The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value


Abstract
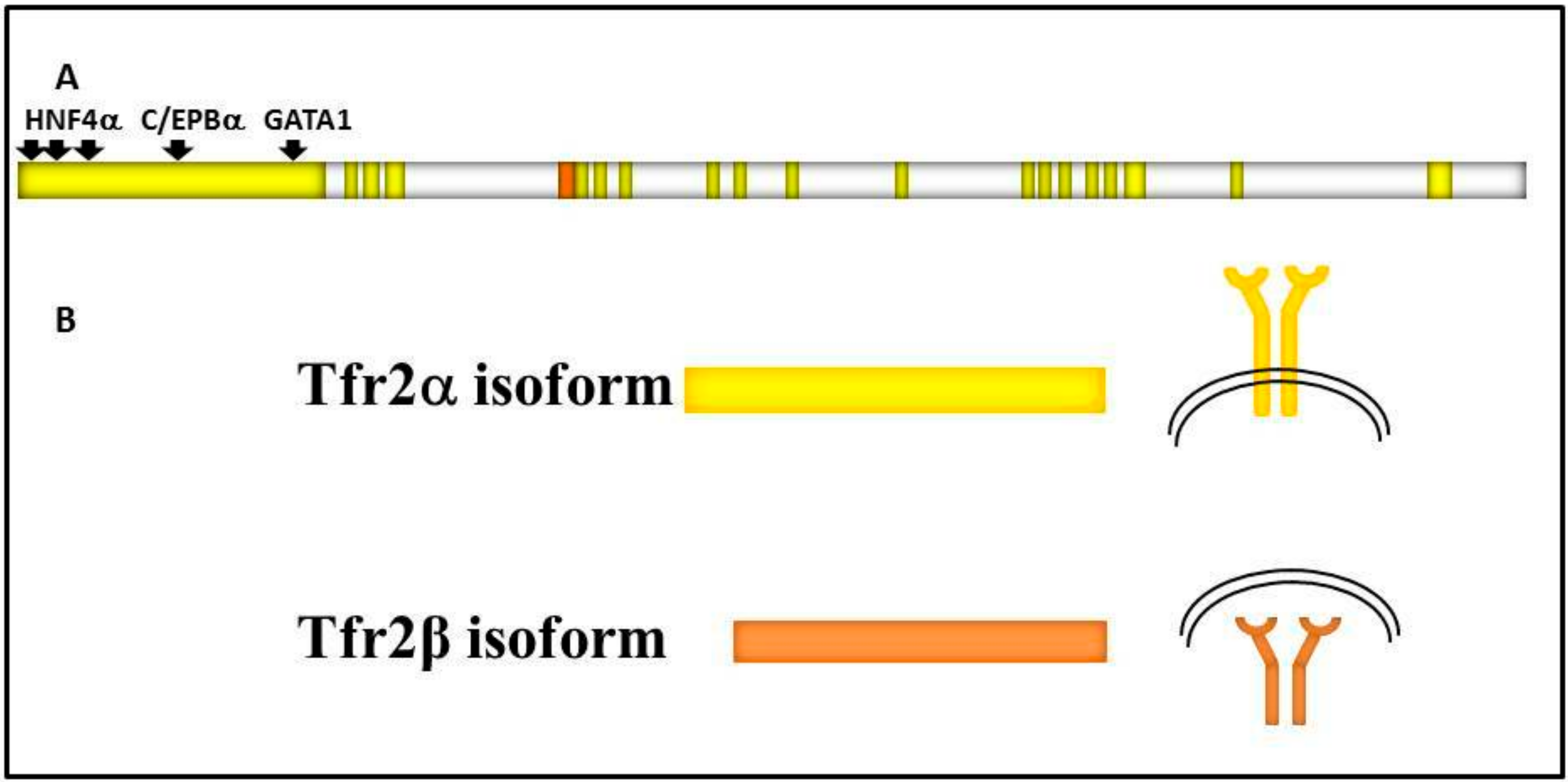
1. Tfr2 Gene and Proteins
1.1. Tfr2 and HFE3
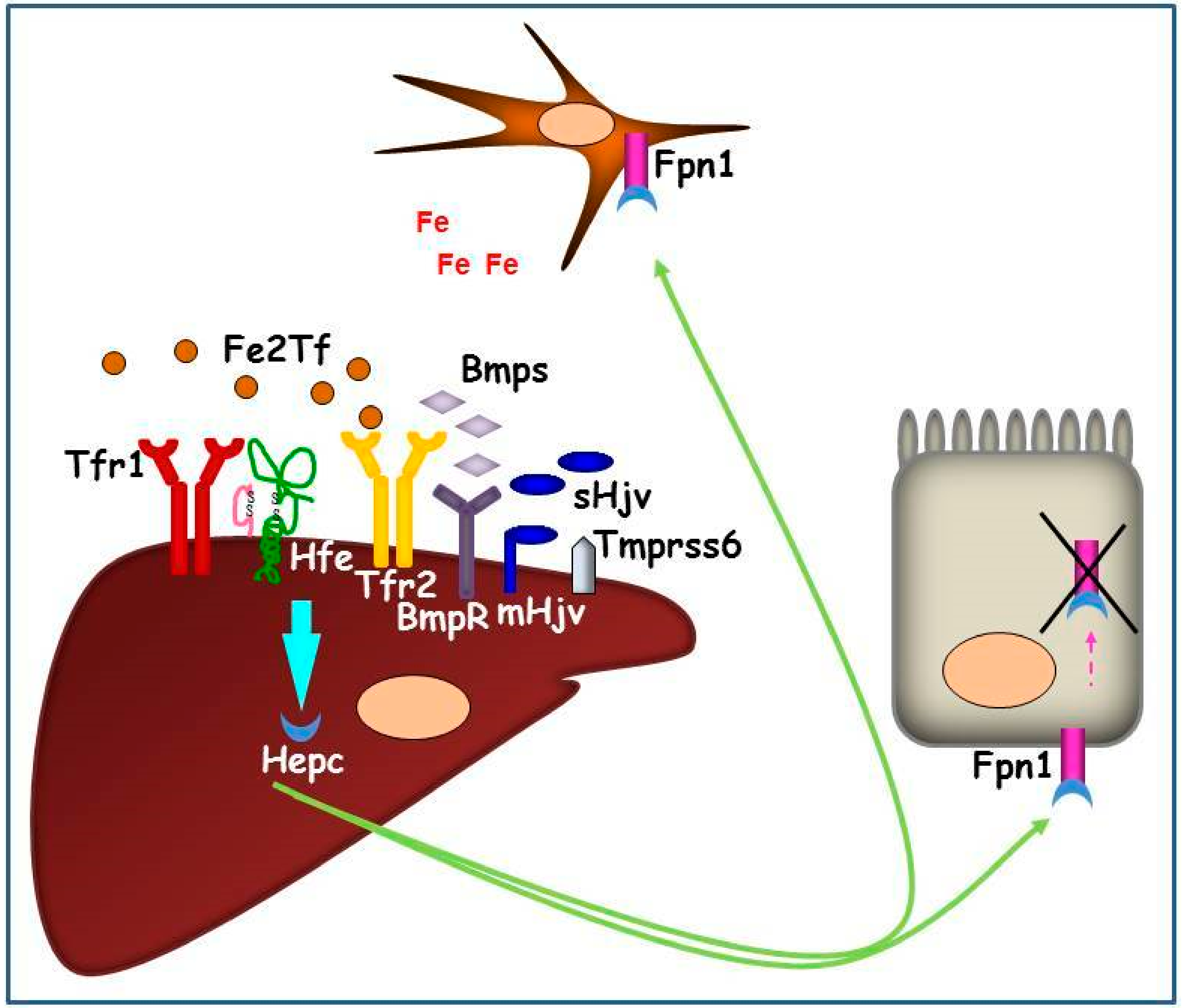
1.2. Systemic Iron Metabolism: The Hepc-Fpn1 Axis and the Proteins Involved in Hepc Regulation
2. Tfr2 in Liver
3. TFR2 Mouse Models
4. Tfr2 in Extrahepatic Tissues
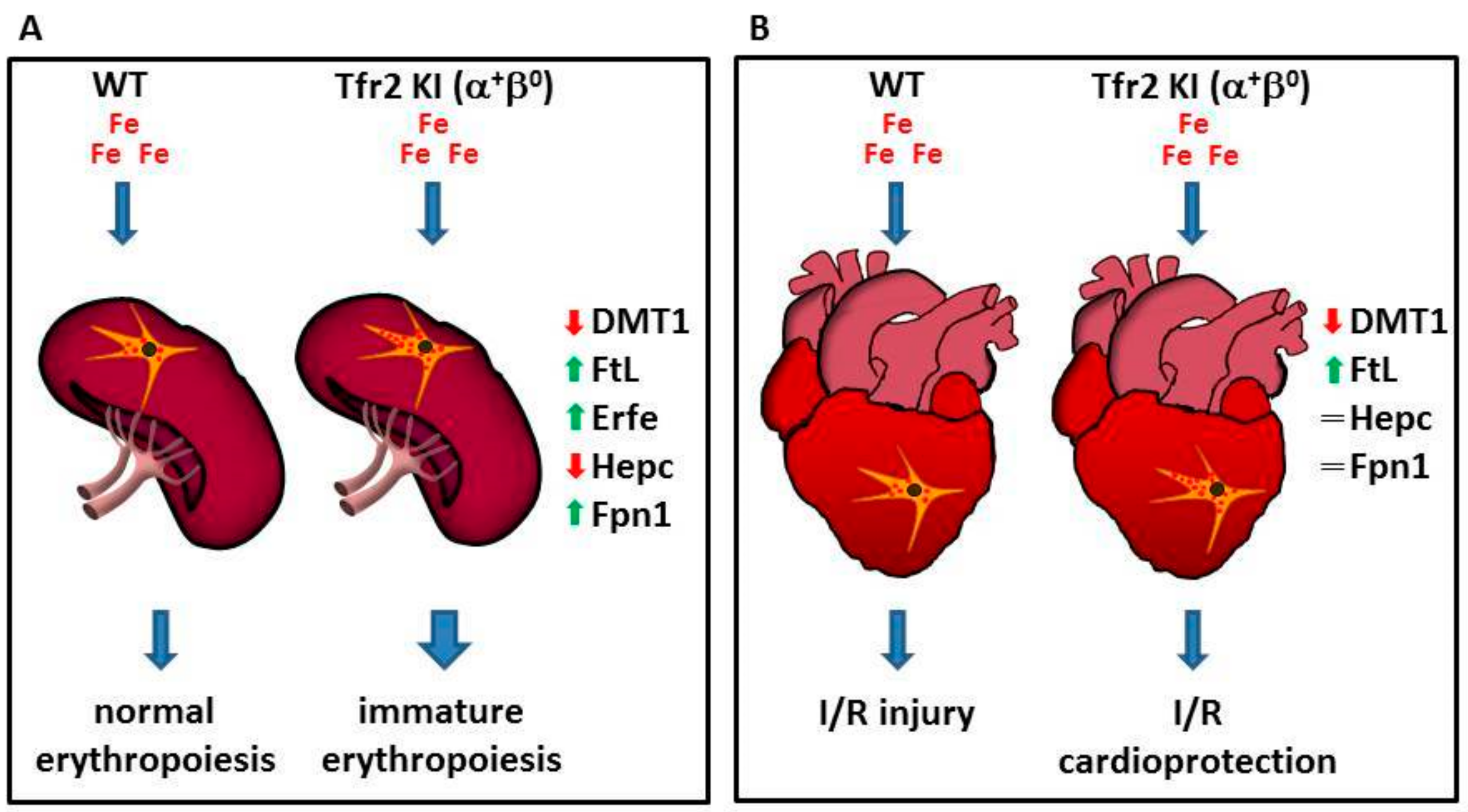
4.1. Tfr2 in the Erythropoietic Compartment
4.2. Tfr2β in the Heart
4.3. Tfr2 in the Central Nervous System (CNS)
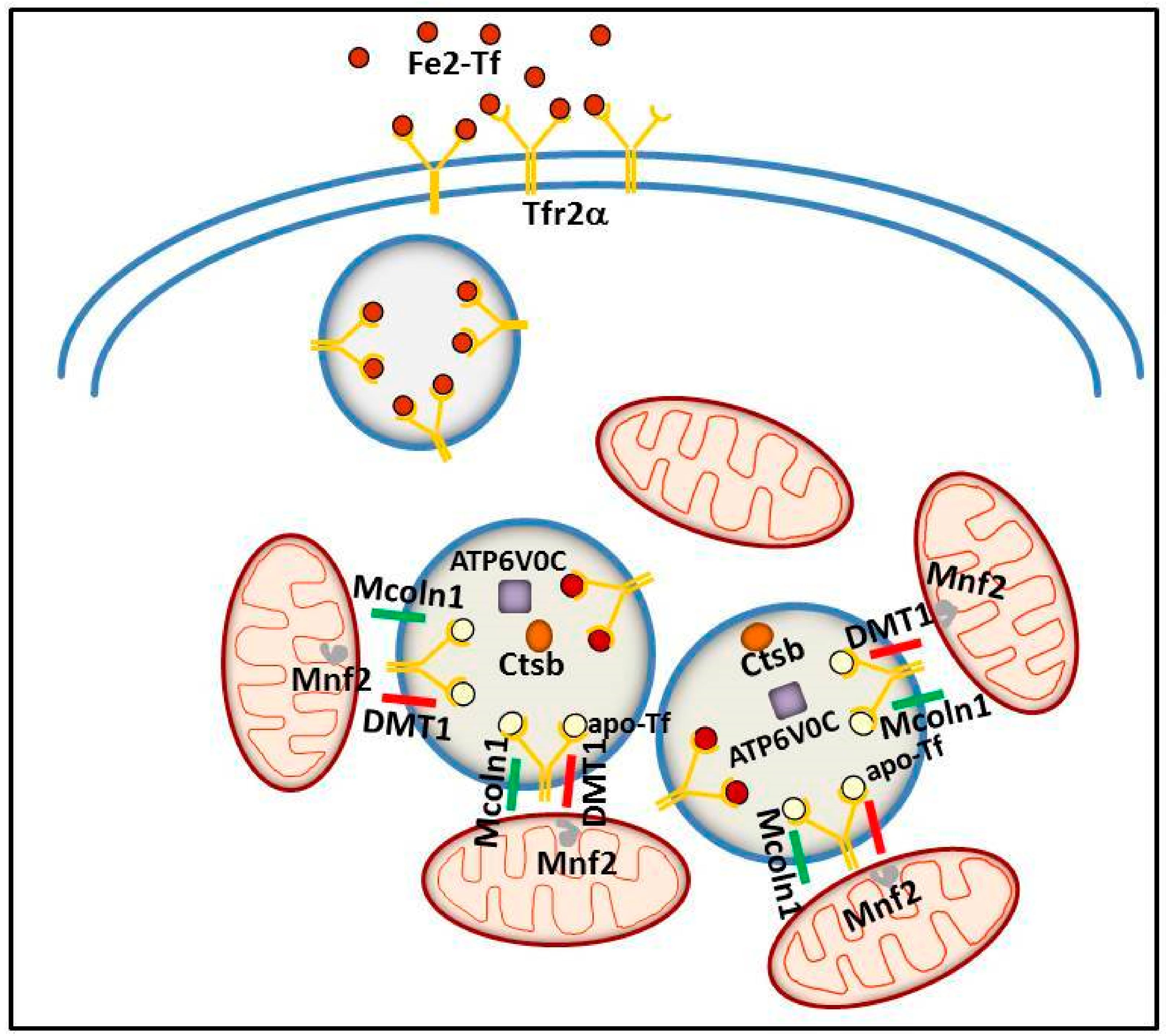
5. Tfr2 in Intracellular Iron Trafficking
6. Tfr2 in Other Diseases
6.1. Tfr2 in Cancer
6.2. Tfr2 in Alzheimer’s Disease (AD)
7. Tfr2 as a Therapeutic Target
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kawabata, H. Transferrin and transferrin receptors update. Free Radic. Biol. Med. 2018. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Yang, R.; Hirama, T.; Vuong, P.T.; Kawano, S.; Gombart, A.F.; Koeffler, H.P. Molecular cloning of transferrin receptor 2. A new member of the transferrin receptor-like family. J. Biol. Chem. 1999, 274, 20826–20832. [Google Scholar] [CrossRef] [PubMed]
- Mastroberardino, P.G.; Hoffman, E.K.; Horowitz, M.P.; Betarbet, R.; Taylor, G.; Cheng, D.; Na, H.M.; Gutekunst, C.A.; Gearing, M.; Trojanowski, J.Q.; et al. A novel transferrin/TfR2-mediated mitochondrial iron transport system is disrupted in Parkinson’s disease. Neurobiol. Dis. 2009, 34, 417–431. [Google Scholar] [CrossRef] [PubMed]
- Pagani, A.; Vieillevoye, M.; Nai, A.; Rausa, M.; Ladli, M.; Lacombe, C.; Mayeux, P.; Verdier, F.; Camaschella, C.; Silvestri, L. Regulation of cell surface transferrin receptor-2 by iron-dependent cleavage and release of a soluble form. Haematologica 2015, 100, 458–465. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Germain, R.S.; Ikezoe, T.; Tong, X.; Green, E.M.; Gombart, A.F.; Koeffler, H.P. Regulation of expression of murine transferrin receptor 2. Blood 2001, 98, 1949–1954. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, S.; Ogawa, M.; Muckenthaler, M.U.; Mizui, Y.; Sasaki, S.; Fujimura, T.; Takizawa, M.; Ariga, N.; Ozaki, H.; Sakaguchi, M.; et al. Hepatocyte Nuclear Factor 4α controls iron metabolism and regulates transferrin receptor 2 in mouse liver. J. Biol. Chem. 2015, 290, 30855–30865. [Google Scholar] [CrossRef] [PubMed]
- Muckenthaler, M.U.; Galy, B.; Hentze, M.W. Systemic iron homeostasis and the iron-responsive element/iron-regulatory protein (IRE/IRP) regulatory network. Annu. Rev. Nutr. 2008, 28, 197–213. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Enns, C.A. CD81 promotes both the degradation of transferrin receptor 2 (TfR2) and the Tfr2-mediated maintenance of hepcidin expression. J. Biol. Chem. 2015, 290, 7841–7850. [Google Scholar] [CrossRef] [PubMed]
- Camaschella, C.; Roetto, A.; Calì, A.; De Gobbi, M.; Garozzo, G.; Carella, M.; Majorano, N.; Totaro, A.; Gasparini, P. The gene TFR2 is mutated in a new type of haemochromatosis mapping to 7q22. Nat. Genet. 2000, 25, 14–15. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P.; Cavey, T.; Ropert, M.; Guggenbuhl, P.; Loréal, O. Genetic hemochromatosis: Pathophysiology; diagnostic and therapeutic management. Presse Med. 2017, 46, e288–e295. [Google Scholar] [CrossRef] [PubMed]
- De Gobbi, M.; Roetto, A. TFR2-Related Hereditary Hemochromatosis. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2005; pp. 1993–2018. [Google Scholar]
- Biasiotto, G.; Belloli, S.; Ruggeri, G.; Zanella, I.; Gerardi, G.; Corrado, M.; Gobbi, E.; Albertini, A.; Arosio, P. Identification of new mutations of the HFE, hepcidin, and transferrin receptor 2 genes by denaturing HPLC analysis of individuals with biochemical indications of iron overload. Clin. Chem. 2003, 49, 1981–1988. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Totaro, A.; Piperno, A.; Piga, A.; Longo, F.; Garozzo, G.; Calì, A.; De Gobbi, M.; Gasparini, P.; Camaschella, C. New mutations inactivating transferrin receptor 2 in hemochromatosis type 3. Blood 2001, 97, 2555–2560. [Google Scholar] [CrossRef] [PubMed]
- Bardou-Jacquet, E.; Cunat, S.; Beaumont-Epinette, M.P.; Kannengiesser, C.; Causse, X.; Sauvion, S.; Pouliquen, B.; Deugnier, Y.; David, V.; Loréal, O.; et al. Variable age of onset and clinical severity in transferrin receptor 2 related haemochromatosis: Novel observations. Br. J. Haematol. 2013, 162, 278–281. [Google Scholar] [CrossRef] [PubMed]
- Majore, S.; Milano, F.; Binni, F.; Stuppia, L.; Cerrone, A.; Tafuri, A.; De Bernardo, C.; Palka, G.; Grammatico, P. Homozygous p.M172K mutation of the TFR2 gene in an Italian family with type 3 hereditary hemochromatosis and early onset iron overload. Haematologica 2006, 91, ECR33. [Google Scholar] [PubMed]
- Finberg, K.E. Regulation of systemic iron homeostasis. Curr. Opin. Hematol. 2013, 20, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Hunter, H.N.; Fulton, D.B.; Ganz, T.; Vogel, H.J. The solution structure of human hepcidin; a peptide hormone with antimicrobial activity that is involved in iron uptake and hereditary hemochromatosis. J. Biol. Chem. 2002, 277, 37597–37603. [Google Scholar] [CrossRef] [PubMed]
- Ganz, T.; Nemeth, E. Hepcidin and iron homeostasis. Biochim. Biophys. Acta 2012, 1823, 1434–1443. [Google Scholar] [CrossRef] [PubMed]
- Courselaud, B.; Pigeon, C.; Inoue, Y.; Inoue, J.; Gonzalez, F.J.; Leroyer, P.; Gilot, D.; Boudjema, K.; Guguen-Guillouzo, C.; Brissot, P.; et al. C/EBPalpha regulates hepatic transcription of hepcidin;an antimicrobial peptide and regulator of iron metabolism. Cross-talk between C/EBP pathway and iron metabolism. J. Biol. Chem. 2002, 277, 41163–41170. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Zumbrennen-Bullough, K.B.; Core, A.B.; Wang, C.Y.; Nairz, M.; Bouley, R.; Swirski, F.K.; Babitt, J.L. Endothelial cells produce bone morphogenetic protein 6 required for iron homeostasis in mice. Blood 2017, 129, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Steinbicker, A.U.; Bartnikas, T.B.; Lohmeyer, L.K.; Leyton, P.; Mayeur, C.; Kao, S.M.; Pappas, A.E.; Peterson, R.T.; Bloch, D.B.; Yu, P.B.; et al. Perturbation of hepcidin expression by BMP type I receptor deletion induces iron overload in mice. Blood 2011, 118, 4224–4230. [Google Scholar] [CrossRef] [PubMed]
- Mayeur, C.; Leyton, P.A.; Kolodziej, S.A.; Yu, B.; Bloch, K.D. BMP type II receptors have redundant roles in the regulation of hepatic hepcidin gene expression and iron metabolism. Blood 2014, 124, 2116–2123. [Google Scholar] [CrossRef] [PubMed]
- Enns, C.A.; Ahmed, R.; Zhang, A.S. Neogenin interacts with matriptase-2 to facilitate hemojuvelin cleavage. J. Biol. Chem. 2012, 287, 35104–35117. [Google Scholar] [CrossRef] [PubMed]
- Canali, S.; Wang, C.Y.; Zumbrennen-Bullough, K.B.; Bayer, A.; Babitt, J.L. Bone morphogenetic protein 2 controls iron homeostasis in mice independent of Bmp6. Am. J. Hematol. 2017, 92, 1204–1213. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, P.J.; Toran, P.T.; Giannetti, A.M.; Bjorkman, P.J.; Andrews, N.C. The transferrin receptor modulates Hfe-dependent regulation of hepcidin expression. Cell Metab. 2008, 7, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Chen, J.; Kramer, M.; Tsukamoto, H.; Zhang, A.S.; Enns, C.A. Interaction of the hereditary hemochromatosis protein HFE with transferrin receptor 2 is required for transferrin-induced hepcidin expression. Cell Metab. 2009, 9, 217–227. [Google Scholar] [CrossRef] [PubMed]
- D’Alessio, F.; Hentze, M.; Muckenthaler, M.U. The hemochromatosis proteins HFE; TfR2; and HJV form a membrane-associated protein complex for hepcidin regulation. J. Hepatol. 2012, 57, 1052–1060. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.F.; Summerville, L.; Crampton, E.M.; Frazer, D.M.; Anderson, G.J.; Subramaniam, V.N. Combined deletion of Hfe and transferrin receptor 2 in mice leads to marked dysregulation of hepcidin and iron overload. Hepatology 2009, 50, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Poli, M.; Luscieti, S.; Gandini, V.; Maccarinelli, F.; Finazzi, D.; Silvestri, L.; Roetto, A.; Arosio, P. Transferrin receptor 2 and HFE regulate furin expression via mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/Erk) signaling. Implications for transferrin- dependent hepcidin regulation. Haematologica 2010, 95, 1832–1840. [Google Scholar] [CrossRef] [PubMed]
- Corradini, E.; Rozier, M.; Meynard, D.; Odhiambo, A.; Lin, H.Y.; Feng, Q.; Migas, M.C.; Britton, R.S.; Babitt, J.L.; Fleming, R.E. Iron regulation of hepcidin despite attenuated Smad1,5,8 signaling in mice without transferrin receptor 2 or Hfe. Gastroenterology 2011, 141, 1907–1914. [Google Scholar] [CrossRef] [PubMed]
- Silvestri, L.; Pagani, A.; Nai, A.; De Domenico, I.; Kaplan, J.; Camaschella, C. The serine protease matriptase-2 (TMPRSS6) inhibits hepcidin activation by cleaving membrane hemojuvelin. Cell Metab. 2008, 8, 502–511. [Google Scholar] [CrossRef] [PubMed]
- Meynard, D.; Vaja, V.; Sun, C.C.; Corradini, E.; Chen, S.; López-Otín, C.; Grgurevic, L.; Hong, C.C.; Stirnberg, M.; Gütschow, M.; et al. Regulation of TMPRSS6 by BMP6 and iron in human cells and mice. Blood 2011, 118, 747–756. [Google Scholar] [CrossRef] [PubMed]
- Nicolas, G.; Chauvet, C.; Viatte, L.; Danan, J.L.; Bigard, X.; Devaux, I.; Beaumont, C.; Kahn, A.; Vaulont, S. The gene encoding the iron regulatory peptide hepcidin is regulated by anemia; hypoxia; and inflammation. J. Clin. Investig. 2002, 110, 1037–1044. [Google Scholar] [CrossRef] [PubMed]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef] [PubMed]
- Papanikolaou, G.; Pantopoulos, K. Systemic iron homeostasis and erythropoiesis. IUBMB Life 2017, 69, 399–413. [Google Scholar] [CrossRef] [PubMed]
- Wrighting, D.M.; Andrews, N.C. Interleukin-6 induces hepcidin expression through STAT3. Blood 2006, 108, 3204–3209. [Google Scholar] [CrossRef] [PubMed]
- Armitage, A.E.; Eddowes, L.A.; Gileadi, U.; Cole, S.; Spottiswoode, N.; Selvakumar, T.A.; Ho, L.P.; Townsend, A.R.; Drakesmith, H. Hepcidin regulation by innate immune and infectious stimuli. Blood 2011, 118, 4129–4139. [Google Scholar] [CrossRef] [PubMed]
- Piperno, A.; Galimberti, S.; Mariani, R.; Pelucchi, S.; Ravasi, G.; Lombardi, C.; Bilo, G.; Revera, M.; Giuliano, A.; Faini, A.; et al. Modulation of hepcidin production during hypoxia-induced erythropoiesis in humans in vivo: Data from the HIGHCARE project. Blood 2011, 117, 2953–2959. [Google Scholar] [CrossRef] [PubMed]
- Talbot, N.P.; Lakhal, S.; Smith, T.G.; Privat, C.; Nickol, A.H.; Rivera-Ch, M.; León-Velarde, F.; Dorrington, K.L.; Mole, D.R.; Robbins, P.A. Regulation of hepcidin expression at high altitude. Blood 2012, 119, 857–860. [Google Scholar] [CrossRef] [PubMed]
- Maurer, E.; Gütschow, M.; Stirnberg, M. Matriptase-2 (TMPRSS6) is directly up-regulated by hypoxia inducible factor-1: Identification of a hypoxia-responsive element in the TMPRSS6 promoter region. Biol. Chem. 2012, 393, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, H.; Fleming, R.E.; Gui, D.; Moon, S.Y.; Saitoh, T.; O’Kelly, J.; Umehara, Y.; Wano, Y.; Said, J.W.; Koeffler, H.P. Expression of hepcidin is down-regulated in TfR2 mutant mice manifesting a phenotype of hereditary hemochromatosis. Blood 2005, 105, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Nemeth, E.; Roetto, A.; Garozzo, G.; Ganz, T.; Camaschella, C. Hepcidin is decreased in TFR2 hemochromatosis. Blood 2005, 105, 1803–1806. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; She, E.; Gelbart, T.; Truksa, J.; Lee, P.; Xia, Y.; Khovananth, K.; Mudd, S.; Mann, N.; Moresco, E.M.; et al. The serine protease TMPRSS6 is required to sense iron deficiency. Science 2008, 320, 1088–1092. [Google Scholar] [CrossRef] [PubMed]
- Finberg, K.E.; Heeney, M.M.; Campagna, D.R.; Aydinok, Y.; Pearson, H.A.; Hartman, K.R.; Mayo, M.M.; Samuel, S.M.; Strouse, J.J.; Markianos, K.; et al. Mutations in TMPRSS6 cause iron-refractory iron deficiency anemia (IRIDA). Nat. Genet. 2008, 40, 569–571. [Google Scholar] [CrossRef] [PubMed]
- Folgueras, A.R.; de Lara, F.M.; Pendás, A.M.; Garabaya, C.; Rodríguez, F.; Astudillo, A.; Bernal, T.; Cabanillas, R.; López-Otín, C.; Velasco, G. Membrane-bound serine protease matriptase-2 (Tmprss6) is an essential regulator of iron homeostasis. Blood 2008, 112, 2539–2545. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Raggi, C.; Deaglio, S.; Sposi, N.M.; Stafsnes, M.; Fecchi, K.; Parolini, I.; Malavasi, F.; Peschle, C.; Sargiacomo, M.; et al. TfR2 localizes in lipid raft domains and is released in exosomes to activate signal transduction along the MAPK pathway. J. Cell Sci. 2006, 119, 4486–4498. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wang, J.; Meyers, K.R.; Enns, C.A. Transferrin-directed internalization and cycling of transferrin receptor 2. Traffic 2009, 10, 1488–1501. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Enns, C.A. Diferric transferrin regulates transferrin receptor 2 protein stability. Blood 2004, 104, 4287–4293. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Enns, C.A. The cytoplasmic domain of transferrin receptor 2 dictates its stability and response to holo-transferrin in Hep3B cells. J. Biol. Chem. 2007, 282, 6201–6209. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.B.; Chen, J.; Murchison, N.; Green, F.A.; Enns, C.A. Transferrin receptor 2: Evidence for ligand-induced stabilization and redirection to a recycling pathway. Mol. Biol. Cell 2007, 18, 743–754. [Google Scholar] [CrossRef] [PubMed]
- Goswami, T.; Andrews, N.C. Hereditary hemochromatosis protein; HFE; interaction with transferrin receptor 2 suggests a molecular mechanism for mammalian iron sensing. J. Biol. Chem. 2006, 281, 28494–28498. [Google Scholar] [CrossRef] [PubMed]
- Valore, E.V.; Ganz, T. Posttranslational processing of hepcidin in human hepatocytes is mediated by the prohormone convertase furin. Blood Cells Mol. Dis. 2008, 40, 132–138. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Ahmann, J.R.; Migas, M.C.; Waheed, A.; Koeffler, H.P.; Kawabata, H.; Britton, R.S.; Bacon, B.R.; Sly, W.S. Targeted mutagenesis of the murine transferrin receptor-2 gene produces hemochromatosis. Proc. Natl. Acad. Sci. USA 2002, 99, 10653–10658. [Google Scholar] [CrossRef] [PubMed]
- Wallace, D.F.; Summerville, L.; Subramaniam, V.N. Targeted disruption of the hepatic transferrin receptor 2 gene in mice leads to iron overload. Gastroenterology 2007, 132, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Roetto, A.; Di Cunto, F.; Pellegrino, R.M.; Hirsch, E.; Azzolino, O.; Bondi, A.; Defilippi, I.; Carturan, S.; Miniscalco, B.; Riondato, F.; et al. Comparison of 3 Tfr2-deficient murine models suggests distinct functions for Tfr2-alpha and Tfr2-beta isoforms in different tissues. Blood 2010, 115, 3382–3389. [Google Scholar] [CrossRef] [PubMed]
- Fleming, R.E.; Feng, Q.; Britton, R.S. Knockout mouse models of iron homeostasis. Annu. Rev. Nutr. 2011, 31, 117–137. [Google Scholar] [CrossRef] [PubMed]
- Gutschow, P.; Schmidt, P.J.; Han, H.; Ostland, V.; Bartnikas, T.B.; Pettiglio, M.A.; Herrera, C.; Butler, J.S.; Nemeth, E.; Ganz, T.; et al. A competitive enzyme-linked immunosorbent assay specific for murine hepcidin-1: Correlation with hepatic mRNA expression in established and novel models of dysregulated iron homeostasis. Haematologica 2015, 100, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Latour, C.; Besson-Fournier, C.; Meynard, D.; Silvestri, L.; Gourbeyre, O.; Aguilar-Martinez, P.; Schmidt, P.J.; Fleming, M.D.; Roth, M.P.; Coppin, H. Differing impact of the deletion of hemochromatosis-associated molecules HFE and transferrin receptor-2 on the iron phenotype of mice lacking bone morphogenetic protein 6 or hemojuvelin. Hepatology 2016, 63, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Pellegrino, R.M.; Rausa, M.; Pagani, A.; Boero, M.; Silvestri, L.; Saglio, G.; Roetto, A.; Camaschella, C. The erythroid function of transferrin receptor 2 revealed by Tmprss6 inactivation in different models of transferrin receptor 2 knockout mice. Haematologica 2014, 99, 1016–1021. [Google Scholar] [CrossRef] [PubMed]
- Rishi, G.; Secondes, E.S.; Wallace, D.F.; Subramaniam, V.N. Hematopoietic deletion of transferrin receptor 2 in mice leads to a block in erythroid differentiation during iron-deficient anemia. Am. J. Hematol. 2016, 91, 812–818. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Chiabrando, D.; Messana, E.; Stolte, J.; Turco, E.; Tolosano, E.; Muckenthaler, M.U. Heme controls ferroportin1 (FPN1) transcription involving Bach1; Nrf2 and a MARE/ARE sequence motif at position -7007 of the FPN1 promoter. Haematologica 2010, 95, 1261–1268. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Fiorito, V.; Marro, S.; Silengo, L.; Altruda, F.; Tolosano, E. Cell-specific regulation of Ferroportin transcription following experimentally-induced acute anemia in mice. Blood Cells Mol. Dis. 2013, 50, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Soranzo, N.; Spector, T.D.; Mangino, M.; Kühnel, B.; Rendon, A.; Teumer, A.; Willenborg, C.; Wright, B.; Chen, L.; Li, M.; et al. A genome-wide meta-analysis identifies 22 loci associated with eight hematological parameters in the HaemGen consortium. Nat. Genet. 2009, 41, 1182–1190. [Google Scholar] [CrossRef] [PubMed]
- Auer, P.L.; Teumer, A.; Schick, U.; O’Shaughnessy, A.; Lo, K.S.; Chami, N.; Carlson, C.; de Denus, S.; Dubé, M.P.; Haessler, J.; et al. Rare and low frequency coding variants in CXCR2 and other genes are associated with hematological traits. Nat. Genet. 2014, 46, 629–634. [Google Scholar] [CrossRef] [PubMed]
- Forejtnikovà, H.; Vieillevoye, M.; Zermati, Y.; Lambert, M.; Pellegrino, R.M.; Guihard, S.; Gaudry, M.; Camaschella, C.; Lacombe, C.; Roetto, A.; et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 2010, 116, 5357–5367. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Lidonnici, M.R.; Rausa, M.; Mandelli, G.; Pagani, A.; Silvestri, L.; Ferrari, G.; Camaschella, C. The second transferrin receptor regulates red blood cell production in mice. Blood 2015, 125, 1170–1179. [Google Scholar] [CrossRef] [PubMed]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, S.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.M.; Riondato, F.; Ferbo, L.; Boero, M.; Palmieri, A.; Osella, L.; Pollicino, P.; Miniscalco, B.; Saglio, G.; Roetto, A. Altered Erythropoiesis in Mouse Models of Type 3 Hemochromatosis. Biomed. Res. Int. 2017, 2408941. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Barrientos, T.; Mao, L.; Rockman, H.A.; Sauve, A.A.; Andrews, N.C. Lethal cardiomyopathy in mice lacking transferrin receptor in the heart. Cell Rep. 2015, 13, 533–545. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Wolna, M.; Carr, C.A.; Miller, J.J.; Christian, H.C.; Ball, V.; Santos, A.; Diaz, R.; Biggs, D.; Stillion, R.; et al. Cardiac ferroportin regulates cellular iron homeostasis and is important for cardiac function. Proc. Natl. Acad. Sci. USA 2015, 112, 3164–3169. [Google Scholar] [CrossRef] [PubMed]
- Lakhal-Littleton, S.; Wolna, M.; Chung, Y.J.; Christian, H.C.; Heather, L.C.; Brescia, M.; Ball, V.; Diaz, R.; Santos, A.; Biggs, D.; et al. An essential cell-autonomous role for hepcidin in cardiac iron homeostasis. Elife 2016, 5, e19804. [Google Scholar] [CrossRef] [PubMed]
- Gammella, E.; Recalcati, S.; Rybinska, I.; Buratti, P.; Cairo, G. Iron-induced damage in cardiomyopathy: Oxidative-dependent and independent mechanisms. Oxid. Med. Cell. Longev. 2015. [Google Scholar] [CrossRef] [PubMed]
- Boero, M.; Pagliaro, P.; Tullio, F.; Pellegrino, R.M.; Palmieri, A.; Ferbo, L.; Saglio, G.; De Gobbi, M.; Penna, C.; Roetto, A. A comparative study of myocardial molecular phenotypes of two Tfrβ null mice: Role in ischemia/reperfusion. Biofactors 2015, 41, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Chevion, M.; Leibowitz, S.; Aye, N.N.; Novogrodsky, O.; Singer, A.; Avizemer, O.; Bulvik, B.; Konijn, A.M.; Berenshtein, E. Heart protection by ischemic preconditioning: A novel pathway initiated by iron and mediated by ferritin. J. Mol. Cell. Cardiol. 2008, 45, 839–845. [Google Scholar] [CrossRef] [PubMed]
- Aquino, D.; Bizzi, A.; Grisoli, M.; Garavaglia, B.; Bruzzone, M.G.; Nardocci, N.; Savoiardo, M.; Chiapparini, L. Age-related iron deposition in the basal ganglia: Quantitative analysis in healthy subjects. Radiology 2009, 252, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Pfefferbaum, A.; Adalsteinsson, E.; Rohlfing, T.; Sullivan, E.V. MRI estimates of brain iron concentration in normal aging: Comparison of field-dependent (FDRI) and phase (SWI) methods. Neuroimage 2009, 47, 493–500. [Google Scholar] [CrossRef] [PubMed]
- Carlson, E.S.; Fretham, S.J.; Unger, E.; O’Connor, M.; Petryk, A.; Schallert, T.; Rao, R.; Tkac, I.; Georgieff, M.K. Hippocampus specific iron deficiency alters competition and cooperation between developing memory systems. J. Neurodev. Disord. 2010, 2, 133–143. [Google Scholar] [CrossRef] [PubMed]
- Millichap, J.G. Etiologic classification of attention-deficit/hyperactivity disorder. Pediatrics 2008, 12, e358–e365. [Google Scholar] [CrossRef] [PubMed]
- Benton, D. The influence of dietary status on the cognitive performance of children. Mol. Nutr. Food Res. 2010, 54, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Wang, Y.; Song, N.; Wang, J.; Jiang, H.; Xie, J. New Progress on the Role of Glia in Iron Metabolism and Iron-Induced Degeneration of Dopamine Neurons in Parkinson’s Disease. Front. Mol. Neurosci. 2018, 10, 455. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, R.C.; Kosman, D.J. Mechanistic analysis of iron accumulation by endothelial cells of the BBB. Biometals 2012, 25, 665–675. [Google Scholar] [CrossRef] [PubMed]
- Benarroch, E.E. Brain iron homeostasis and neurodegenerative disease. Neurology 2009, 72, 1436–1440. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.; Dong, X.P.; Wang, F.; Xu, H. Mechanisms of brain iron transport: Insight into neurodegeneration and CNS disorders. Future Med. Chem. 2010, 2, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Jiang, H.; Xie, J.X. Ferroportin1 and hephaestin are involved in the nigral iron accumulation of 6-OHDA-lesioned rats. Eur. J. Neurosci. 2007, 25, 2766–2772. [Google Scholar] [CrossRef] [PubMed]
- Morris, C.M.; Candy, J.M.; Keith, A.B.; Oakley, A.E.; Taylor, G.A.; Pullen, R.G.; Bloxham, C.A.; Gocht, A.; Edwardson, J.A. Brain iron homeostasis. J. Inorg. Biochem. 1992, 47, 257–265. [Google Scholar] [CrossRef]
- Connor, J.R.; Boeshore, K.L.; Benkovic, S.A.; Menzies, S.L. Isoforms of ferritin have a specific cellular distribution in the brain. J. Neurosci. Res. 1994, 37, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Rouault, T.A. Iron metabolism in the CNS: Implications for neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.M.; Fu, L.J.; Duan, X.L.; Crooks, D.R.; Yu, P.; Qian, Z.M.; Di, X.J.; Li, J.; Rouault, T.A.; Chang, Y.Z. Role of hepcidin in murine brain iron metabolism. Cell. Mol. Life Sci. 2010, 67, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Du, F.; Qian, Z.M.; Luo, Q.; Yung, W.H.; Ke, Y. Hepcidin suppresses brain iron accumulation by downregulating iron transport proteins in iron-overloaded rats. Mol. Neurobiol. 2015, 52, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Vela, D. Hepcidin, an emerging and important player in brain iron homeostasis. J. Transl. Med. 2018, 16. [Google Scholar] [CrossRef] [PubMed]
- Moos, T.; Rosengren Nielsen, T.; Skjørringe, T.; Morgan, E.H. Iron trafficking inside the brain. J. Neurochem. 2007, 103, 1730–1740. [Google Scholar] [CrossRef] [PubMed]
- Hänninen, M.M.; Haapasalo, J.; Haapasalo, H.; Fleming, R.E.; Britton, R.S.; Bacon, B.R.; Parkkila, S. Expression of iron-related genes in human brain and brain tumors. BMC Neurosci. 2009, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Acikyol, B.; Graham, R.M.; Trinder, D.; House, M.J.; Olynyk, J.K.; Scott, R.J.; Milward, E.A.; Johnstone, D.M. Brain transcriptome perturbations in the transferrin receptor 2 mutant mouse support the case for brain changes in iron loading disorders, including effects relating to long-term depression and long-term potentiation. Neuroscience 2013, 235, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Pellegrino, R.M.; Boda, E.; Montarolo, F.; Boero, M.; Mezzanotte, M.; Saglio, G.; Buffo, A.; Roetto, A. Transferrin Receptor 2 Dependent Alterations of Brain Iron Metabolism Affect Anxiety Circuits in the Mouse. Sci. Rep. 2016, 6, 30725. [Google Scholar] [CrossRef] [PubMed]
- Adhikari, A. Distributed circuits underlying anxiety. Front. Behav. Neurosci. 2014, 8, 112. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, S.L.; Buchanan, D.D.; Ahmed, I.; Taylor, K.D.; Loriot, M.A.; Sinsheimer, J.S.; Bronstein, J.M.; Elbaz, A.; Mellick, G.D.; Rotter, J.I.; et al. Pooled analysis of iron-related genes in Parkinson’s disease: Association with transferrin. Neurobiol. Dis. 2014, 62, 172–178. [Google Scholar] [CrossRef] [PubMed]
- Khalil, S.; Holy, M.; Grado, S.; Fleming, R.; Kurita, R.; Nakamura, Y.; Goldfarb, A. A specialized pathway for erythroid iron delivery through lysosomal trafficking of transferrin receptor 2. Blood Adv. 2017, 1, 1181–1194. [Google Scholar] [CrossRef] [PubMed]
- Robb, A.D.; Ericsson, M.; Wessling-Resnick, M. Transferrin receptor 2 mediates a biphasic pattern of transferrin uptake associated with ligand delivery to multivesicular bodies. Am. J. Physiol. Cell Physiol. 2004, 287, C1769–C1775. [Google Scholar] [CrossRef] [PubMed]
- Smilevska, T.; Stamatopoulos, K.; Samara, M.; Belessi, C.; Tsompanakou, A.; Paterakis, G.; Stavroyianni, N.; Athanasiadou, I.; Chiotoglou, I.; Hadzidimitriou, A.; et al. Transferrin receptor-1 and 2 expression in chronic lymphocytic leukemia. Leuk. Res. 2006, 30, 183–189. [Google Scholar] [CrossRef] [PubMed]
- Gangat, N.; Patnaik, M.M.; Tefferi, A. Myelodysplastic syndromes: Contemporary review and how we treat. Am. J. Hematol. 2016, 91, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Di Savino, A.; Gaidano, V.; Palmieri, A.; Crasto, F.; Volpengo, A.; Lorenzatti, R.; Scaravaglio, P.; Manello, A.; Nicoli, P.; Gottardi, E.; et al. Clinical significance of TFR2 and EPOR expression in bone marrow cells in myelodysplastic syndromes. Br. J. Haematol. 2017, 176, 491–495. [Google Scholar] [CrossRef] [PubMed]
- Nakamaki, T.; Kawabata, H.; Saito, B.; Matsunawa, M.; Suzuki, J.; Adachi, D.; Tomoyasu, S.; Phillip Koeffler, H. Elevated levels of transferrin receptor 2 mRNA, not transferrin receptor 1 mRNA, are associated with increased survival in acute myeloid leukaemia. Br. J. Haematol. 2004, 125, 42–49. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Oliviero, I.; Deaglio, S.; Mariani, G.; Biffoni, M.; Sposi, N.M.; Malavasi, F.; Peschle, C.; Testa, U. Transferrin receptor 2 is frequently expressed in human cancer cell lines. Blood Cells Mol. Dis. 2007, 39, 82–91. [Google Scholar] [CrossRef] [PubMed]
- Calzolari, A.; Larocca, L.M.; Deaglio, S.; Finisguerra, V.; Boe, A.; Raggi, C.; Ricci-Vitani, L.; Pierconti, F.; Malavasi, F.; De Maria, R.; et al. Transferrin receptor 2 is frequently and highly expressed in glioblastomas. Transl. Oncol. 2010, 3, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural. Transm. 2011, 118, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Crespo, Â.C.; Silva, B.; Marques, L.; Marcelino, E.; Maruta, C.; Costa, S.; Timóteo, A.; Vilares, A.; Couto, F.S.; Faustino, P.; et al. Genetic and biochemical markers in patients with Alzheimer’s disease support a concerted systemic iron homeostasis dysregulation. Neurobiol. Aging 2014, 35, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Artuso, I.; Lidonnici, M.R.; Altamura, S.; Mandelli, G.; Pettinato, M.; Muckenthaler, M.U.; Silvestri, L.; Ferrari, G.; Camaschella, C.; Nai, A. Transferrin Receptor 2 is a potential novel therapeutic target for beta-thalassemia: Evidence from a murine model. Blood 2018. [Google Scholar] [CrossRef] [PubMed]
- Girelli, D.; Nemeth, E.; Swinkels, D.W. Hepcidin in the diagnosis of iron disorders. Blood 2016, 127, 2809–2813. [Google Scholar] [CrossRef] [PubMed]
- Akinc, A.; Chan-Daniels, A.; Sehgal, A.; Foster, D.; Bettencourt, B.R.; Hettinger, J.; Racie, T.; Aubin, J.; Kuchimanchi, S.; Epstein-Barashand, H.; et al. Targeting the hepcidin pathway with RNAi therapeutics for the treatment of anemia. Blood 2011, 21, 688. [Google Scholar]
- Raha, A.A.; Vaishnav, R.A.; Friedland, R.P.; Bomford, A.; Raha-Chowdhury, R. The systemic iron-regulatory proteins hepcidin and ferroportin are reduced in the brain in Alzheimer’s disease. Acta Neuropathol. Commun. 2013, 1, 55. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Kanthasamy, A.G.; Reddy, M.B. EGCG protects against 6-OHDA-induced neurotoxicity in a cell culture model. Parkinsons Dis. 2015. [Google Scholar] [CrossRef] [PubMed]
- Clardy, S.L.; Wang, X.; Boyer, P.J.; Earley, C.J.; Allen, R.P.; Connor, J.R. Is ferroportin–hepcidin signaling altered in restless legs syndrome? J. Neurol. Sci. 2006, 247, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Zheng, M.; Tao, W.; Zou, Y.; Farokhzad, O.C.; Shi, B. Nanotechnology-Based Strategies for siRNA Brain Delivery for Disease Therapy. Trends Biotechnol. 2018, 36, 562–575. [Google Scholar] [CrossRef] [PubMed]
- Couch, J.A.; Yu, Y.J.; Zhang, Y.; Tarrant, J.M.; Fuji, R.N.; Meilandt, W.J.; Solanoy, H.; Tong, R.K.; Hoyte, K.; Luk, W.; et al. Addressing safety liabilities of TfR bispecific antibodies that cross the blood-brain barrier. Sci. Transl. Med. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebrón, J.A.; Björkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef] [PubMed]
- Mazzucchelli, S.; Truffi, M.; Baccarini, F.; Beretta, M.; Sorrentino, L.; Bellini, M.; Rizzuto, M.A.; Ottria, R.; Ravelli, A.; Ciuffreda, P.; et al. H-Ferritin-nanocaged olaparib: A promising choice for both BRCA-mutated and sporadic triple negative breast cancer. Sci. Rep. 2017, 7, 7505. [Google Scholar] [CrossRef] [PubMed]
- Sakurai, K.; Sohda, T.; Ueda, S.; Tanaka, T.; Hirano, G.; Yokoyama, K.; Morihara, D.; Aanan, A.; Takeyama, Y.; Irie, M.; et al. Immunohistochemical demonstration of transferrin receptor 1 and 2 in human hepatocellular carcinoma tissue. Hepatogastroenterology 2014, 61, 426–430. [Google Scholar] [PubMed]
- Voth, B.; Nagasawa, D.T.; Pelargos, P.E.; Chung, L.K.; Ung, N.; Gopen, Q.; Tenn, S.; Kamei, D.T.; Yang, I. Transferrin receptors and glioblastoma multiforme: Current findings and potential for treatment. J. Clin. Neurosci. 2015, 22, 1071–1076. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.P.; Smith, R.A. Drug delivery to mitochondria: The key to mitochondrial medicine. Adv. Drug Deliv. Rev. 2000, 41, 235–250. [Google Scholar] [CrossRef]
- Bürk, K. Friedreich Ataxia: Current status and future prospects. Cerebellum Ataxias 2017. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| HH type | Acronym | Inheritance | Gene | Protein | Function |
|---|---|---|---|---|---|
| HFE1 | HFE-HHC | AR | HFE | Hfe | Hepc regulator |
| HFE2a | HJV-HHC | AR | HJV | Hemojuvelin | Hepc regulator |
| HFE2b | HEPC-HHC | AR | HAMP | Hepc | Fe absorption inhibitor |
| HFE3 | TFR2-HHC | AR | TFR2 | Tfr2 | Hepc regulator |
| HFE4 | FPN1-HHC | AD | SLC40A1 | Fpn1 | Hepc receptor |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roetto, A.; Mezzanotte, M.; Pellegrino, R.M. The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value. Pharmaceuticals 2018, 11, 115. https://doi.org/10.3390/ph11040115
Roetto A, Mezzanotte M, Pellegrino RM. The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value. Pharmaceuticals. 2018; 11(4):115. https://doi.org/10.3390/ph11040115
Chicago/Turabian StyleRoetto, Antonella, Mariarosa Mezzanotte, and Rosa Maria Pellegrino. 2018. "The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value" Pharmaceuticals 11, no. 4: 115. https://doi.org/10.3390/ph11040115
APA StyleRoetto, A., Mezzanotte, M., & Pellegrino, R. M. (2018). The Functional Versatility of Transferrin Receptor 2 and Its Therapeutic Value. Pharmaceuticals, 11(4), 115. https://doi.org/10.3390/ph11040115