Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening

 ,
,  , , , ,
, , , ,  and
and
Abstract
1. Introduction
2. Results and Discussion
2.1. Pharmacokinetic and Toxicological Properties
2.2. Biological Activity Prediction
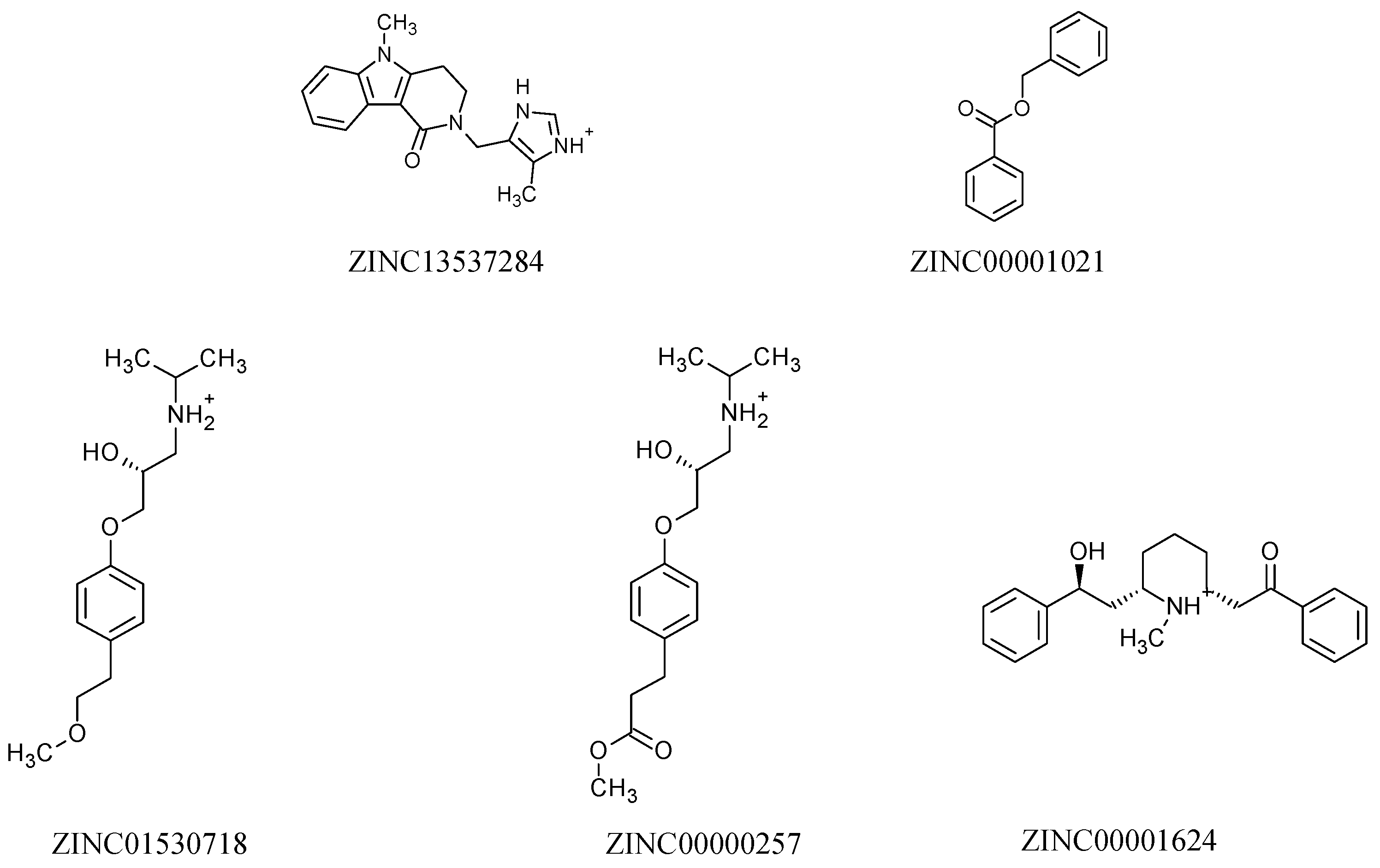
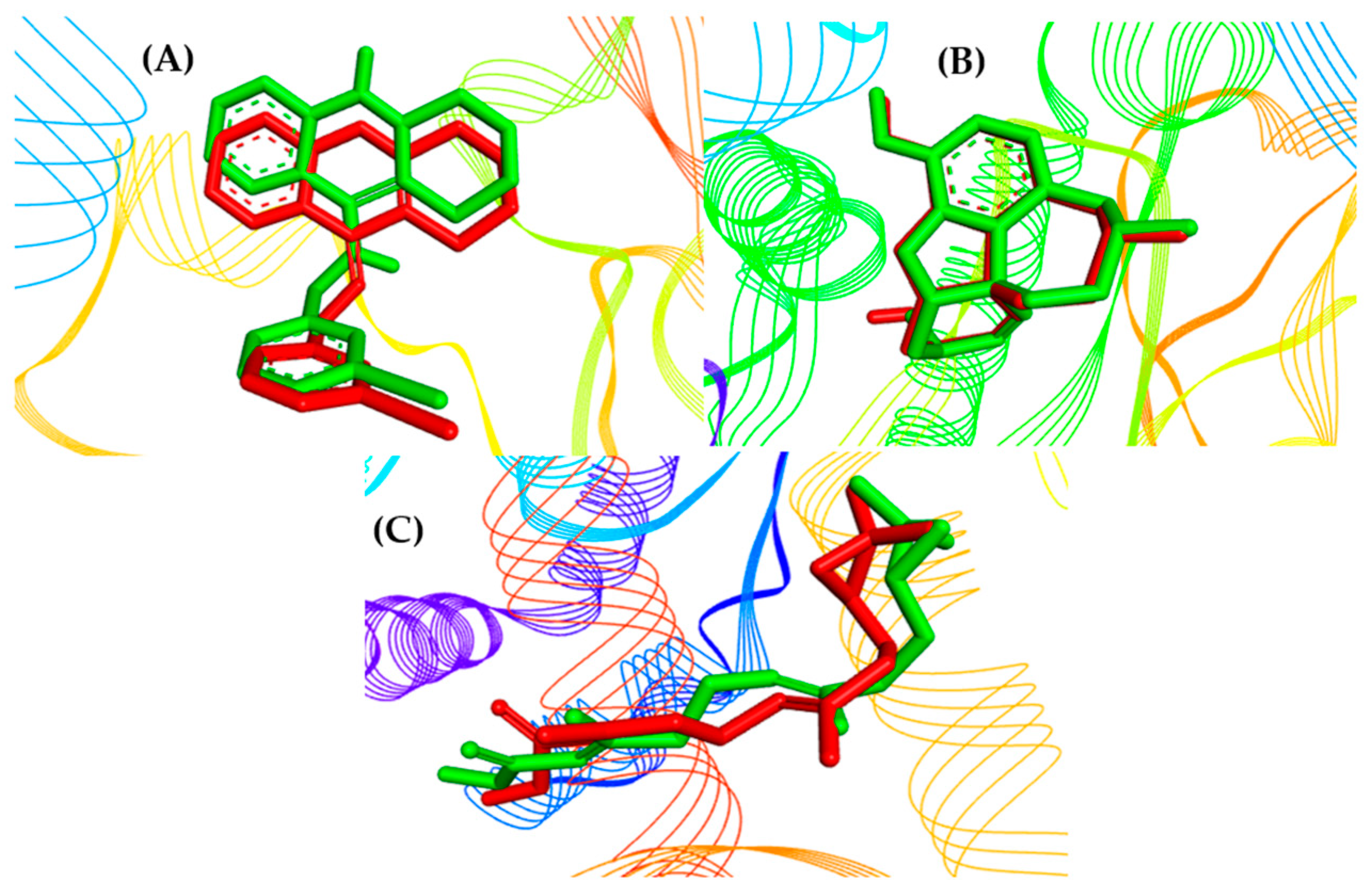
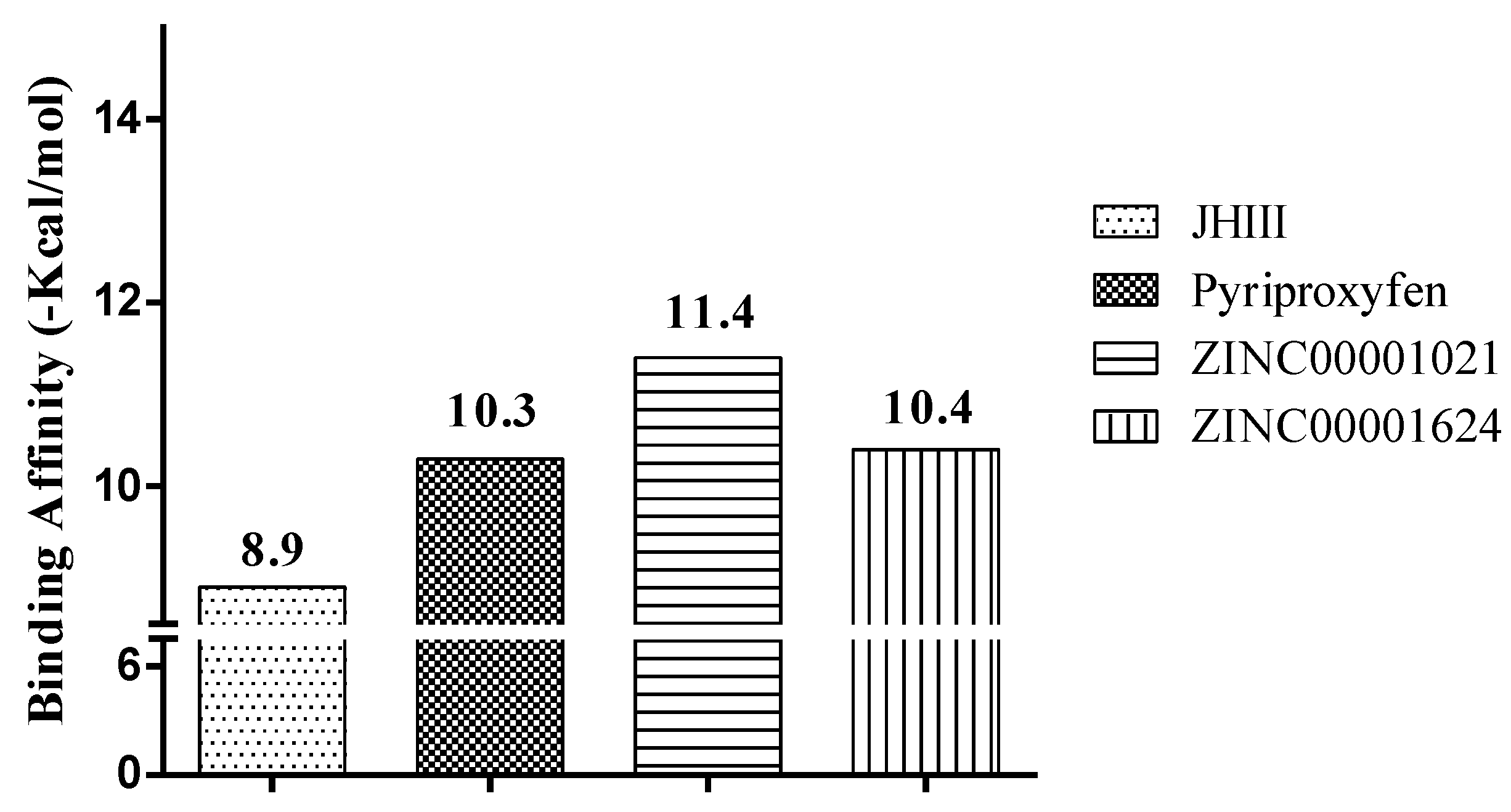
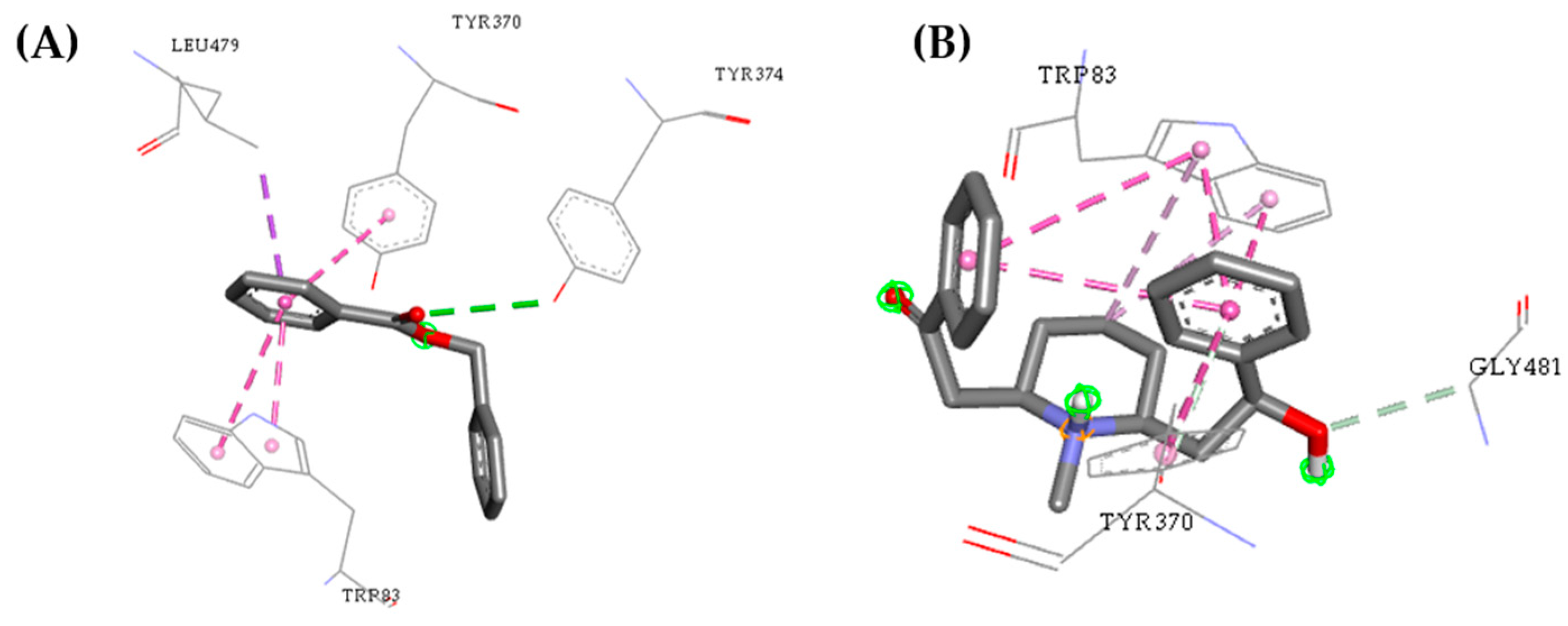
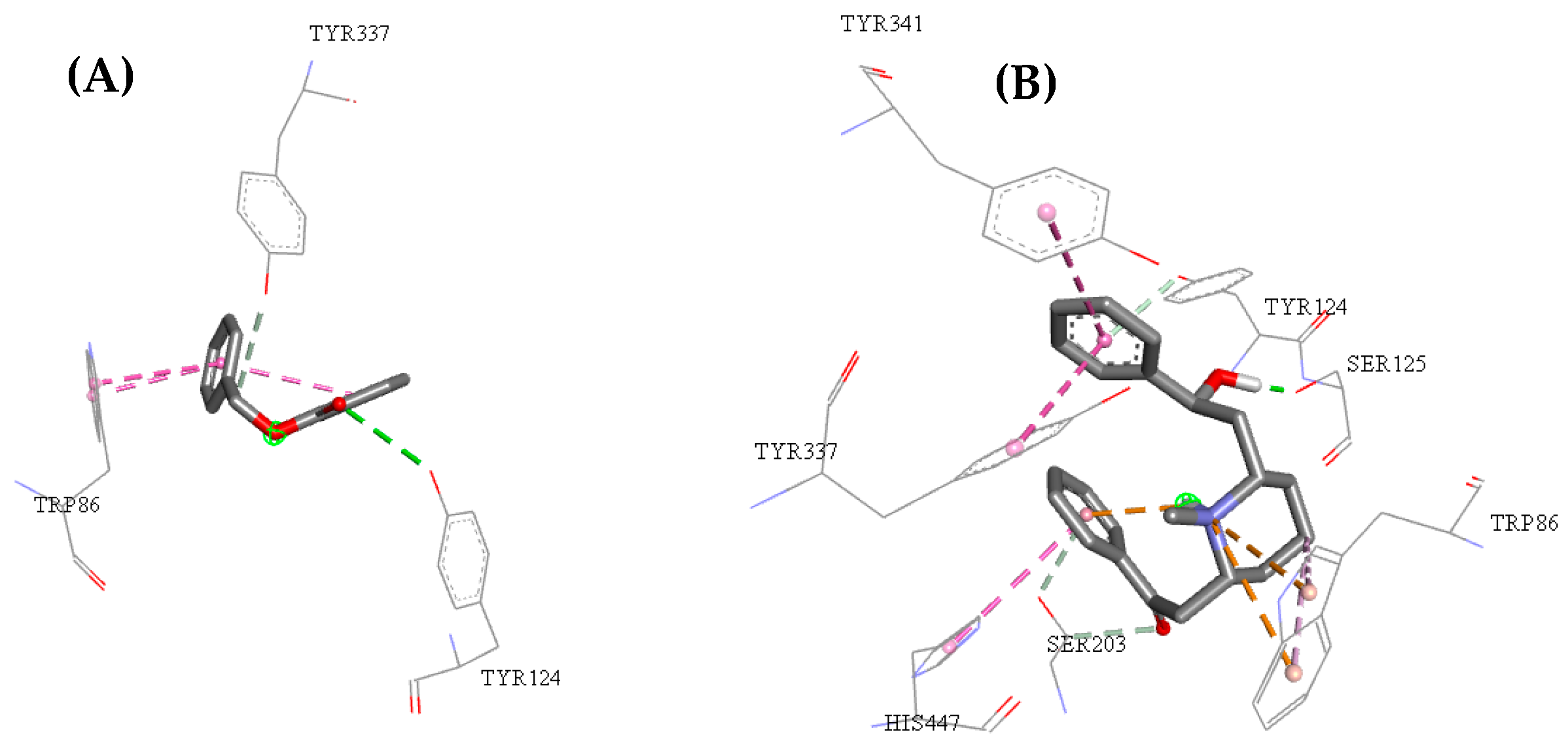
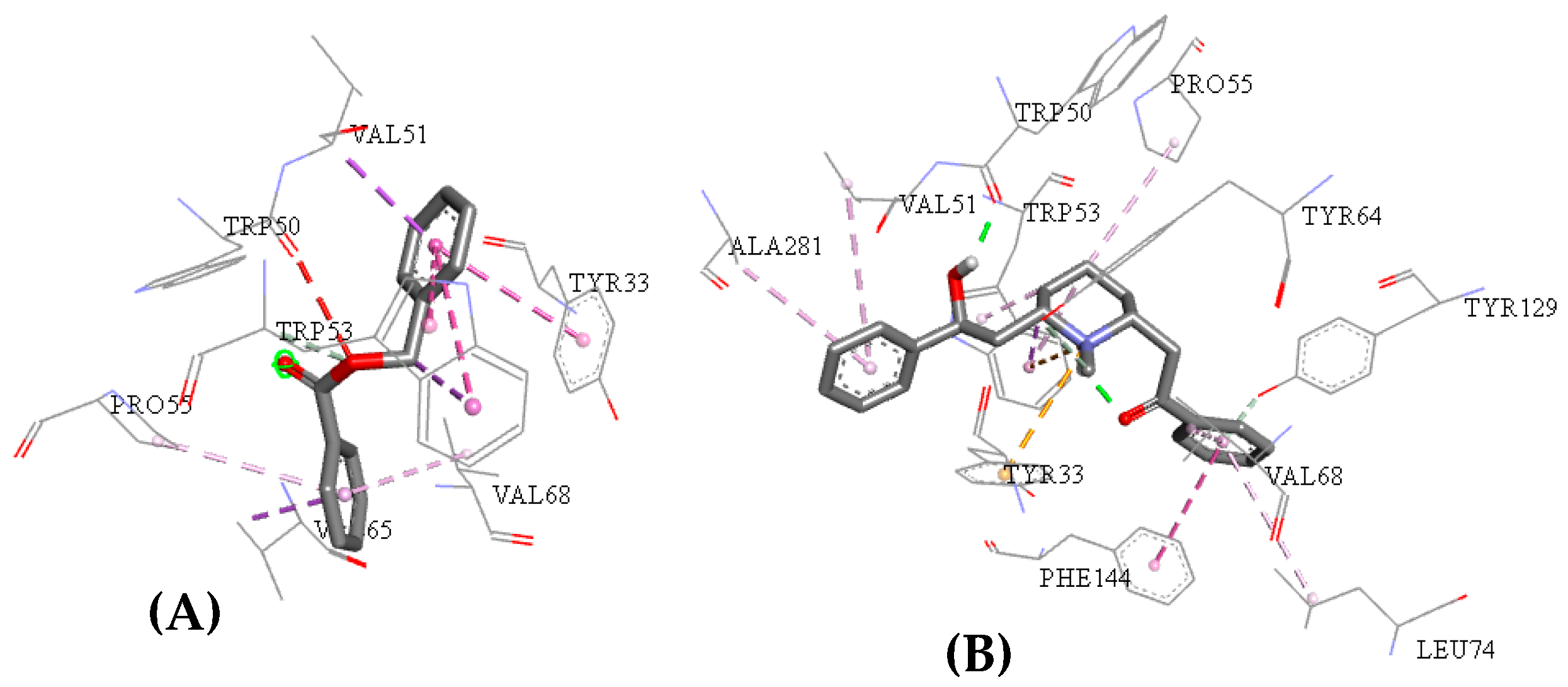
2.3. Molecular Docking Study
2.4. Toxicity Risk Assessment
2.5. Structure–Activity Relationship of the Promising Molecule
3. Materials and Methods
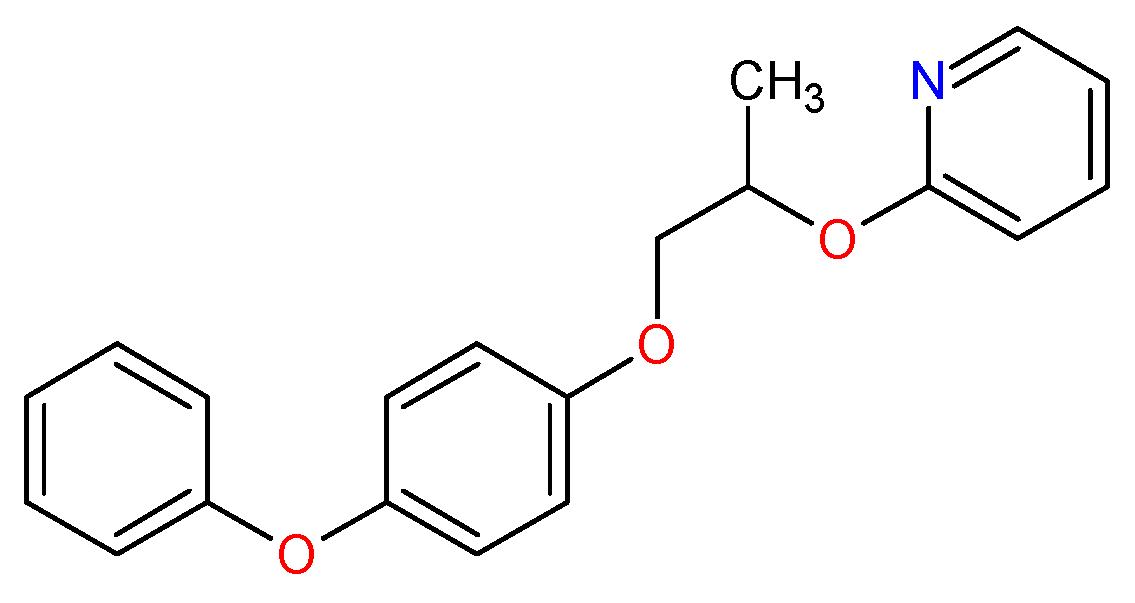
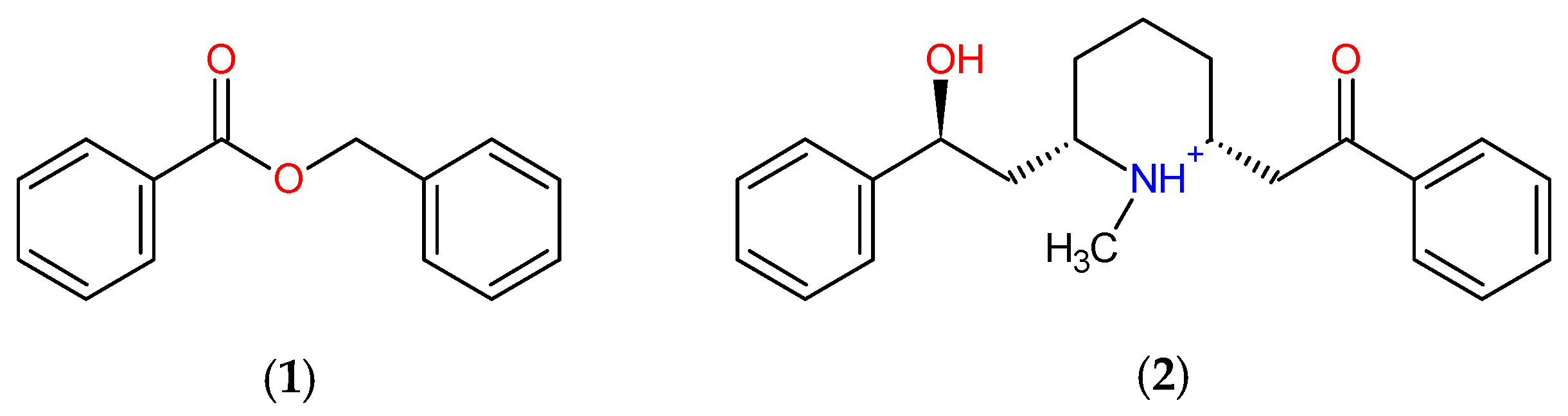
3.1. Template Compound
3.2. Generation of Conformers Library in Database
3.3. Virtual Screening
3.3.1. Rapid Overlay of Chemical Structures (ROCS)
3.3.2. Electrostatic Similarity (EON)
3.4. In silico Pharmacokinetic and Toxicological Properties
3.5. Biological Activity Predictions of the Compounds from Virtual Screening
3.6. Molecular Docking Simulations
3.6.1. Selection of Enzymes and Inhibitors Structures
3.6.2. Docking Study with AutoDock 4.2/Vina 1.1.2 via Graphical Interface PyRx (Version 0.8.30)
3.7. Toxicity Risk Assessment
4. Conclusions
Supplementary Materials
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Diallo, D.; Sall, A.A.; Diagne, C.T.; Faye, O.; Faye, O.; Ba, Y.; Hanley, K.H.; Buenemann, M.; Weaver, S.C.; Diallo, M. Zika Virus Emergence in Mosquitoes in Southeastern Senegal, 2011. PLoS ONE 2014, 9, e109442. [Google Scholar] [CrossRef] [PubMed]
- Lekweiry, K.M.; Salem, M.S.O.A.; Brahim, K.O.; Ould Lemrabott, M.A.O.; Brengues, C.; Faye, O.; Simard, F.; Boukhary, A.O.M.S. Aedes aegypti (Diptera: Culicidae) in Mauritania: First Report on the Presence of the Arbovirus Mosquito Vector in Nouakchott. J. Med. Entomol. 2015, 52, 730–733. [Google Scholar] [CrossRef] [PubMed]
- World Health Organizations. Global Strategy for Dengue Prevention and Control 2012–2020; World Health Organizations: Paris, France, 2012; Volume 43. [Google Scholar]
- Ramos, R.S.; Rodrigues, A.B.L.; Farias, A.L.F.; Simões, R.C.; Pinheiro, M.T.; Ferreira, R.M.A.; Barbosa, L.M.C.; Souto, R.N.P.; Fernandes, J.B.; Santos, L.S.; et al. Chemical Composition and In Vitro Antioxidant, Cytotoxic, Antimicrobial, and Larvicidal Activities of the Essential Oil of Mentha piperita L. (Lamiaceae). Sci. World J. 2017, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ministério da Saúde. Monitoramento dos Casos de Dengue e Febre de Chikungunya até a Semana Epidemiológica (SE) 53 de 2014. Bol. Epidemiol. 2015, 46, 7. [Google Scholar]
- Costa, I.M.P.; Calado, D.C. Incidência dos casos de dengue (2007–2013) e distribuição sazonal de culicídeos (2012–2013) em Barreiras, Bahia Epidemiol. Serv. Saude 2016, 25, 735–744. [Google Scholar] [CrossRef]
- Whitehorn, J.; Farrar, J. Dengue. Br. Med. Bull. 2010, 95, 161–1730. [Google Scholar] [CrossRef] [PubMed]
- Kroupova, A.; Ivascu, A.; Reimao-Pinto, M.M.; Ameres, S.L.; Jinek, M. Structural basis for acceptor RNA substrate selectivity of the 3 terminal uridylyl transferase Tailor. Nucleic Acids Res. 2018, 47, 1030–1042. [Google Scholar] [CrossRef]
- Engdahl, C.; Knutsson, S.; Fredriksson, S.Å.; Linusson, A.; Bucht, G.; Ekström, F. Acetylcholinesterases from the Disease Vectors Aedes aegypti and Anopheles gambiae: Functional Characterization and Comparisons with Vertebrate Orthologues. PLoS ONE 2015, 10, e0138598. [Google Scholar] [CrossRef]
- Botas, G.S.; Cruz, R.A.S.; DE Almeida, F.B.; Duarte, J.L.; Araújo, R.S.; Souto, R.N.P.; Ferreira, R.; Carvalho, J.C.T.; Santos, M.G.; Rocha, L.; et al. Baccharis reticularia DC. and Limonene Nanoemulsions: Promising Larvicidal Agents for Aedes aegypti (Diptera: Culicidae) Control. Molecules 2017, 22, 1990. [Google Scholar] [CrossRef]
- Blenau, W.; Rademacher, E.; Baumann, A. Plant essential oils and formamidines as insecticides/acaricides: What are the molecular targets? Apidologie 2012, 43, 334–347. [Google Scholar] [CrossRef]
- Ostettmann, K.; Borloz, A.; Urbain, A.; Marston, A. Natural Product Inhibitors of Acetylcholinesterase. Curr. Org. Chem. 2006, 10, 825–847. [Google Scholar] [CrossRef]
- Miyazawa, M.; Tougo, H.; Ishihara, M. Inhibition of acetylcholinesterase activity by essential oil from Citrus paradisi. Nat. Prod. Lett. 2001, 15, 205–210. [Google Scholar] [CrossRef] [PubMed]
- Savelev, S.; Okello, E.; Perry, N.S.L.; Wilkins, R.M.; Perry, E.K. Synergistic and antagonistic interactions of anticholinesterase terpenoids in Salvia lavandulaefolia essential oil. Pharmacol. Biochem. Behav. 2003, 75, 661–668. [Google Scholar] [CrossRef]
- Ahmadi, F.; Jamali, N.; Moradian, R.; Astinchap, B. Binding Studies of Pyriproxyfen to DNA by Multispectroscopic Atomic Force Microscopy and Molecular Modeling Methods. DNA Cell Biol. 2012, 31, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Ponlawat, A.; Fansiri, T.; Kurusarttra, S.; Pongsir, I.A.; Mccardle, P.W.; Evans, B.P.; Richardson, J.H. Development and evaluation of a pyriproxyfen treated Device to control the dengue vector, Aedes aegypti (l.) (diptera:culicidae). J. Trop. Med. Public Health 2013, 44, 167–178. [Google Scholar]
- Braga, I.A.; Valle, D. Aedes aegypti: Inseticidas, mecanismos de ação e resistência. Epidemiol. Serv. Saúde 2007, 16, 279–293. [Google Scholar] [CrossRef]
- Kim, I.H.; Pham, V.; Jablonka, W.; Goodman, W.G.; Ribeiro, J.M.C.; Andersen, J.F. A mosquito hemolymph odorant-binding protein Family member specifically binds juvenile hormone. J. Biol. Chem. 2017, 292, 15329–15339. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.J.; Goh, K.S. Environmental fate and properties of pyriproxyfen. J. Pestic. Sci 2008, 33, 339–350. [Google Scholar] [CrossRef]
- Olmstead, A.W.; Leblanc, G.A. Insecticidal Juvenile Hormone Analogs Stimulate the Production of Male Offspring in the Crustacean Daphnia magna. Environ. Health Perspect. 2003, 111, 919–924. [Google Scholar] [CrossRef] [PubMed]
- Paul, A.; Harrington, L.C.; Scott, J.G. Evaluation of Novel Insecticides for Control of Dengue Vector Aedes aegypti (Diptera: Culicidae). J. Med. Entomol. 2006, 43, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Harburguer, L.V.; Seccacini, E.; Masuh, H.; Audino, P.G.; Zerbaa, E.; Licastro, S. Thermal behaviour and biological activity against Aedes aegypti (Diptera: Culicidae) of permethrin and pyriproxyfen in a smoke-generating formulation. Pest. Manag Sci. 2009, 65, 1208–1214. [Google Scholar] [CrossRef] [PubMed]
- Harwood, J.F.; Helmey, W.L.; Turnwall, B.B.; Justice, K.D.; Muhammed Farooq, M.; Richardson, A.G. Controlling Aedes aegypti in Cryptic Environments with Manually Carried Ultra-Low Volume and Mist Blower Pesticide Applications. J. Am. Mosq. Control. Assoc. 2016, 32, 217–223. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv Ver. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gaddaguti, V.; Rao, T.V.; Rao, A.P. Potential mosquito repellent compounds of Ocimum species against 3N7H and 3Q8I of Anopheles gambiae. 3 Biotech 2016, 6, 26. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, W.L.; Duffy, E.M. Prediction of drug solubility from structure. Adv. Drug Deliv Ver. 2002, 54, 355–366. [Google Scholar] [CrossRef]
- Itoh, T. Control of DF/DHF Vector, Aedes Mosquito, with Insecticides. Trop. Med. 1994, 35, 259–267. [Google Scholar]
- Andrighetti, M.T.M.; Cerone, F.; Rigueti, M.; Galvani, K.C.; Macoris, M.L.G. Effect of pyriproxyfen in Aedes aegypti populations with different levels of susceptibility to the organophosphate temephos. Dengue Bull. 2008, 32, 186–198. [Google Scholar]
- Zimmerman, H.J. Hepatotoxicity: The Adverse Effects of Drugs and other Chemicals on the Liver; Appleton-Century-Crofts: New York, NY, USA, 1999. [Google Scholar]
- International Agency for Research on Cancer (IARC). Combined Estrogen-Progestogen Contraceptives and Combined Estrogen-Progestogen Menopausal Therapy; IARC Monographs on the Evaluation of Carcinogenic Risks to Humans; World Health Organization: Paris, France, 2007; Volume 91, p. 543. Available online: http://monographs.iarc.fr/ENG/Publications/corrigenda.php (accessed on 6 December 2018).
- Marchand, B.; Barbier, P.; Ducomb, G.; Foussereau, J.; Martin, P.; Benezra, C. Allergic contact dermatitis to various salols (phenyl salicylates). A structure-activity relationship study in man and in animal (guinea pig). Arch. Dermatol. Res. 1982, 272, 61–66. [Google Scholar] [CrossRef]
- Barratt, M.D.; Basketter, D.A.; Roberts, D.W. Skin sensitization structure-activity relationships for phenyl benzoates. Toxicol. In Vitro 1994, 8, 823–826. [Google Scholar] [CrossRef]
- Payne, M.P.; Walsh, P.T. Structure-activity relationships for skin sensitization potential: Development of structural alerts for use in knowledge-based toxicity prediction systems. J. Chem. Inf. Comput. Sci. 1994, 34, 154–161. [Google Scholar] [CrossRef]
- Cronin, M.T.D.; Basketter, D.A. Multivariate QSAR analysis of a skin sensitization database. Sar Qsar Environ. Res. 1994, 2, 159–179. [Google Scholar] [CrossRef]
- Opdyke, D.L.J. Monographs on fragrance raw materials: Dihydrocoumarin. Food Cosmet. Toxicol. 1974, 12, 521–522. [Google Scholar] [CrossRef]
- Anstead, G.M.; Carlson, K.E.; Katzenellenbogen, J.A. The estradiol pharmacophore: Ligand structure-estrogen receptor binding affinity relationships and a model for the receptor binding site. Steroids 1997, 62, 268–303. [Google Scholar] [CrossRef]
- Fang, H.; Tong, W.; Shi, L.M.; Blair, R.; Perkins, R.; Branham, W.; Hass, B.S.; Xie, Q.; Dial, S.L.; Moland, C.L.; et al. Structure-activity relationships for a large diverse set of natural, synthetic, and environmental estrogens. Chem. Res. Toxicol. 2001, 14, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Greene, R.R.; Burrill, M.W.; Ivy, A.C. Experimental intersexuality: The paradoxical effects of estrogens on the sexual development of the female rat. Anat. Rec. 1939, 74, 429–438. [Google Scholar] [CrossRef]
- Greene, R.R.; Burrill, M.W.; Ivy, A.C. Experimental intersexuality: The effects of estrogens on the antenatal sexual development of the rat. Am. J. Anat. 1940, 67, 305–345. [Google Scholar] [CrossRef]
- Yasuda, Y.; Kihara, T.; Tanimura, T.; Nishimura, H. Gonadal dysgenesis induced by prenatal exposure to ethinyl estradiol in mice. Teratology 1985, 32, 219–227. [Google Scholar] [CrossRef]
- Tan, E.L.; Brimer, P.A.; Schenley, R.L.; Hsie, A.W. Mutagenicity and cytotoxicity of dimethyl and monomethyl sulfates in the CHO/HGPRT system. J. Toxicol. Environ. Health 1983, 11, 373–380. [Google Scholar] [CrossRef] [PubMed]
- Barber, E.D.; Donish, W.H.; Mueller, K.R. A procedure for the quantitative measurement of the mutagenicity of volatile liquids in the Ames Salmonella/microsome assay. Mutat. Res. 1981, 90, 31–48. [Google Scholar] [CrossRef]
- Eriksson, L.; Hellberg, S.; Johansson, E.; Jonsson, J.; Sjostrom, M.; Wold, S.; Berglind, R.; Karlsson, B. A strategy for ranking environmentally occurring chemicals. Part VI. QSARs for the mutagenic effects of halogenated aliphatics. Acta Chem. Scand. 1991, 45, 935–944. [Google Scholar] [CrossRef]
- Perry, M.D.; Ng, C.A.; Mann, S.A.; Sadrieh, A.; Imtiaz, M.; Hill, A.P.; Vandenberg, J.I. Getting to the heart of hERG K+ channel gating. J. Physiol. 2015, 593, 2575–2585. [Google Scholar] [CrossRef] [PubMed]
- Lagunin, A.; Filimonov, D.A.; Poroikov, V.V. Multi-targeted natural products evaluation based on biological activity prediction with PASS. Cur. Phar. Des. 2010, 16, 1703–1717. [Google Scholar] [CrossRef]
- Gowtham, U.; Jayakanthan, M.D.; Sundar, S. Molecular docking studies of dithionitrobenzoic acid and its related compounds to protein disulfide isomerase: Computational screening of inhibitors to HIV-1 entry. BMC Bioinform. 2008, 9, S14. [Google Scholar] [CrossRef]
- Hevener, K.E.; Zhao, W.; Ball, D.M.; Babaoglu, K.; Qi, J.; White, S.W.; Lee, R.E. Validation of molecular docking softwares for virtual screening against dihydropteroate synthase. J. Chem. Inform. Model 2009, 49, 444–460. [Google Scholar] [CrossRef] [PubMed]
- Harel, M.; Kryger, G.; Rosenberry, T.L.; Mallender, W.D.; Lewis, T.; Fletcher, R.J.; Guss, J.M.; Silman, I.; Sussman, J.L. Three-Dimensional Structures of Drosophila Melanogaster Acetylcholinesterase and of its Complexes with Two Potent Inhibitors. Protein Sci. 2000, 9, 1063. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Wang, Y.H.; Liu, H.T.; Xu, Y.X.; Wei, Z.G.; Chen, Y.H.; Shen, W.D. Genotyping of acetylcholinesterase in insects. Pestic. Biochem. Physiol. 2010, 98, 19–25. [Google Scholar] [CrossRef]
- Anderson, J.A.; Coats, J.R. Acetylcholinesterase inhibition by nootkatone and carvacrol in arthropods. Pestic. Biochem. Physiol. 2012, 102, 124–128. [Google Scholar] [CrossRef]
- Atanasova, M.; Stavrakov, G.; Philipova, I.; Zheleva, D.; Yordanov, N.; Doytchinova, I. Galantamine derivatives with indole moiety: Docking, design, synthesis and acetylcholinesterase inhibitory activity. Bioorganic Med. Chem. 2015, 17, 5382–5389. [Google Scholar] [CrossRef] [PubMed]
- Chigurupati, S.; Selvaraj, M.; Mani, V.; Selvarajan, K.K.; Mohammade, J.I.; Kaveti, B.; Bera, H.; Palanimuthu, V.R.; Teh, L.K.; Salleh, M.Z. Identification of novel acetylcholinesterase inhibitors: Indolopyrazoline derivatives and molecular docking studies. Bioorganic Chem. 2016, 67, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Fournier, D.; Mutero, A.; Pralavorio, M.; Bride, J.M. Drosophila acetylcholinesterase: Mechanisms of resistance to organophosphates. Chem. Biol. Interact. 1993, 87, 233–238. [Google Scholar] [CrossRef]
- Gnagey, A.L.; Forte, M.; Rosenberry, T.L. Isolation and characterization of acetylcholinesterase from Drosophila. J. Biol. Chem. 1987, 262, 13290–13298. [Google Scholar] [PubMed]
- Meriç, A. Molecular modelling of 2-iminothiazoles as insecticidal activity. Bulg. Chem. Commun. 2017, 49, 5–14. [Google Scholar]
- Noriega, F.G.; Ribeirob, J.M.C.; Koenerc, J.F.; Valenzuelab, J.G.; Hernandez-Martinezd, S.; Phamb, V.M.; Feyereisenc, R. Comparative genomics of insect juvenile hormone biosynthesis. Insect Biochem. Mol. Biol. 2006, 36, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Timms, B.G.; Howdeshell, K.L.; Barton, L.; Bradley, S.; Richter, C.A.; Vom Saal, F.S. Estrogenic chemicals in plastic and oral contraceptives disrupt development of the fetal mouse prostate and urethra. Proc. Natl. Acad. Sci. USA 2005, 102, 7014–7019. [Google Scholar] [CrossRef] [PubMed]
- Coscollà, C.; Vicent, Y.; Martí, P.; Pastor, A. Analysis of currently used pesticides in fine airborne particulate matter (PM 2.5) by pressurized liquid extraction and liquid chromatography–tandem mass spectrometry. J. Chromatogr. A 2008, 1200, 100–107. [Google Scholar] [CrossRef]
- Yang, Y.; Dong, B.; Xu, H.; Zheng, X.; Heong, K.L.; Lu, Z. Susceptibility to Insecticides and Ecological Fitness in Resistant Rice Varieties of Field Nilaparvata lugens Stål Population Free from Insecticides in Laboratory. Rice Sci. 2014, 21, 181–186. [Google Scholar] [CrossRef]
- World Health Organization. The WHO Recommended Classification of Pesticides by Hazard and Guidelines to Classification; World Health Organization: Paris, France, 2009. [Google Scholar]
- Drwal, M.N.; Banerjee, P.; Dunkel, M.; Wettig, M.R.; Preissner, R. ProTox: A web server for the in silico prediction of rodent oral toxicity. Nucleic Acids Res. 2014, 42, W53–W58. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, P.; Eckert, A.O.; Schrey, A.K.; Preissner, R. ProTox-II: A webserver for the prediction of toxicity of chemicals. Nucleic Acids Res. 2018, 46, W257–W263. [Google Scholar] [CrossRef]
- Banerjee, P.; Dehnbostel, F.O.; Preissner, R. Prediction is a Balancing Act: Importance of Sampling Methods to Balance Sensitivity and Specificity of Predictive Models based on Imbalanced Chemical Data Sets. Front. Chem. 2018, 6, 362. [Google Scholar] [CrossRef]
- Vale, A.; Lotti, M. Organophosphorus and carbamate insecticide poisoning. Handb. Clin. Neurol. 2015, 131, 149–168. [Google Scholar] [CrossRef]
- Pizova, H.; Havelkova, M.; Stepankova, S.; Bak, A.; Kauerova, T.; Kozik, V.; Oravec, M.; Imramovsky, A.; Kollar, P.; Bobal, P.; et al. Proline-Based Carbamates as Cholinesterase Inhibitors. Molecules 2017, 22, 1969. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, R.J.; Kogan, V.; Shelton, J.F.; Delwiche, L.; Hansen, R.L.; Ozonoff, S.; Ma, C.C.; McCanlies, E.C.; Bennett, D.H.; Hertz-Picciotto, I.; et al. Combined Prenatal Pesticide Exposure and Folic Acid Intake in Relation to Autism Spectrum Disorder. Environ. Health Perspect. 2017, 125, 097007. [Google Scholar] [CrossRef] [PubMed]
- Beránková, M.; Hojerová, J.; Melegová, L. Exposure of amateur gardeners to pesticides via the non-gloved skin per day. Food Chem. Toxicol. 2017, 108 Pt A, 224–235. [Google Scholar] [CrossRef]
- Simões, M.O. Farmacognosia: Da planta ao medicamento, 5th ed.; Editora da UFRGS: Porto Alegre, Brazil, 2003. [Google Scholar]
- Tan, L.T.H.; Yin, L.H.L.; Yin, W.F.; Chan, C.K.; Kadir, H.A.; Chan, K.G.; Goh, B.H. Traditional Uses, Phytochemistry, and Bioactivities of Cananga odorata (Ylang-Ylang). Evid. Based Complement. Alternat. Med. 2015, 1–30. [Google Scholar] [CrossRef]
- Mossa, A.T.H. Green pesticides: Essential oils as biopesticides in insect-pest management. J. Environ. Sci Technol. 2016, 9, 354–378. [Google Scholar] [CrossRef]
- Mostafiz, M.M.; Jhan, P.K.; Shim, J.-K.; Lee, K.Y. Methyl benzoate exhibits insecticidal and repellent activities against Bemisia tabaci (Gennadius) (Hemiptera: Aleyrodidae). PLoS ONE 2018, 13, e0208552. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, J.E.C. Nicotinoid Insecticides and the Nicotinic Acetylcholine Receptor (eBook); Springer: Berlin/Heidelberg, Germany, 1999; pp. 1–299. [Google Scholar]
- Harriott, N.D.; Williams, J.P.; Smith, E.B.; Bozigian, H.P.; Grigoriadis, D.E. VMAT2 Inhibitors and the Path to Ingrezza (Valbenazine). Prog. Med. Chem. 2018, 87–111. [Google Scholar] [CrossRef]
- Kang, G.; Kim, J.; Park, H.; Kim, T.H. Crystal structure of pyriproxyfen. Acta Cryst. 2015, 71, 588. [Google Scholar] [CrossRef]
- Hawkins, P.C.D.; Nicholls, A. Conformer generation with OMEGA: Learning from the data set and the analysis of failures. J. Chem. Inf. Modeling 2012, 52, 2919–2936. [Google Scholar] [CrossRef]
- Silva, R.C.; Poiani, J.G.C.; Ramos, R.S.; Costa, J.S.; Silva, C.H.T.P.; Brasil, D.S.B.; Santos, C.B.R. Ligand- and structure-based virtual screening from 16-(N, N-diisobutylaminomethyl)-6α hydroxyivouacapan-7β,17β-lactone compound with potential anti-prostate câncer activity. J. Serb. Chem. Soc. 2018, 83, 1–23. [Google Scholar] [CrossRef]
- Naylor, E.; Arredouani, A.; Vasudevan, S.R.; Lewis, A.M.; Parkesh, R.; Mizote, A.; Rosen, D.; Thomas, J.M.; Izumi, M.; Ganesan, A.; et al. Identification of a chemical probe for NAADP by virtual screening. Nat. Chem. Biol. 2009, 5, 220–226. [Google Scholar] [CrossRef] [PubMed]
- Schrödinger. QikProp: Rapid ADME Predictions of Drug Candidates; Versão 3.4; Schrödinger: New York, NY, USA, 2011. [Google Scholar]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur. J. Pharm. Sci. 2000, 10, 195–204. [Google Scholar] [CrossRef]
- Testa, B.; Balmat, A.L.; Long, A.; Judson, P. Predicting drug metabolism—An evaluation of the expert system METEOR. Chem. Biodivers. 2005, 2, 872–885. [Google Scholar] [CrossRef] [PubMed]
- Sanderson, D.M.; Earnshaw, C.G. Computer prediction of possible toxic action from chemical structure; the DEREK system. Hum. Exp. Toxicol. 1991, 10, 261–273. [Google Scholar] [CrossRef] [PubMed]
- Rindings, J.E.; Barratt, M.D.; Cary, R. Computer prediction of possible toxic action from chemical structure, an update of the DEREK system. Toxicology 1996, 106, 267–279. [Google Scholar] [CrossRef]
- Lhasa Limited. Derek Nexus; Versão 10.0.2; Lhasa Limited: Leeds, UK, 2007. [Google Scholar]
- Goel, R.K.; Singh, D.; Lagunin, A.; Poroikov, V. PASS-assisted exploration of new therapeutic potential of natural products. Med. Chem. Res. 2011, 20, 1509–1514. [Google Scholar] [CrossRef]
- Ferreira, J.V.; Chaves, G.A.; Marino, B.L.B.; Sousa, K.P.A.; Souza, L.R.; Brito, M.F.B.; Texeira, H.R.C.; Silva, C.H.T.P.; Santos, C.B.R.; Hage-Melim, L.I.S. Cannabinoid type 1 receptor (CB1) bioligand with therapeutic potential for withdrawal syndrome in chemical dependents Cannabis sativa. Chemmedchem 2017, 12, 1408–1416. [Google Scholar] [CrossRef] [PubMed]
- Burkert, U.; Allinger, N.L. Molecular Mechanics. ACS Monograph 177; American Chemical Society: Washington, DC, USA, 1982. [Google Scholar]
- Kirchmair, J.; Göller, A.H.; Lang, D.; Kunze, J.; Testa, B.; Wilson, I.D.; Glen, R.C.; Schneider, G. Predicting drug metabolism: Experiment and/or computation? Nat. Rev. Drug Discov. 2015, 14, 387–404. [Google Scholar] [CrossRef]
- Cheung, J.; Rudolph, M.J.; Burshteyn, F.; Cassidy, M.S.; Gary, E.N.; Love, J.; Franklin, M.C.; Height, J.J. Structures of Human Acetylcholinesterase in Complex with Pharmacologically Important Ligands. J. Med. Chem. 2012, 55, 10282–10286. [Google Scholar] [CrossRef]
- Padilha, E.C.; Serafim, R.B.; Sarmiento, D.Y.R.; Santos, C.F.; Santos, C.B.R.; Silva, C.H.T.P. New PPARα/γ/δ optimal activator rationally designed by computational methods. J. Braz. Chem. Soc. 2016, 27, 1636–1647. [Google Scholar] [CrossRef]
- Pereira, A.L.E.; Santos, G.B.F.; Franco, M.S.F.; Federico, L.B.; Silva, C.H.T.P.; Santos, C.B.R. Molecular modeling and statistical analysis in the design of derivatives of human dipeptidyl peptidase IV. J. Biomol. Struct. Dyn. 2018, 36, 318–334. [Google Scholar] [CrossRef] [PubMed]
- Santos, C.B.R.; Ramos, R.S.; Ortiz, B.L.S.; Silva, G.M.; Giuliatti, S.; Balderas-Lopez, J.L.; Navarrete, A.; Carvalho, J.C.T. Oil from the fruits of Pterodon emarginatus Vog.: A traditional anti-inflammatory. Study combining in vivo and in silico. J. Ethnopharmacol. 2018, 222, 107–120. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.; Neto, M.; Silva, L.; Ramos, R.S.; Costa, J.C.; Brasil, D.; Lobato, C.; Costa, G.; Bittencourt, J.; Silva, C.; et al. Identification of Novel Protein Kinase Receptor Type 2 Inhibitors Using Pharmacophore and Structure-Based Virtual Screening. Molecules 2018, 23, 453. [Google Scholar] [CrossRef] [PubMed]
- Costa, J.S.; Costa, K.L.; Cruz, J.V.; Ramos, R.S.; Silva, L.B.; Brasil, D.S.B.; Silva, C.H.T.P.; Santos, C.B.R.; Macêdo, W.J.C. Virtual Screening and Statistical Analysis in the Design of New Caffeine Analogues Molecules with Potential Epithelial Anticancer Activity. Curr. Pharm. Des. 2017, 23, 1–19. [Google Scholar] [CrossRef]
- Studio Discovery, Insight, I; Accelrys Software Inc.: San Diego, CA, USA, 2009.












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Molecules | a Star | b CNS | c MW | d QP logKp | e HBD | f HBA | g R5 | h R3 |
|---|---|---|---|---|---|---|---|---|
| Normal range | 0–5 | −2 to +2 | <500 | −8 to −1 | <5 | <10 | Max. 4 | Max. 3 |
| Pyriproxyfen | 1 | 1 | 321.4 | 0.8 | 0 | 4 | 1 | 0 |
| ZINC11616655 | 0 | 0 | 376.5 | −1.6 | 1 | 3 | 0 | 1 |
| ZINC13537284 | 0 | 0 | 294.4 | −1.7 | 2 | 5 | 0 | 0 |
| ZINC00073711 | 0 | 1 | 201.2 | −1.8 | 1 | 3 | 0 | 0 |
| ZINC00001021 | 0 | 0 | 212.2 | −0.6 | 0 | 2 | 0 | 0 |
| ZINC11616399 | 0 | 0 | 384.5 | −2.3 | 0 | 4 | 0 | 1 |
| ZINC01530753 | 0 | 1 | 356.1 | −2.3 | 0 | 4 | 0 | 0 |
| ZINC01530718 | 0 | 1 | 267.4 | −3.0 | 3 | 4 | 0 | 0 |
| ZINC11616398 | 0 | 0 | 384.5 | −2.2 | 0 | 4 | 0 | 1 |
| ZINC00000257 | 0 | 0 | 295.4 | −4.1 | 1 | 4 | 0 | 0 |
| ZINC04363405 | 0 | 0 | 414.6 | −2.9 | 0 | 4 | 0 | 1 |
| ZINC03831238 | 0 | 0 | 299.4 | −3.3 | 4 | 3 | 0 | 0 |
| ZINC12504271 | 0 | 0 | 366.8 | −2.4 | 0 | 3 | 0 | 1 |
| ZINC00538483 | 0 | 1 | 371.9 | −3.4 | 1 | 6 | 0 | 0 |
| ZINC00001624 | 0 | 1 | 337.5 | −2.9 | 2 | 3 | 0 | 0 |
| Molecules | Prediction | Alert |
|---|---|---|
| Pyriproxyfen | - | No alert |
| ZINC11616655 | Hepatotoxicity | Plausible |
| Skin sensitization | ||
| Teratogenicity | ||
| Estrogenicity | ||
| ZINC13537284 | - | No alert |
| ZINC00073711 | Hepatotoxicity | Plausible |
| Teratogenicity | ||
| ZINC00001021 | - | No alert |
| ZINC11616399 | Hepatotoxicity | Plausible |
| Skin sensitization | ||
| Teratogenicity | ||
| Estrogenicity | ||
| ZINC01530753 | Carcinogenicity | Plausible |
| Chromosome damage | ||
| Skin sensitization | ||
| ZINC01530718 | - | No alert |
| ZINC11616398 | Hepatotoxicity | Plausible |
| Skin sensitization | ||
| Teratogenicity | ||
| Estrogenicity | ||
| ZINC00000257 | - | No alert |
| ZINC04363405 | Skin sensitization | Plausible |
| ZINC03831238 | hERG channel inhibition | Plausible |
| Skin sensitization | ||
| ZINC12504271 | Skin sensitization | Plausible |
| ZINC00538483 | hERG channel inhibition | Plausible |
| Skin sensitization | ||
| ZINC00001624 | - | No alert |
| Molecules | Pa a | Pi b | Biological Activity |
|---|---|---|---|
| Pyriproxyfen | 0.586 | 0.003 | Insecticide |
| I40 | 0.025 | 0.005 | Acetylcholine transporter inhibitor |
| GNT | 0.376 | 0.154 | Acetylcholine neuromuscular blocking agent |
| JHIII | 0.336 | 0.011 | Insecticide |
| ZINC13537284 | - | - | - |
| ZINC00001021 | 0.444 | 0.005 | Insecticide |
| ZINC01530718 | - | - | - |
| ZINC00000257 | - | - | - |
| ZINC00001624 | 0.450 | 0.005 | Acetylcholine antagonist |
| 0.433 | 0.044 | Acetyl esterase inhibitor |
| Molecules | Predicted LD50 (mg/kg) | Predicted Toxicity Class [a] |
|---|---|---|
| Pyriproxyfen (Control) | 2000 | IV |
| I40 | 200 | III |
| GNT | 19 | II |
| JHIII | 5000 | IV |
| ZINC00001021 | 1000 | IV |
| ZINC00001624 | 349 | III |
| Enzyme | Inhibitor | Coordinates of the Grid Center | Grid Size (Points) |
|---|---|---|---|
| AChE (PDB code: 1QON) | 9-(3-Iodobenzylamino)-1,2,3,4-Tetrahydroacridine | X = 33.4862 Y = 67.9151 Z = 9.4399 | 35 x 34 y 31 z |
| AChE (PDB code: 4EY6) | (−)-galanthamine | X = 9.090 Y = −60.485 Z = −23.703 | 32 x 38 y 36 z |
| Juvenile hormone (PDB code: 5V13) | methyl (2E,6E)-9-[(2R)-3,3-dimethyloxiran-2-yl]-3,7-dimethylnona-2,6-dienoate | X = −213.788 Y = 1.653 Z = 352.848 | 40 x 44 y 36 z |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramos, R.d.S.; Costa, J.d.S.; Silva, R.C.; da Costa, G.V.; Rodrigues, A.B.L.; Rabelo, É.d.M.; Souto, R.N.P.; Taft, C.A.; Silva, C.H.T.d.P.d.; Rosa, J.M.C.; et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals 2019, 12, 20. https://doi.org/10.3390/ph12010020
Ramos RdS, Costa JdS, Silva RC, da Costa GV, Rodrigues ABL, Rabelo ÉdM, Souto RNP, Taft CA, Silva CHTdPd, Rosa JMC, et al. Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals. 2019; 12(1):20. https://doi.org/10.3390/ph12010020
Chicago/Turabian StyleRamos, Ryan da Silva, Josivan da Silva Costa, Rai Campos Silva, Glauber Vilhena da Costa, Alex Bruno Lobato Rodrigues, Érica de Menezes Rabelo, Raimundo Nonato Picanço Souto, Carlton Anthony Taft, Carlos Henrique Tomich de Paula da Silva, Joaquín Maria Campos Rosa, and et al. 2019. "Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening" Pharmaceuticals 12, no. 1: 20. https://doi.org/10.3390/ph12010020
APA StyleRamos, R. d. S., Costa, J. d. S., Silva, R. C., da Costa, G. V., Rodrigues, A. B. L., Rabelo, É. d. M., Souto, R. N. P., Taft, C. A., Silva, C. H. T. d. P. d., Rosa, J. M. C., Santos, C. B. R. d., & Macêdo, W. J. d. C. (2019). Identification of Potential Inhibitors from Pyriproxyfen with Insecticidal Activity by Virtual Screening. Pharmaceuticals, 12(1), 20. https://doi.org/10.3390/ph12010020