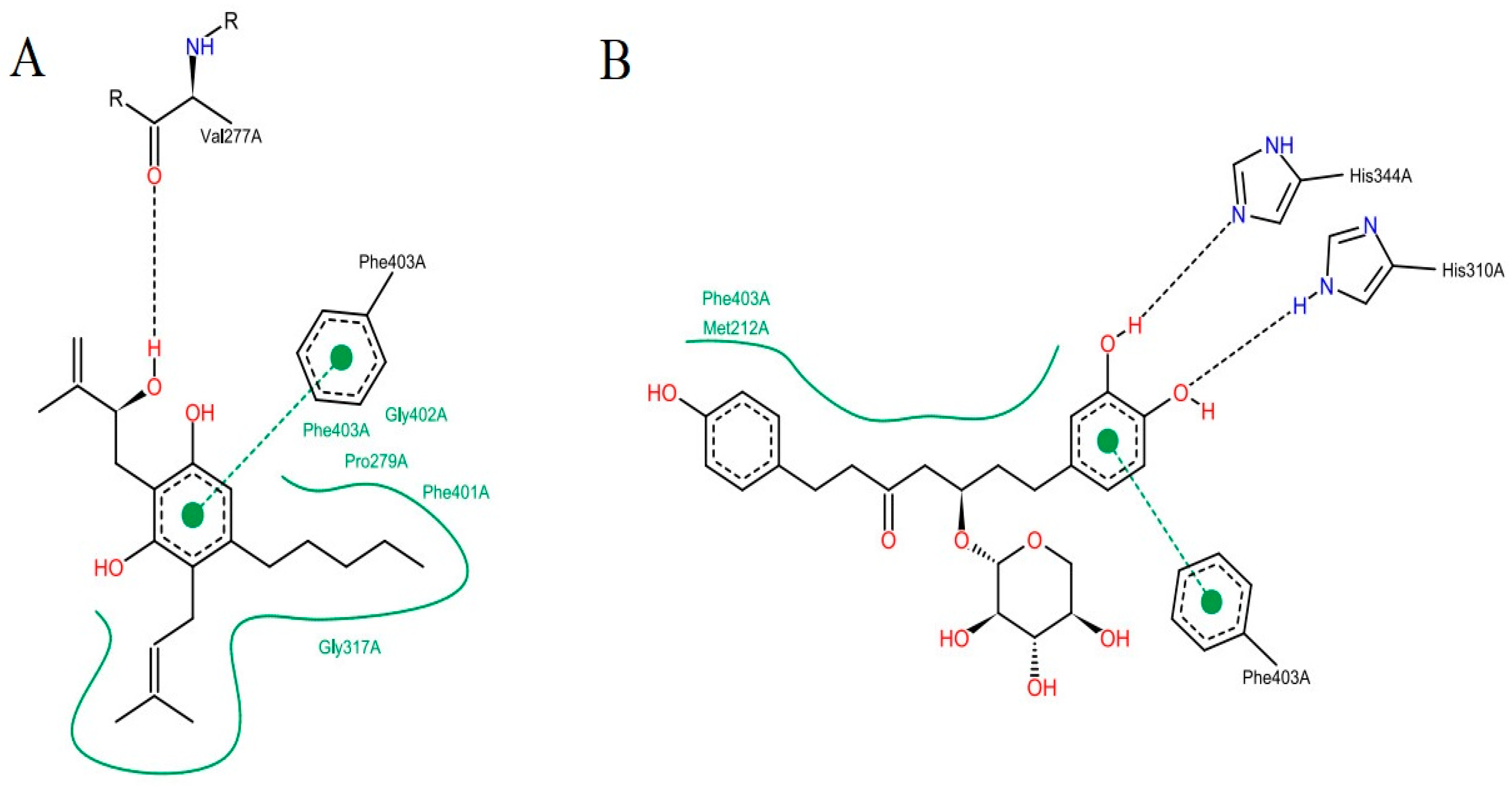
2.4.1. Hydrogen Bonds Established between Receptor-Ligands
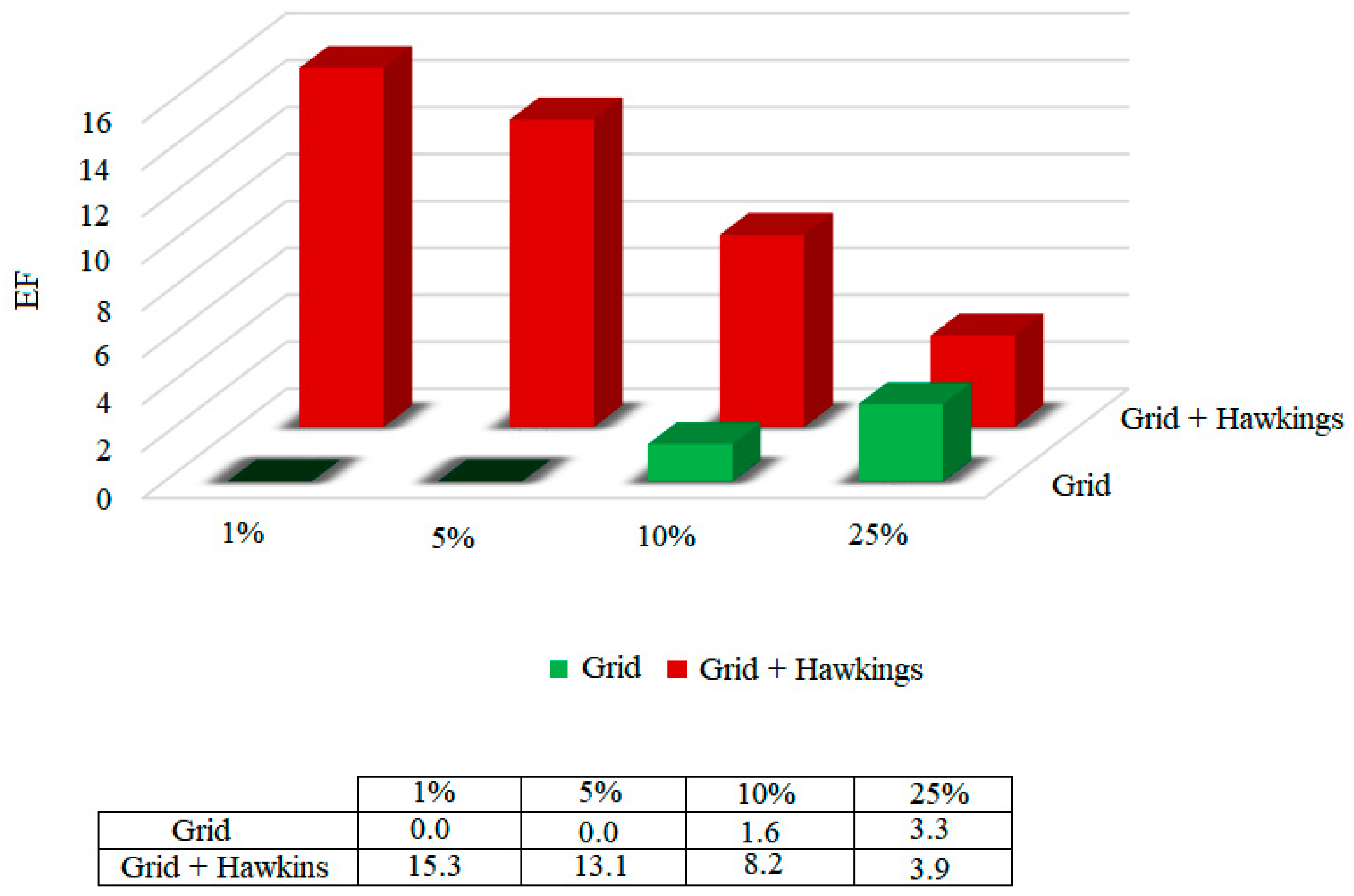
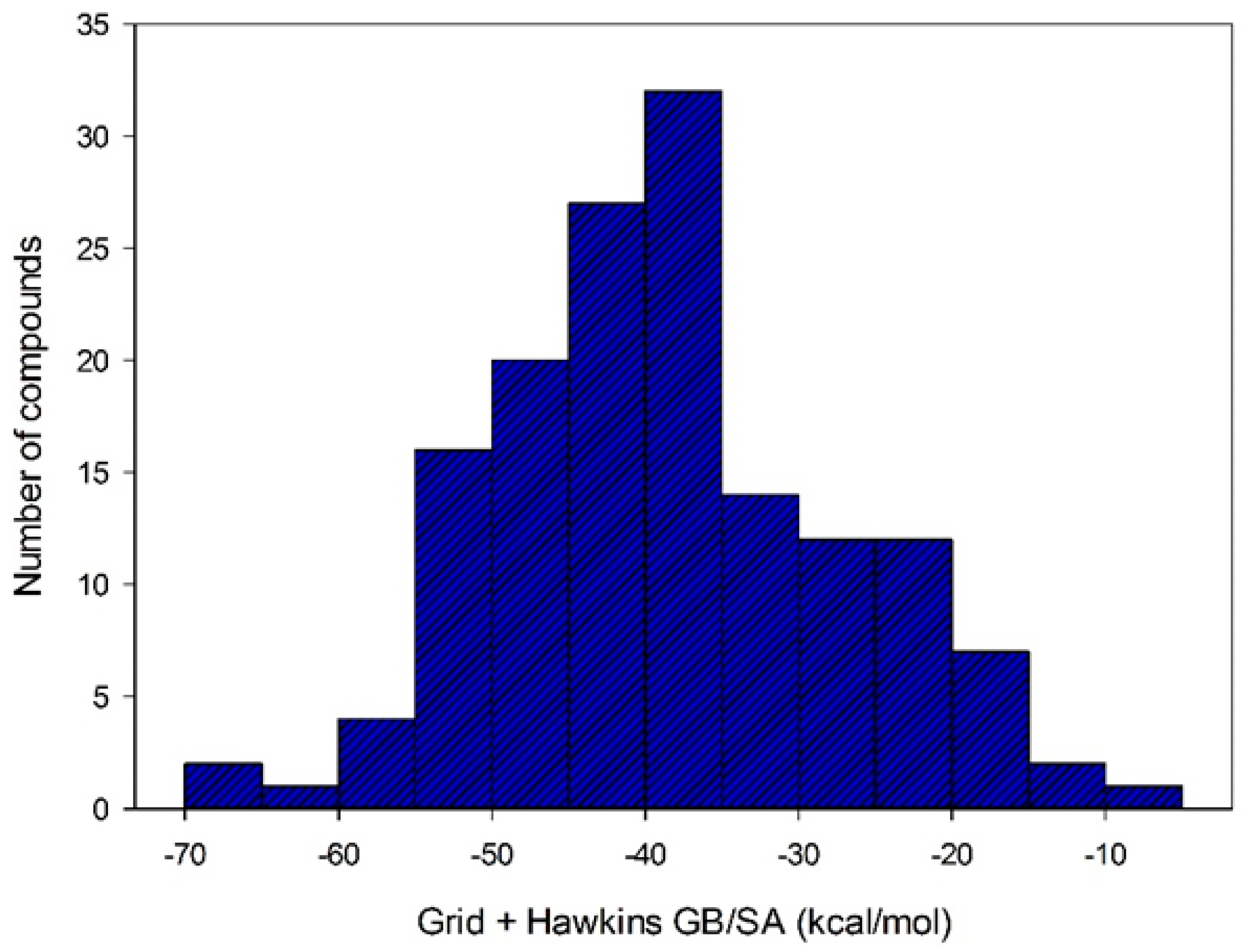
2.4.2. Bind Free Energy KasA-Ligands
3. Materials and Methods
3.1. Dataset
3.2. Pharmacophore Models Construction
Pharmacophore-Based Virtual Screening
3.3. Docking-Based Virtual Screening
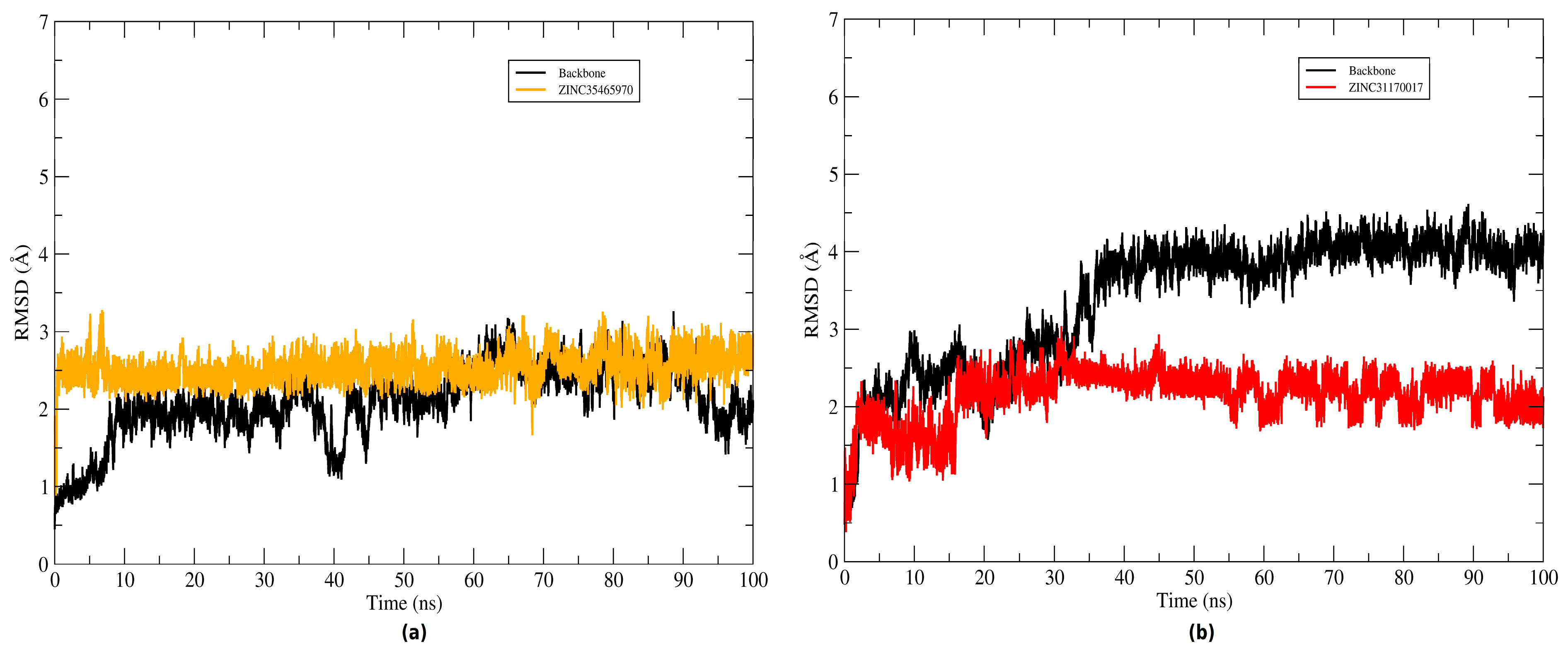
3.4. Molecular Dynamics (MD) Simulations
Binding Free Energy Calculations
4. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mello, F.C.Q.; Silva, D.R.; Dalcolmo, M.P. Tuberculosis: Where are we? J. Bras. Pneumol. 2018, 44, 82. [Google Scholar] [CrossRef]
- World Health Organization. Global Tuberculosis Report; WHO: Geneva, Switzerland, 2017. [Google Scholar]
- Santos, D.S.; Vasconselos, I.B.; Meyer, E.; Sales, F.A.M.; Moreira, I.S.; Basso, L.A. The mode of inhibition of Mycobacterium tuberculosis wild-type and isoniazid-resistant 2-trans-enoyl-ACP(CoA) reductase enzymes by an inorganic complex. Anti-Infect. Agents Med. Chem. 2008, 7, 50–62. [Google Scholar] [CrossRef]
- Moreira, T.R.; Zandonade, E.; Maciel, E.L.N. Risco de infecção tuberculosa em agentes comunitários de saúde. Rev. Saúde Pública 2010, 44, 332–338. [Google Scholar] [CrossRef]
- Antonova, A.V.; Gryadunov, V.A.; Zimenkov, D.V. Molecular mechanisms of drug tolerance in Mycobacterium tuberculosis. Mol. Biol. (Mosk.) 2018, 52, 435–450. [Google Scholar] [CrossRef]
- Ananthan, S.; Faaleolea, E.R.; Goldman, R.C.; Hobrath, J.V.; Kwong, C.D.; Laughon, B.E.; Maddry, J.A.; Mehta, A.; Rasmussen, L.; Reynolds, R.C.; et al. High-throughput screening for inhibitors of Mycobacterium tuberculosis H37Rv. Tuberculosis 2009, 89, 334–353. [Google Scholar] [CrossRef]
- Ikram, A.I.; Satti, M.L.; Lalani, F.K.; Zaman, G.; Gardezi, A.H.; Ahmed, P. Evaluation of nitrate reductase assay for early detection of multi and extensively drug resistance tuberculosis in our setup. J. Coll. Physicians Surg. Pak. 2018, 28, 22–25. [Google Scholar] [CrossRef]
- Miranda, T.F.N.; Santos Junior, M.C. Identificação de potenciais inibidores da beta-cetoacil sintase do Mycobacterium tuberculosis por triagem virtual. Rev. Virtual. Quim. 2016, 8, 1740–1756. [Google Scholar] [CrossRef]
- Alves, V.M.; Braga, R.C.; Muratov, E.N.; Andrade, C.H. Quimioinformática: Uma introdução. Quím. Nova 2018, 41, 202–212. [Google Scholar] [CrossRef]
- Kumar, V.; Krishna, S.A.; Siddiqi, M.I. Virtual screening strategies: Recent advances in the identification and design of anti-cancer agents. Methods 2014, 71, 64–70. [Google Scholar] [CrossRef]
- Silva, R.C.; Poiani, J.G.C.; Ramos, R.S.; Costa, J.S.; Silva, C.H.T.P.; Brasil, D.S.B.; Santos, C.B.R. Ligand- and structure-based virtual screening from 16-(N,N-diisobutylaminomethyl)-6α-hydroxyivouacapan-7β,17β-lactone compound with potential anti-prostate cancer activity. J. Serb. Chem. Soc. 2018, 83, 1–23. [Google Scholar] [CrossRef]
- Bhatt, A.; Molle, V.; Besra, G.S.; Jacobs, W.R., Jr.; Kremer, L. The Mycobacterium tuberculosis FAS-II condensing enzymes: Their role in mycolic acid biosynthesis, acid-fastness, pathogenesis and in future drug development. Mol. Microbiol. 2007, 64, 1442–1454. [Google Scholar] [CrossRef]
- Zhang, H.J.; Li, Z.L.; Zhu, H.L. Advances in the Research of β-Ketoacyl-ACP Synthase III (FabH) Inhibitors. Curr. Med. Chem. 2012, 19, 1225–1237. [Google Scholar] [CrossRef]
- Brandão, H.N.; David, J.P.; Couto, R.D.; Nascimento, J.A.P.; David, J.M. Química e farmacologia de quimioterápicos antineoplásicos derivados de plantas. Quím. Nova 2010, 33, 1359–1369. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Cordier, C.; Morton, D.; Murrison, S.; Nelson, A.; O’leary-steele, C. Natural products as an inspiration in the diversity-oriented synthesis of bioactive compound libraries. Nat. Prod. Rep. 2008, 25, 719–737. [Google Scholar] [CrossRef]
- Braga, R.C.; Andrade, C.H. Assessing the Performance of 3D Pharmacophore Models in Virtual Screening: How Good are They? Curr. Top. Med. Chem. 2013, 13, 1127–1138. [Google Scholar] [CrossRef]
- Ferreira, R.S.; Oliva, G.; Andricopulo, A.D. Integração das técnicas de triagem virtual e triagem biológica automatizada em alta escala: Oportunidades e desafios em P&D de fármacos. Quím. Nova 2011, 34, 1770–1778. [Google Scholar] [CrossRef]
- Caballero, J. 3D-QSAR (CoMFA and CoMSIA) and pharmacophore (GALAHAD) studies on the differential inhibition of aldose reductase by flavonoid compounds. J. Mol. Graph. Model. 2010, 29, 363–371. [Google Scholar] [CrossRef]
- Dorfman, R.J.; Smith, K.M.; Masek, B.B.; Clark, R.D. A knowledge-based approach to generating diverse but energetically representative ensembles of ligand conformers. J. Comput. Aided Mol. Des. 2007, 22, 681–691. [Google Scholar] [CrossRef]
- Eyunni, S.K.; Gangapuran, M.; Redda, K.K. In-vitro antiproliferative activity of new tetrahydroisoquinolines (THIQs) on ishikawa cells and their 3D pharmacophore models. Lett. Drug Des. Discov. 2014, 11, 428–436. [Google Scholar] [CrossRef]
- Rella, M.; Rushworth, C.A.; Guy, J.L.; Turner, A.J.; Langer, T.; Jackson, R.M. Structure-based pharmacophore design and virtual screening for novel angiotensin converting enzyme 2 inhibitors. J. Chem. Inf. Model. 2006, 46, 708–716. [Google Scholar] [CrossRef]
- Chen, Z.; Myint, Z.; Xie, Q. New QSAR prediction models derived from GPCR CB2-antagonistic triaryl bis-sulfone analogues by a combined molecular morphological and pharmacophoric approach. SAR QSAR Environ. Res. 2011, 22, 525–544. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. Software news and update AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comp. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Lang, P.T.; Kuntz, I.; Allen, W.J.; Balius, T.; Brozell, S.; Jiang, L.; Mcgee, T.D., Jr.; Moustakas, D.; Mukherjee, D.; Zhou, Y.; et al. DOCK 6.0 Users Manual. The Official UCSF DOCK Web-Site. Fev 2015. Available online: http://dock.compbio. ucsf.edu/DOCK_6/dock6_manual.htm (accessed on 5 February 2015).
- Wong, C.F. Flexible ligand flexible protein docking in protein kinases systems. Biochim. Biophym. Acta 2008, 1784, 244–251. [Google Scholar] [CrossRef]
- Nicholls, A. What do we know and when do we know it? J. Comput. Aided Mol. Des. 2008, 22, 239–255. [Google Scholar] [CrossRef]
- Fawcett, T. An introduction to ROC analysis. Pattern Recogn. Lett. 2006, 27, 861–874. [Google Scholar] [CrossRef]
- Matsubara, E.T. Relações entre Ranking, Análise ROC e Calibração em Aprendizado de Máquina. Ph.D. Thesis, USP, São Carlos, Brazil, 2008. [Google Scholar]
- Triballeau, N.; Acher, F.; Brabet, I.; Pin, J.P.; Bertrand, H.O. Virtual screening workflow development guided by the “receiver operating characteristic” curve approach. Application to high-throughput docking on metabotropic glutamate receptor subtype 4. J. Med. Chem. 2008, 48, 2534–2547. [Google Scholar] [CrossRef]
- Jain, A.N.; Nicholls, A. Recommendations for evaluation of computational methods. J. Comput. Aided Mol. Des. 2008, 22, 133–139. [Google Scholar] [CrossRef]
- Irwin, J.J.; Sterling, T.; Mysinger, M.M.; Bolstad, E.S.; Coleman, R.G. ZINC: A Free Tool to Discover Chemistry for Biology. J. Chem. Inf. Model. 2012, 52, 1757–1768. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem Substance and Compound databases. Nucleic Acids Res. 2016, 4, D1202–D1213. [Google Scholar] [CrossRef]
- Pence, H.E.; Willians, A. ChemSpider: An online chemical information resource. J. Chem. Educ. 2010, 87, 1123–1124. [Google Scholar] [CrossRef]
- Bissantz, C.; Kuhn, B.; Stahl, M. A Medicinal Chemist’s Guide to Molecular Interactions. J. Med. Chem. Persp. 2010, 53, 5061–5084. [Google Scholar] [CrossRef]
- Schiebel, J.; Kapilashrami, K.; Fekete, A.; Bommineni, G.R.; Schaefe, R.; Mueller, M.J.; Tonge, P.J. Structural Basis for the Recognition of Mycolic Acid Precursors by KasA, a Condensing Enzyme and Drug Target from Mycobacterium Tuberculosis. J. Biol. Chem. 2013, 288, 34190–34204. [Google Scholar] [CrossRef]
- Chemaxon. Marvin Sketch, version 15.4.20; ChemAxon: Budapest, Hungary, 2015.
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef]
- Tripos Inc. SYBYL-X, Discovery Software for Computational Chemistry and Molecular Modelling, Princeton, NJ, USA, 2012.
- Jakalian, A.; Jack, D.B.; Bayly, C.I. Fast, efficient generation of high-quality atomic charges. AM1-BCC model: II. Parameterization and validation. J. Comput. Chem. 2002, 23, 1623–1641. [Google Scholar] [CrossRef]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef]
- Ferrin, T.E.; Huang, C.C.; Jarvis, L.E.; Langridge, R. The Midas display system. J. Mol. Graf. 1988, 6, 13–27. [Google Scholar] [CrossRef]
- Kuntz, I.D.; Blaney, J.M.; Oatley, S.J.; Langridge, R.; Ferrin, T.E. A geometric approach to macromolecule–ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [Google Scholar] [CrossRef]
- Shoichet, B.K.; Kuntz, I.D.; Bodian, D.L. Molecular docking using shape descriptors. J. Comput. Chem. 1992, 13, 380–397. [Google Scholar] [CrossRef]
- Meng, E.C.; Shoichet, B.K.; Kuntz, I.D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar] [CrossRef]
- Udatha, D.B.R.K.; Sugaya, N.; Olsson, L.; Panagiotou, G. How well do the substrates KISS the enzyme? Molecular docking program selection for feruloyl esterases. Sci. Rep. 2012, 2, 323. [Google Scholar] [CrossRef]
- Cruz, J.V.; Neto, M.F.A.; Silva, L.B.; Ramos, R.d.S.; Costa, J.d.S.; Brasil, D.S.B.; Lobato, C.C.; da Costa, G.V.; Bittencourt, J.D.A.H.M.; da Silva, C.H.T.P.; et al. Identification of novel protein kinase receptor type 2 inhibitors using pharmacophore and structure-based virtual screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef]
- Feher, M. Consensus scoring for protein–ligand interactions. Drug Discov. Today 2006, 11, 421–428. [Google Scholar] [CrossRef]
- Stierand, K.; Maaß, P.C.; Rarey, M. Molecular complexes at a glance: Automated generation of two-dimensional complex diagrams. Bioinformatics 2006, 22, 1710–1716. [Google Scholar] [CrossRef]
- Cornell, W.D.; Cieplak, P.; Bayly, C.I.; Kollman, P.A. Application of RESP charges to calculate conformational energies, hydrogen bond energies, and free energies of solvation. J. Am. Chem. Soc. 1993, 115, 9620–9631. [Google Scholar] [CrossRef]
- Wang, J.; Wolf, R.M.; Caldwell, J.W.; Kollman, P.A.; Case, D.A. Development and testing of a general amber force field. J. Comput. Chem. 2004, 25, 1157–1174. [Google Scholar] [CrossRef]
- Ponder, J.W.; Case, D.A. Force fields for protein simulations. Adv. Protein Chem. 2003, 66, 27–85. [Google Scholar]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Salomon-Ferrer, R.; Case, D.A.; Walker, R.C. An overview of the Amber biomolecular simulation package. Wiley Interdiscip. Rev. Comput. Mol. Sci. 2013, 3, 198–210. [Google Scholar] [CrossRef]
- Cruz, J.N.; Costa, J.F.S.; Khayat, A.S.; Kuca, K.; Barros, C.A.L.; Neto, A.M.J.C. Molecular dynamics simulation and binding free energy studies of novel leads belonging to the benzofuran class inhibitors of Mycobacterium tuberculosis Polyketide Synthase 13. J. Biomol. Struct. Dyn. 2018, 1–12. [Google Scholar] [CrossRef]
- Izaguirre, J.A.; Catarello, D.P.; Wozniak, J.M.; Skeel, R.D. Langevin stabilization of molecular dynamics. J. Chem. Phys. 2001, 114, 2090–2098. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Ryckaert, J.P.; Ryckaert, J.P.; Ciccotti, G.; Berendsen, H.J.C.C. Numerical integration of the cartesian equations of motion of a system with constraints: Molecular dynamics of n-alkanes. J. Comput. Phys. 1977, 23, 327–341. [Google Scholar] [CrossRef]
- Chen, F.; Liu, H.; Sun, H.; Pan, P.; Li, Y.; Li, D.; Hou, T. Assessing the performance of the MM/PBSA and MM/GBSA methods. 6. Capability to predict protein-protein binding free energies and re-rank binding poses generated by protein-protein docking. Phys. Chem. Chem. Phys. 2016, 18, 22129–22139. [Google Scholar] [CrossRef]
- de Oliveira, M.S.; da Cruz, J.N.; Gomes Silva, S.; da Costa, W.A.; de Sousa, S.H.B.; Bezerra, F.W.F.; Teixeira, E.; da Silva, N.J.N.; de Aguiar Andrade, E.H.; de Jesus Chaves Neto, A.M.; et al. Phytochemical profile, antioxidant activity, inhibition of acetylcholinesterase and interaction mechanism of the major components of the Piper divaricatum essential oil obtained by supercritical CO2. J. Supercrit. Fluids 2019, 145, 74–84. [Google Scholar] [CrossRef]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Models | Strain Energy (kcal/mol) | Hbond | Mol-qry |
|---|---|---|---|
| 2 | 7.53 | 650.30 | 112.40 |
| 8 | 7.58 | 613.30 | 114.60 |
| 1 | 8.02 | 619.30 | 127.70 |
| 5 | 9.05 | 497.20 | 122.60 |
| 10 | 9.54 | 478.10 | 125.10 |
| 4 | 10.52 | 520.90 | 128.40 |
| 7 | 45.88 | 504.00 | 126.20 |
| 9 | 67.80 | 550.70 | 132.00 |
| 6 | 69.55 | 482.60 | 136.70 |
| 3 | 496.79 | 655.80 | 133.10 |
| Molecule | Structure | QFIT |
|---|---|---|
| ZINC35465970 |  | 66.76 |
| ZINC15959689 |  | 62.97 |
| ZINC16032930 |  | 62.07 |
| ZINC31161132 |  | 59.99 |
| ZINC72320274 |  | 59.86 |
| MOLECULE | CONSENSUS |
|---|---|
| ZINC35465970 | 122.10 |
| ZINC31170017 | 108.57 |
| ZINC12659549 | 108.52 |
| ZINC08453820 | 107.44 |
| ZINC15959689 | 107.28 |
| Acceptor | Hydrogen Donor | Donor | Occupancy (%) a | Average Distance (Å) |
|---|---|---|---|---|
| ZINC35465970 | ||||
| ZINC35465970_416@O25 | GLN_170@HE22 | GLN_170@NE2 | 30.46 | 3.17 |
| ZINC35465970_416@O25 | HIS_344@HE2 | HIS_344@NE2 | 28.75 | 3.03 |
| ZINC35465970_416@O24 | LYS_339@HZ3 | LYS_339@NZ | 21.69 | 2.89 |
| ZINC35465970_416@O24 | LYS_339@HZ1 | LYS_339@NZ | 21.19 | 2.89 |
| ZINC35465970_416@O24 | LYS_339@HZ2 | LYS_339@NZ | 20.01 | 2.90 |
| ZINC31170017 | ||||
| MET_212@O | ZINC31170017_416@H62 | ZINC31170017_416@O37 | 71.27 | 2.81 |
| ARG_233@O | ZINC31170017_416@H56 | ZINC31170017_416@O22 | 49.69 | 3.01 |
| Compound | ΔEvdW | ΔEele | ΔGGB | ΔGNP | ΔGbind |
|---|---|---|---|---|---|
| ZINC35465970 | −45.21 | −11.93 | 32.31 | −6.06 | −30.90 |
| ZINC31170017 | −35.86 | −11.48 | 25.04 | −5.18 | −27.49 |
| Molecule | Structure | Ki (µM) |
|---|---|---|
| 1 |  | 0.46 |
| 2 |  | 0.90 |
| 3 |  | 1.90 |
| 4 |  | 7.10 |
| 5 |  | 16.00 |
| 6 |  | 34.00 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, V.d.S.; Araújo, J.S.C.; Silva, R.C.; da Costa, G.V.; Cruz, J.N.; De A. Neto, M.F.; Campos, J.M.; Santos, C.B.R.; Leite, F.H.A.; Junior, M.C.S. In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals 2019, 12, 36. https://doi.org/10.3390/ph12010036
Pinto VdS, Araújo JSC, Silva RC, da Costa GV, Cruz JN, De A. Neto MF, Campos JM, Santos CBR, Leite FHA, Junior MCS. In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals. 2019; 12(1):36. https://doi.org/10.3390/ph12010036
Chicago/Turabian StylePinto, Vinícius de S., Janay S. C. Araújo, Rai C. Silva, Glauber V. da Costa, Jorddy N. Cruz, Moysés F. De A. Neto, Joaquín M. Campos, Cleydson B. R. Santos, Franco H. A. Leite, and Manoelito C. S. Junior. 2019. "In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations" Pharmaceuticals 12, no. 1: 36. https://doi.org/10.3390/ph12010036
APA StylePinto, V. d. S., Araújo, J. S. C., Silva, R. C., da Costa, G. V., Cruz, J. N., De A. Neto, M. F., Campos, J. M., Santos, C. B. R., Leite, F. H. A., & Junior, M. C. S. (2019). In Silico Study to Identify New Antituberculosis Molecules from Natural Sources by Hierarchical Virtual Screening and Molecular Dynamics Simulations. Pharmaceuticals, 12(1), 36. https://doi.org/10.3390/ph12010036