New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases



Abstract
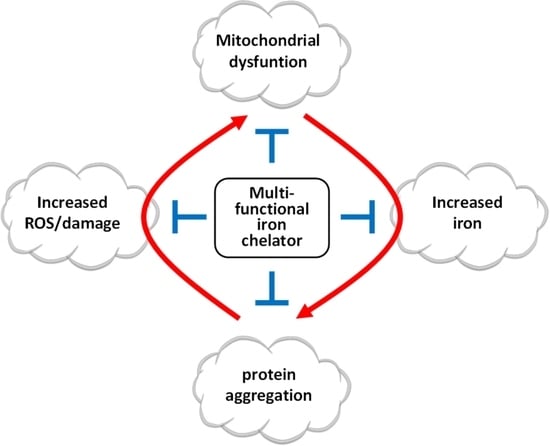
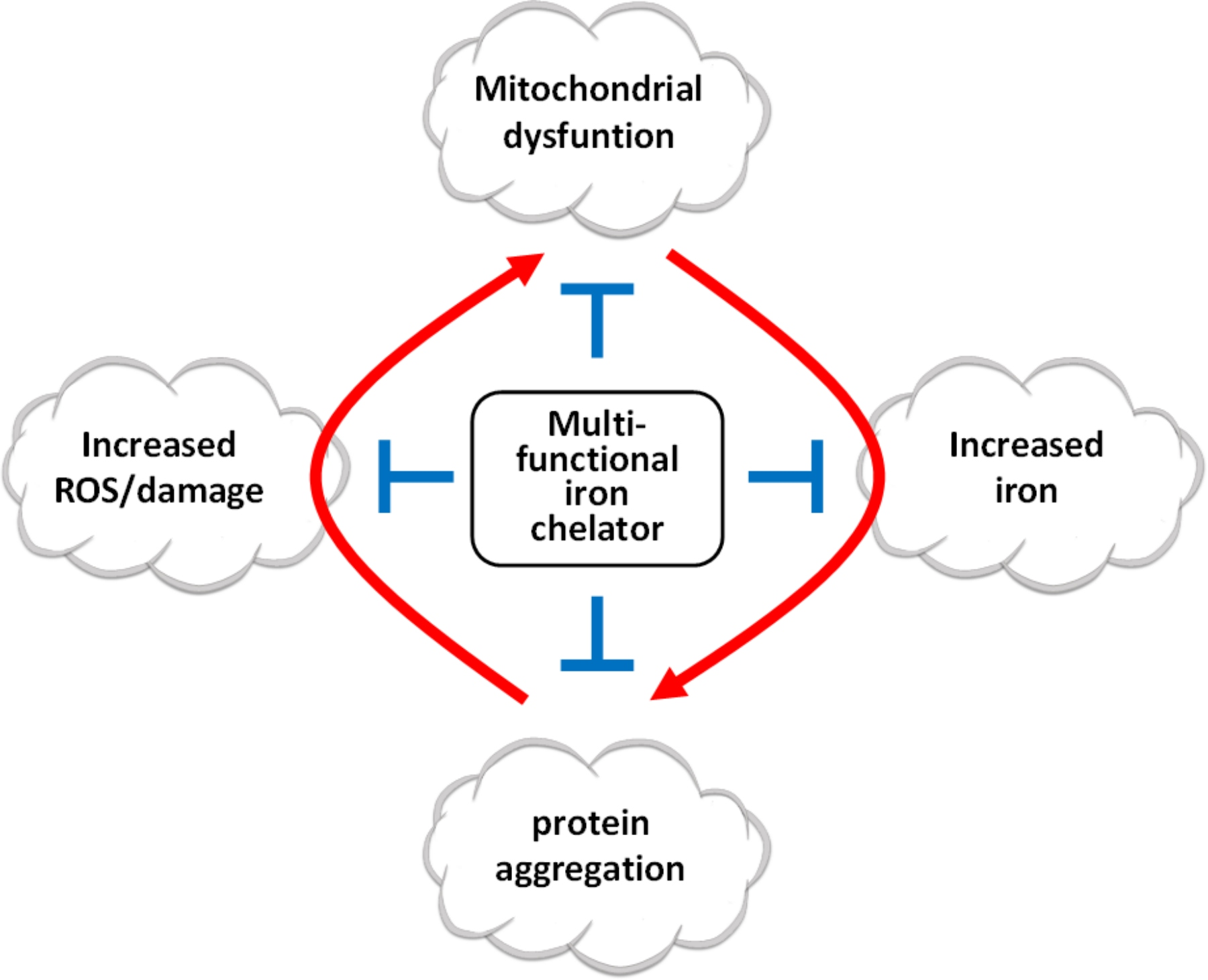
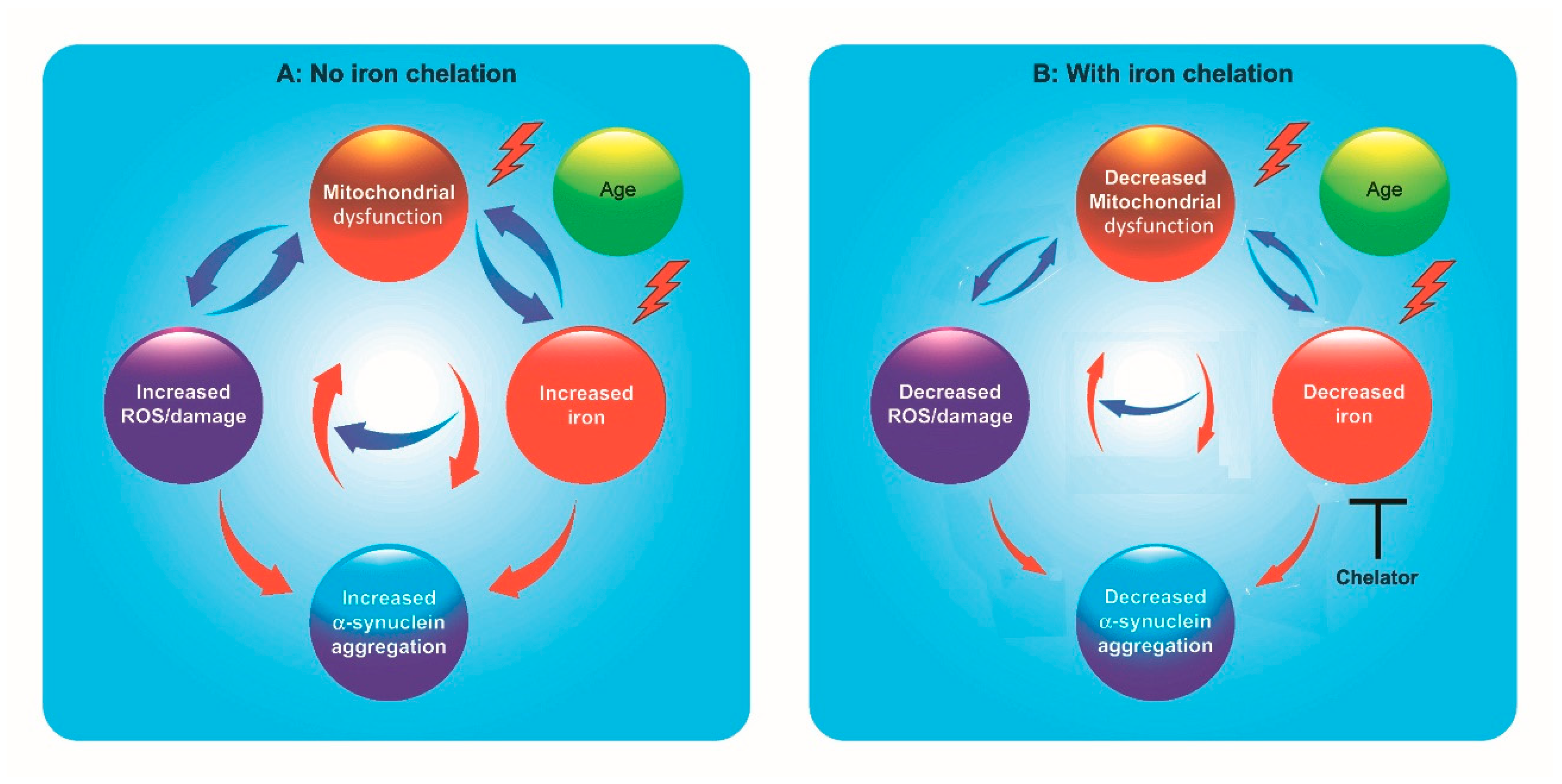
1. Introduction
2. Neurodegenerative Diseases with an Iron Accumulation Component
3. Clinical Trials Using Iron Chelation
3.1. Parkinson’s Disease
3.2. Friedreich’s Ataxia
3.3. Neurodegeneration with Brain Iron Accumulation (NBIA) Disorders
3.4. Huntington Disease
3.5. Alzheimer’s Disease
4. Potential Risks of Iron Chelation Therapy
5. New Multifunctional Iron/Copper Chelators with Therapeutic Capacity
5.1. Epigallocatechin-3-Gallate (EGCG)
5.2. MAO-B Inhibitor Hybrids
5.3. Glucose Hybrids
5.4. Acetyl Cholinesterase Inhibitor Hybrids
5.5. Dopamine Receptor Agonist Hybrids
5.6. Curcumin Hybrids
5.7. Benzothiazole–3-Hydroxy-4-Pyridine Hybrids
5.8. MAO-B Inhibitors
5.9. Histamine H3 Receptor Antagonists
5.10. Coumarin Hybrids
6. Conclusions
Author Contributions
Acknowledgments
Conflicts of Interest
References
- Hallgren, B.; Sourander, P. The effect of age on the non-haemin iron in the human brain. J. Neurochem. 1958, 3, 41–51. [Google Scholar] [CrossRef] [PubMed]
- Zecca, L.; Youdim, M.B.; Riederer, P.; Connor, J.R.; Crichton, R.R. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004, 5, 863–873. [Google Scholar] [CrossRef] [PubMed]
- Crichton, R.R.; Dexter, D.T.; Ward, R.J. Brain iron metabolism and its perturbation in neurological diseases. J. Neural Transm. 2011, 118, 301–314. [Google Scholar] [CrossRef] [PubMed]
- Garry, P.J.; Goodwin, J.S.; Hunt, W.C. Iron status and anemia in the elderly: New findings and a review of previous studies. J. Am. Geriatr. Soc. 1983, 31, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Zucca, F.A.; Duyn, J.H.; Crichton, R.R.; Zecca, L. The role of iron in brain ageing and neurodegenerative disorders. Lancet Neurol. 2014, 13, 1045–1060. [Google Scholar] [CrossRef]
- Nunez, M.T.; Urrutia, P.; Mena, N.; Aguirre, P.; Tapia, V.; Salazar, J. Iron toxicity in neurodegeneration. Biometals 2012, 25, 761–776. [Google Scholar] [CrossRef] [PubMed]
- Apostolakis, S.; Kypraiou, A.M. Iron in neurodegenerative disorders: Being in the wrong place at the wrong time? Rev. Neurosci. 2017, 28, 893–911. [Google Scholar] [CrossRef] [PubMed]
- Munoz, Y.; Carrasco, C.M.; Campos, J.D.; Aguirre, P.; Nunez, M.T. Parkinson’s Disease: The Mitochondria-Iron Link. Parkinson Dis. 2016, 2016, 7049108. [Google Scholar]
- Carocci, A.; Catalano, A.; Sinicropi, M.S.; Genchi, G. Oxidative stress and neurodegeneration: The involvement of iron. Biometals 2018. [Google Scholar] [CrossRef] [PubMed]
- Kolnagou, A.; Kontoghiorghe, C.N.; Kontoghiorghes, G.J. New targeted therapies and diagnostic methods for iron overload diseases. Front. Biosci. 2018, 10, 1–20. [Google Scholar]
- Kwiatkowski, J.L. Management of transfusional iron overload—Differential properties and efficacy of iron chelating agents. J. Blood Med. 2011, 2, 135–149. [Google Scholar] [CrossRef] [PubMed]
- Poggiali, E.; Cassinerio, E.; Zanaboni, L.; Cappellini, M.D. An update on iron chelation therapy. Blood Transfus. 2012, 10, 411–422. [Google Scholar] [PubMed]
- Saliba, A.N.; Harb, A.R.; Taher, A.T. Iron chelation therapy in transfusion-dependent thalassemia patients: Current strategies and future directions. J. Blood Med. 2015, 6, 197–209. [Google Scholar] [PubMed]
- Flaten, T.P.; Aaseth, J.; Andersen, O.; Kontoghiorghes, G.J. Iron mobilization using chelation and phlebotomy. J. Trace Elem. Med. Boil. 2012, 26, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.; Martin, M.; Schwartz, E. Depletion of excessive liver iron stores with desferrioxamine. Br. J. Haematol. 1984, 58, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Han, Y.H.; Kang, B.M.; Mun, C.W.; Lee, S.J.; Baik, S.K. Quantitative assessment of subcortical atrophy and iron content in progressive supranuclear palsy and parkinsonian variant of multiple system atrophy. J. Neurol. 2013, 260, 2094–2101. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Ben-Shachar, D.; Riederer, P. Is Parkinson’s disease a progressive siderosis of substantia nigra resulting in iron and melanin induced neurodegeneration? Acta Neurol. Scand. Suppl. 1989, 126, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Savoiardo, M. Differential diagnosis of Parkinson’s disease and atypical parkinsonian disorders by magnetic resonance imaging. Neurol. Sci. 2003, 24 (Suppl. S1), S35–S37. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Lyoo, C.H.; Ahn, S.J.; Rinne, J.O.; Lee, M.S. Brain regional iron contents in progressive supranuclear palsy. Park. Relat. Disord. 2017, 45, 28–32. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, B.; Ferrer, I.; Gil, F.; Hilfiker, S. Biomonitorization of iron accumulation in the substantia nigra from Lewy body disease patients. Toxicol. Rep. 2017, 4, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Lao-Kaim, N.P.; Loane, C.; Politis, M.; Roussakis, A.A.; Valle-Guzman, N.; Kefalopoulou, Z.; Paul-Visse, G.; Widner, H.; Xing, Y.; et al. Motor associations of iron accumulation in deep grey matter nuclei in Parkinson’s disease: A cross-sectional study of iron-related magnetic resonance imaging susceptibility. Eur. J. Neurol. 2017, 24, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Cummings, J.; Perlman, S.; Hance, D.B.; Mintz, J. Increased basal ganglia iron levels in Huntington disease. Arch. Neurol. 1999, 56, 569–574. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.A.; Harris, P.L.; Sayre, L.M.; Perry, G. Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. Proc. Natl. Acad. Sci. USA 1997, 94, 9866–9868. [Google Scholar] [CrossRef] [PubMed]
- Perry, G.; Taddeo, M.A.; Petersen, R.B.; Castellani, R.J.; Harris, P.L.; Siedlak, S.L.; Cash, A.D.; Liu, Q.; Nunomura, A.; Atwood, C.S.; et al. Adventiously-bound redox active iron and copper are at the center of oxidative damage in Alzheimer disease. Biometals 2003, 16, 77–81. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.J.R.; Ayton, S.; Bush, A.I. Iron and Alzheimer’s Disease: An Update on Emerging Mechanisms. J. Alzheimer’s Dis. 2018, 64, S379–S395. [Google Scholar] [CrossRef] [PubMed]
- Bartzokis, G.; Sultzer, D.; Cummings, J.; Holt, L.E.; Hance, D.B.; Henderson, V.W.; Mintz, J. In vivo evaluation of brain iron in Alzheimer disease using magnetic resonance imaging. Arch. Gen. Psychiatry 2000, 57, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Bulk, M.; Abdelmoula, W.M.; Nabuurs, R.J.A.; van der Graaf, L.M.; Mulders, C.W.H.; Mulder, A.A.; Jost, C.R.; Koster, A.J.; van Buchem, M.A.; Natte, R.; et al. Postmortem MRI and histology demonstrate differential iron accumulation and cortical myelin organization in early- and late-onset Alzheimer’s disease. Neurobiol. Aging 2018, 62, 231–242. [Google Scholar] [CrossRef] [PubMed]
- Chiang, S.; Kovacevic, Z.; Sahni, S.; Lane, D.J.; Merlot, A.M.; Kalinowski, D.S.; Huang, M.L.; Richardson, D.R. Frataxin and the molecular mechanism of mitochondrial iron-loading in Friedreich’s ataxia. Clin. Sci. 2016, 130, 853–870. [Google Scholar] [CrossRef] [PubMed]
- Fermin-Delgado, R.; Roa-Sanchez, P.; Speckter, H.; Perez-Then, E.; Rivera-Mejia, D.; Foerster, B.; Stoeter, P. Involvement of globus pallidus and midbrain nuclei in pantothenate kinase-associated neurodegeneration: Measurement of T2 and T2* time. Clin. Neuroradiol. 2013, 23, 11–15. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; De Grandis, E.; Barzaghi, C.; Mascaretti, M.; Garavaglia, B.; Zanotto, E.; Morana, G.; Biancheri, R. Early-onset neurodegeneration with brain iron accumulation due to PANK2 mutation. Brain Dev. 2012, 34, 536–538. [Google Scholar] [CrossRef] [PubMed]
- Swaiman, K.F. Hallervorden-Spatz syndrome and brain iron metabolism. Arch. Neurol. 1991, 48, 1285–1293. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, S.J.; Kurian, M.A.; Hogarth, P. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2018, 147, 293–305. [Google Scholar] [PubMed]
- Poplawska-Domaszewicz, K.; Florczak-Wyspianska, J.; Kozubski, W. Update on neurodegeneration with brain iron accumulation. Neurol. I Neurochir. Polska 2014, 48, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Tonekaboni, S.H.; Mollamohammadi, M. Neurodegeneration with brain iron accumulation: An overview. Iran. J. Child Neurol. 2014, 8, 1–8. [Google Scholar] [PubMed]
- Wiethoff, S.; Houlden, H. Neurodegeneration with brain iron accumulation. Handb. Clin. Neurol. 2017, 145, 157–166. [Google Scholar] [PubMed]
- Moreau, C.; Duce, J.A.; Rascol, O.; Devedjian, J.C.; Berg, D.; Dexter, D.; Cabantchik, Z.I.; Bush, A.I.; Devos, D. Iron as a therapeutic target for Parkinson’s disease. Mov. Disord. 2018, 33, 568–574. [Google Scholar] [CrossRef] [PubMed]
- Lehmann, W.D.; Heinrich, H.C. Impaired phenylalanine-tyrosine conversion in patients with iron-deficiency anemia studied by a L-(2H5)phenylalanine-loading test. Am. J. Clin. Nutr. 1986, 44, 468–474. [Google Scholar] [CrossRef] [PubMed]
- Ohnishi, T. Iron-sulfur clusters/semiquinones in complex I. Biochim. Biophys. Acta 1998, 1364, 186–206. [Google Scholar] [CrossRef]
- Stiban, J.; So, M.; Kaguni, L.S. Iron-Sulfur Clusters in Mitochondrial Metabolism: Multifaceted Roles of a Simple Cofactor. Biochem. 2016, 81, 1066–1080. [Google Scholar] [CrossRef] [PubMed]
- Segura-Aguilar, J.; Metodiewa, D.; Welch, C.J. Metabolic activation of dopamine o-quinones to o-semiquinones by NADPH cytochrome P450 reductase may play an important role in oxidative stress and apoptotic effects. Biochim. Biophys. Acta 1998, 1381, 1–6. [Google Scholar] [CrossRef]
- Arriagada, C.; Paris, I.; Sanchez de las Matas, M.J.; Martinez-Alvarado, P.; Cardenas, S.; Castaneda, P.; Graumann, R.; Perez-Pastene, C.; Olea-Azar, C.; Couve, E.; et al. On the neurotoxicity mechanism of leukoaminochrome o-semiquinone radical derived from dopamine oxidation: Mitochondria damage, necrosis, and hydroxyl radical formation. Neurobiol. Dis. 2004, 16, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Zoccarato, F.; Toscano, P.; Alexandre, A. Dopamine-derived dopaminochrome promotes H(2)O(2) release at mitochondrial complex I: Stimulation by rotenone, control by Ca(2+), and relevance to Parkinson disease. J. Biol. Chem. 2005, 280, 15587–15594. [Google Scholar] [CrossRef] [PubMed]
- Uranga, R.M.; Salvador, G.A. Unraveling the Burden of Iron in Neurodegeneration: Intersections with Amyloid Beta Peptide Pathology. Oxidative Med. Cell. Longev. 2018, 2018, 2850341. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.; Mena, N.; Núñez, M.T. Iron dyshomeostasis in Parkinson’s disease. J. Neural Transm. Suppl. 2006, 205–213. [Google Scholar]
- Mena, N.P.; Bulteau, A.L.; Salazar, J.; Hirsch, E.C.; Núñez, M.T. Effect of mitochondrial complex I inhibition on Fe-S cluster protein activity. Biochem. Biophys. Res. Commun. 2011, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Aguirre, P.; Tapia, V.; Carrasco, C.M.; Mena, N.P.; Nunez, M.T. Cell death induced by mitochondrial complex I inhibition is mediated by Iron Regulatory Protein 1. Biochim. Biophys. Acta 2017. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.J. alpha-synuclein aggregation: A link between mitochondrial defects and Parkinson’s disease? Antioxid. Redox Signal. 2003, 5, 337–348. [Google Scholar] [CrossRef] [PubMed]
- Betarbet, R.; Canet-Aviles, R.M.; Sherer, T.B.; Mastroberardino, P.G.; McLendon, C.; Kim, J.H.; Lund, S.; Na, H.M.; Taylor, G.; Bence, N.F.; et al. Intersecting pathways to neurodegeneration in Parkinson’s disease: Effects of the pesticide rotenone on DJ-1, alpha-synuclein, and the ubiquitin-proteasome system. Neurobiol. Dis. 2006, 22, 404–420. [Google Scholar] [CrossRef] [PubMed]
- Esteves, A.R.; Arduino, D.M.; Silva, D.F.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial Dysfunction: The Road to Alpha-Synuclein Oligomerization in PD. Parkinson Dis. 2011, 2011, 693761. [Google Scholar] [CrossRef] [PubMed]
- Karmacharya, M.B.; Hada, B.; Park, S.R.; Choi, B.H. Low-Intensity Ultrasound Decreases alpha-Synuclein Aggregation via Attenuation of Mitochondrial Reactive Oxygen Species in MPP(+)-Treated PC12 Cells. Mol. Neurobiol. 2017, 54, 6235–6244. [Google Scholar] [CrossRef] [PubMed]
- Hashimoto, M.; Rockenstein, E.; Crews, L.; Masliah, E. Role of protein aggregation in mitochondrial dysfunction and neurodegeneration in Alzheimer’s and Parkinson’s diseases. Neuromol. Med. 2003, 4, 21–36. [Google Scholar] [CrossRef]
- Reeve, A.K.; Ludtmann, M.H.; Angelova, P.R.; Simcox, E.M.; Horrocks, M.H.; Klenerman, D.; Gandhi, S.; Turnbull, D.M.; Abramov, A.Y. Aggregated alpha-synuclein and complex I deficiency: Exploration of their relationship in differentiated neurons. Cell Death Dis. 2015, 6, e1820. [Google Scholar] [CrossRef] [PubMed]
- Rocha, E.M.; De Miranda, B.; Sanders, L.H. Alpha-synuclein: Pathology, mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurobiol. Dis. 2018, 109, 249–257. [Google Scholar] [CrossRef] [PubMed]
- Faustini, G.; Bono, F.; Valerio, A.; Pizzi, M.; Spano, P.; Bellucci, A. Mitochondria and alpha-Synuclein: Friends or Foes in the Pathogenesis of Parkinson’s Disease? Genes 2017, 8, 377. [Google Scholar] [CrossRef] [PubMed]
- Mullin, S.; Schapira, A. alpha-Synuclein and mitochondrial dysfunction in Parkinson’s disease. Mol. Neurobiol. 2013, 47, 587–597. [Google Scholar] [CrossRef] [PubMed]
- Belaidi, A.A.; Bush, A.I. Iron neurochemistry in Alzheimer’s disease and Parkinson’s disease: Targets for therapeutics. J. Neurochem. 2015. [Google Scholar] [CrossRef] [PubMed]
- Mena, N.P.; Urrutia, P.J.; Lourido, F.; Carrasco, C.M.; Núñez, M.T. Mitochondrial iron homeostasis and its dysfunctions in neurodegenerative disorders. Mitochondrion 2015, 21, 92–105. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Arab, N.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta 2017, 1864, 399–430. [Google Scholar] [CrossRef] [PubMed]
- Murphy, C.J.; Oudit, G.Y. Iron-overload cardiomyopathy: Pathophysiology, diagnosis, and treatment. J. Card. Fail. 2010, 16, 888–900. [Google Scholar] [CrossRef] [PubMed]
- Fisher, S.A.; Brunskill, S.J.; Doree, C.; Chowdhury, O.; Gooding, S.; Roberts, D.J. Oral deferiprone for iron chelation in people with thalassaemia. Cochrane Database Syst. Rev. 2013. [Google Scholar] [CrossRef] [PubMed]
- Gulati, V.; Harikrishnan, P.; Palaniswamy, C.; Aronow, W.S.; Jain, D.; Frishman, W.H. Cardiac involvement in hemochromatosis. Cardiol. Rev. 2014, 22, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Brissot, P. Optimizing the diagnosis and the treatment of iron overload diseases. Expert Rev. Gastroenterol. Hepatol. 2016, 10, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Belmont, A.; Kwiatkowski, J.L. Deferiprone for the treatment of transfusional iron overload in thalassemia. Expert Rev. Hematol. 2017, 10, 493–503. [Google Scholar] [CrossRef] [PubMed]
- Bollig, C.; Schell, L.K.; Rucker, G.; Allert, R.; Motschall, E.; Niemeyer, C.M.; Bassler, D.; Meerpohl, J.J. Deferasirox for managing iron overload in people with thalassaemia. Cochrane Database Syst. Rev. 2017, 8, Cd007476. [Google Scholar] [CrossRef] [PubMed]
- Aydinok, Y. Iron Chelation Therapy as a Modality of Management. Hematol. Oncol. Clin. N. Am. 2018, 32, 261–275. [Google Scholar] [CrossRef] [PubMed]
- Diez-Lopez, C.; Comin-Colet, J.; Gonzalez-Costello, J. Iron overload cardiomyopathy: From diagnosis to management. Curr. Opin. Cardiol. 2018, 33, 334–340. [Google Scholar] [CrossRef] [PubMed]
- Ballas, S.K.; Zeidan, A.M.; Duong, V.H.; DeVeaux, M.; Heeney, M.M. The effect of iron chelation therapy on overall survival in sickle cell disease and beta-thalassemia: A systematic review. Am. J. Hematol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Ward, R.J.; Dexter, D.T.; Crichton, R.R. Neurodegenerative diseases and therapeutic strategies using iron chelators. J. Trace Elem. Med. Boil. 2015, 31, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Devos, D.; Moreau, C.; Devedjian, J.C.; Kluza, J.; Petrault, M.; Laloux, C.; Jonneaux, A.; Ryckewaert, G.; Garçon, G.; Rouaix, N.; et al. Targeting chelatable iron as a therapeutic modality in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 195–210. [Google Scholar] [CrossRef] [PubMed]
- Dusek, P.; Schneider, S.A.; Aaseth, J. Iron chelation in the treatment of neurodegenerative diseases. J. Trace Elem. Med. Boil. 2016. [Google Scholar] [CrossRef] [PubMed]
- Shvartsman, M.; Kikkeri, R.; Shanzer, A.; Cabantchik, Z.I. Non-transferrin-bound iron reaches mitochondria by a chelator-inaccessible mechanism: Biological and clinical implications. Am. J. Physiol. Cell Physiol. 2007, 293, C1383–C1394. [Google Scholar] [CrossRef] [PubMed]
- Grolez, G.; Moreau, C.; Sablonniere, B.; Garcon, G.; Devedjian, J.C.; Meguig, S.; Gele, P.; Delmaire, C.; Bordet, R.; Defebvre, L.; et al. Ceruloplasmin activity and iron chelation treatment of patients with Parkinson’s disease. BMC Neurol. 2015, 15, 74. [Google Scholar] [CrossRef] [PubMed]
- Martin-Bastida, A.; Ward, R.J.; Newbould, R.; Piccini, P.; Sharp, D.; Kabba, C.; Patel, M.C.; Spino, M.; Connelly, J.; Tricta, F.; et al. Brain iron chelation by deferiprone in a phase 2 randomised double-blinded placebo controlled clinical trial in Parkinson’s disease. Sci. Rep. 2017, 7, 1398. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M. Friedreich’s ataxia: Clinical aspects and pathogenesis. Semin. Neurol. 1999, 19, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Gordon, N. Friedreich’s ataxia and iron metabolism. Brain Dev. 2000, 22, 465–468. [Google Scholar] [CrossRef]
- Richardson, D.R.; Mouralian, C.; Ponka, P.; Becker, E. Development of potential iron chelators for the treatment of Friedreich’s ataxia: Ligands that mobilize mitochondrial iron. Biochim. Biophys. Acta 2001, 1536, 133–140. [Google Scholar] [CrossRef]
- Richardson, D.R. Friedreich’s ataxia: Iron chelators that target the mitochondrion as a therapeutic strategy? Expert Opin. Investig. Drugs 2003, 12, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Boddaert, N.; Le Quan Sang, K.H.; Rotig, A.; Leroy-Willig, A.; Gallet, S.; Brunelle, F.; Sidi, D.; Thalabard, J.C.; Munnich, A.; Cabantchik, Z.I. Selective iron chelation in Friedreich ataxia: Biologic and clinical implications. Blood 2007, 110, 401–408. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Hausmann, L. Deferiprone for the treatment of Friedreich’s ataxia. J. Neurochem. 2013, 126 (Suppl. S1), 142–146. [Google Scholar] [CrossRef] [PubMed]
- Velasco-Sanchez, D.; Aracil, A.; Montero, R.; Mas, A.; Jimenez, L.; O’Callaghan, M.; Tondo, M.; Capdevila, A.; Blanch, J.; Artuch, R.; et al. Combined therapy with idebenone and deferiprone in patients with Friedreich’s ataxia. Cerebellum 2011, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Elincx-Benizri, S.; Glik, A.; Merkel, D.; Arad, M.; Freimark, D.; Kozlova, E.; Cabantchik, I.; Hassin-Baer, S. Clinical Experience With Deferiprone Treatment for Friedreich Ataxia. J. Child Neurol. 2016, 31, 1036–1040. [Google Scholar] [CrossRef] [PubMed]
- Pandolfo, M.; Arpa, J.; Delatycki, M.B.; Le Quan Sang, K.H.; Mariotti, C.; Munnich, A.; Sanz-Gallego, I.; Tai, G.; Tarnopolsky, M.A.; Taroni, F.; et al. Deferiprone in Friedreich ataxia: A 6-month randomized controlled trial. Ann. Neurol. 2014, 76, 509–521. [Google Scholar] [CrossRef] [PubMed]
- Arpa, J.; Sanz-Gallego, I.; Rodriguez-de-Rivera, F.J.; Dominguez-Melcon, F.J.; Prefasi, D.; Oliva-Navarro, J.; Moreno-Yanguela, M. Triple therapy with deferiprone, idebenone and riboflavin in Friedreich’s ataxia—Open-label trial. Acta Neurol. Scand. 2014, 129, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Limongi, J.C. Neurodegeneration with brain iron accumulation. Arq. Neuro-Psiquiatr. 2016, 74, 517–518. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Kurian, M.A.; Hayflick, S.J. Neurodegeneration with Brain Iron Accumulation: Genetic Diversity and Pathophysiological Mechanisms. Annu. Rev Genom. Hum. Genet. 2015, 16, 257–279. [Google Scholar] [CrossRef] [PubMed]
- Salomao, R.P.; Pedroso, J.L.; Gama, M.T.; Dutra, L.A.; Maciel, R.H.; Godeiro-Junior, C.; Chien, H.F.; Teive, H.A.; Cardoso, F.; Barsottini, O.G. A diagnostic approach for neurodegeneration with brain iron accumulation: Clinical features, genetics and brain imaging. Arq. Neuro-Psiquiatr. 2016, 74, 587–596. [Google Scholar] [CrossRef] [PubMed]
- Gregory, A.; Hayflick, S.J. Pantothenate Kinase-Associated Neurodegeneration. In Genereviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2018. [Google Scholar]
- Vakili, S.; Drew, A.L.; Von Schuching, S.; Becker, D.; Zeman, W. Hallervorden-Spatz syndrome. Arch. Neurol. 1977, 34, 729–738. [Google Scholar] [CrossRef] [PubMed]
- Schaffert, D.A.; Johnsen, S.D.; Johnson, P.C.; Drayer, B.P. Magnetic resonance imaging in pathologically proven Hallervorden-Spatz disease. Neurology 1989, 39, 440–442. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Dickson, A.C. Iron in the Hallervorden-Spatz syndrome. Pediatr. Neurol. 2001, 25, 148–155. [Google Scholar] [CrossRef]
- Gregory, A.; Kurian, M.A.; Maher, E.R.; Hogarth, P.; Hayflick, S.J. PLA2G6-Associated Neurodegeneration. In Genereviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Kurian, M.A.; Hayflick, S.J. Pantothenate kinase-associated neurodegeneration (PKAN) and PLA2G6-associated neurodegeneration (PLAN): Review of two major neurodegeneration with brain iron accumulation (NBIA) phenotypes. Int. Rev. Neurobiol. 2013, 110, 49–71. [Google Scholar] [PubMed]
- Simonati, A.; Trevisan, C.; Salviati, A.; Rizzuto, N. Neuroaxonal dystrophy with dystonia and pallidal involvement. Neuropediatrics 1999, 30, 151–154. [Google Scholar] [CrossRef] [PubMed]
- Wakabayashi, K.; Fukushima, T.; Koide, R.; Horikawa, Y.; Hasegawa, M.; Watanabe, Y.; Noda, T.; Eguchi, I.; Morita, T.; Yoshimoto, M.; et al. Juvenile-onset generalized neuroaxonal dystrophy (Hallervorden-Spatz disease) with diffuse neurofibrillary and lewy body pathology. Acta Neuropathol. 2000, 99, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Hogarth, P.; Gregory, A.; Kruer, M.C.; Sanford, L.; Wagoner, W.; Natowicz, M.R.; Egel, R.T.; Subramony, S.H.; Goldman, J.G.; Berry-Kravis, E.; et al. New NBIA subtype: Genetic, clinical, pathologic, and radiographic features of MPAN. Neurology 2013, 80, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Schulte, E.C.; Claussen, M.C.; Jochim, A.; Haack, T.; Hartig, M.; Hempel, M.; Prokisch, H.; Haun-Junger, U.; Winkelmann, J.; Hemmer, B.; et al. Mitochondrial membrane protein associated neurodegenration: A novel variant of neurodegeneration with brain iron accumulation. Mov. Disord. 2013, 28, 224–227. [Google Scholar] [CrossRef] [PubMed]
- Hartig, M.; Prokisch, H.; Meitinger, T.; Klopstock, T. Mitochondrial membrane protein-associated neurodegeneration (MPAN). Int. Rev. Neurobiol. 2013, 110, 73–84. [Google Scholar] [PubMed]
- Deutschlander, A.; Konno, T.; Ross, O.A. Mitochondrial membrane protein-associated neurodegeneration. Parkinsonism Relat. Disord. 2017, 39, 1–3. [Google Scholar] [CrossRef] [PubMed]
- Haack, T.B.; Hogarth, P.; Gregory, A.; Prokisch, H.; Hayflick, S.J. BPAN: The only X-linked dominant NBIA disorder. Int. Rev. Neurobiol. 2013, 110, 85–90. [Google Scholar] [PubMed]
- Verhoeven, W.M.; Egger, J.I.; Koolen, D.A.; Yntema, H.; Olgiati, S.; Breedveld, G.J.; Bonifati, V.; van de Warrenburg, B.P. Beta-propeller protein-associated neurodegeneration (BPAN), a rare form of NBIA: Novel mutations and neuropsychiatric phenotype in three adult patients. Parkinsonism Relat. Disord. 2014, 20, 332–336. [Google Scholar] [CrossRef] [PubMed]
- Evers, C.; Seitz, A.; Assmann, B.; Opladen, T.; Karch, S.; Hinderhofer, K.; Granzow, M.; Paramasivam, N.; Eils, R.; Diessl, N.; et al. Diagnosis of CoPAN by whole exome sequencing: Waking up a sleeping tiger’s eye. Am. J. Med. Genet. Part A 2017. [Google Scholar] [CrossRef] [PubMed]
- Annesi, G.; Gagliardi, M.; Iannello, G.; Quattrone, A.; Iannello, G.; Quattrone, A. Mutational analysis of COASY in an Italian patient with NBIA. Parkinsonism Relat. Disord. 2016, 28, 150–151. [Google Scholar] [CrossRef] [PubMed]
- Dusi, S.; Valletta, L.; Haack, T.B.; Tsuchiya, Y.; Venco, P.; Pasqualato, S.; Goffrini, P.; Tigano, M.; Demchenko, N.; Wieland, T.; et al. Exome sequence reveals mutations in CoA synthase as a cause of neurodegeneration with brain iron accumulation. Am. J. Hum. Genet. 2014, 94, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Kruer, M.C.; Paisan-Ruiz, C.; Boddaert, N.; Yoon, M.Y.; Hama, H.; Gregory, A.; Malandrini, A.; Woltjer, R.L.; Munnich, A.; Gobin, S.; et al. Defective FA2H leads to a novel form of neurodegeneration with brain iron accumulation (NBIA). Ann. Neurol. 2010, 68, 611–618. [Google Scholar] [CrossRef] [PubMed]
- Garone, C.; Pippucci, T.; Cordelli, D.M.; Zuntini, R.; Castegnaro, G.; Marconi, C.; Graziano, C.; Marchiani, V.; Verrotti, A.; Seri, M.; et al. FA2H-related disorders: A novel c.270+3A>T splice-site mutation leads to a complex neurodegenerative phenotype. Dev. Med. Child Neurol. 2011, 53, 958–961. [Google Scholar] [CrossRef] [PubMed]
- Najim al-Din, A.S.; Wriekat, A.; Mubaidin, A.; Dasouki, M.; Hiari, M. Pallido-pyramidal degeneration, supranuclear upgaze paresis and dementia: Kufor-Rakeb syndrome. Acta Neurol. Scand. 1994, 89, 347–352. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, A.; Heimbach, A.; Grundemann, J.; Stiller, B.; Hampshire, D.; Cid, L.P.; Goebel, I.; Mubaidin, A.F.; Wriekat, A.L.; Roeper, J.; et al. Hereditary parkinsonism with dementia is caused by mutations in ATP13A2, encoding a lysosomal type 5 P-type ATPase. Nat. Genet. 2006, 38, 1184–1191. [Google Scholar] [CrossRef] [PubMed]
- Schneider, S.A.; Paisan-Ruiz, C.; Quinn, N.P.; Lees, A.J.; Houlden, H.; Hardy, J.; Bhatia, K.P. ATP13A2 mutations (PARK9) cause neurodegeneration with brain iron accumulation. Mov. Disord. 2010, 25, 979–984. [Google Scholar] [CrossRef] [PubMed]
- Yazaki, M.; Yoshida, K.; Nakamura, A.; Furihata, K.; Yonekawa, M.; Okabe, T.; Yamashita, N.; Ohta, M.; Ikeda, S. A novel splicing mutation in the ceruloplasmin gene responsible for hereditary ceruloplasmin deficiency with hemosiderosis. J. Neurol. Sci. 1998, 156, 30–34. [Google Scholar] [CrossRef]
- Nittis, T.; Gitlin, J.D. The copper-iron connection: Hereditary aceruloplasminemia. Semin. Hematol. 2002, 39, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Curtis, A.R.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef] [PubMed]
- Levi, S.; Rovida, E. Neuroferritinopathy: From ferritin structure modification to pathogenetic mechanism. Neurobiol. Dis. 2015, 81, 134–143. [Google Scholar] [CrossRef] [PubMed]
- McNeill, A.; Birchall, D.; Hayflick, S.J.; Gregory, A.; Schenk, J.F.; Zimmerman, E.A.; Shang, H.; Miyajima, H.; Chinnery, P.F. T2* and FSE MRI distinguishes four subtypes of neurodegeneration with brain iron accumulation. Neurology 2008, 70, 1614–1619. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Saggar, K.; Kaur, M.; Pannu, D.S. Magnetic resonance imaging in pantothenate kinase-2-associated neurodegeneration. J. Pediatr. Neurosci. 2012, 7, 27–29. [Google Scholar] [CrossRef] [PubMed]
- Zorzi, G.; Zibordi, F.; Chiapparini, L.; Bertini, E.; Russo, L.; Piga, A.; Longo, F.; Garavaglia, B.; Aquino, D.; Savoiardo, M.; et al. Iron-related MRI images in patients with pantothenate kinase-associated neurodegeneration (PKAN) treated with deferiprone: Results of a phase II pilot trial. Mov. Disord. 2011, 26, 1756–1759. [Google Scholar] [CrossRef] [PubMed]
- Abbruzzese, G.; Cossu, G.; Balocco, M.; Marchese, R.; Murgia, D.; Melis, M.; Galanello, R.; Barella, S.; Matta, G.; Ruffinengo, U.; et al. A pilot trial of deferiprone for neurodegeneration with brain iron accumulation. Haematologica 2011, 96, 1708–1711. [Google Scholar] [CrossRef] [PubMed]
- Cossu, G.; Abbruzzese, G.; Matta, G.; Murgia, D.; Melis, M.; Ricchi, V.; Galanello, R.; Barella, S.; Origa, R.; Balocco, M.; et al. Efficacy and safety of deferiprone for the treatment of pantothenate kinase-associated neurodegeneration (PKAN) and neurodegeneration with brain iron accumulation (NBIA): Results from a four years follow-up. Parkinsonism Relat. Disord. 2014, 20, 651–654. [Google Scholar] [CrossRef] [PubMed]
- Rohani, M.; Razmeh, S.; Shahidi, G.A.; Alizadeh, E.; Orooji, M. A pilot trial of deferiprone in pantothenate kinase-associated neurodegeneration patients. Neurol. Int. 2017, 9, 7279. [Google Scholar] [CrossRef] [PubMed]
- Investigators, H.S.G.R.H. Safety, tolerability, and efficacy of PBT2 in Huntington’s disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2015, 14, 39–47. [Google Scholar]
- Ritchie, C.W.; Bush, A.I.; Mackinnon, A.; Macfarlane, S.; Mastwyk, M.; MacGregor, L.; Kiers, L.; Cherny, R.; Li, Q.X.; Tammer, A.; et al. Metal-protein attenuation with iodochlorhydroxyquin (clioquinol) targeting Abeta amyloid deposition and toxicity in Alzheimer disease: A pilot phase 2 clinical trial. Arch. Neurol. 2003, 60, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Lannfelt, L.; Blennow, K.; Zetterberg, H.; Batsman, S.; Ames, D.; Harrison, J.; Masters, C.L.; Targum, S.; Bush, A.I.; Murdoch, R.; et al. Safety, efficacy, and biomarker findings of PBT2 in targeting Abeta as a modifying therapy for Alzheimer’s disease: A phase IIa, double-blind, randomised, placebo-controlled trial. Lancet Neurol. 2008, 7, 779–786. [Google Scholar] [CrossRef]
- Winter, W.E.; Bazydlo, L.A.; Harris, N.S. The molecular biology of human iron metabolism. Lab. Med. 2014, 45, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Kontoghiorghes, G.J.; Kolnagou, A.; Peng, C.T.; Shah, S.V.; Aessopos, A. Safety issues of iron chelation therapy in patients with normal range iron stores including thalassaemia, neurodegenerative, renal and infectious diseases. Expert Opin. Drug Saf. 2010, 9, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, E.; Takeshige, K.; Oishi, T.; Murai, Y.; Minakami, S. 1-Methyl-4-phenylpyridinium (MPP+) induces NADH-dependent superoxide formation and enhances NADH-dependent lipid peroxidation in bovine heart submitochondrial particles. Biochem. Biophys. Res. Commun. 1990, 170, 1049–1055. [Google Scholar] [CrossRef]
- Shukla, A.; Agarwal, K.N.; Shukla, G.S. Latent iron deficiency alters gamma-aminobutyric acid and glutamate metabolism in rat brain. Experientia 1989, 45, 343–345. [Google Scholar] [CrossRef] [PubMed]
- Li, D. Effects of iron deficiency on iron distribution and gamma-aminobutyric acid (GABA) metabolism in young rat brain tissues. Hokkaido Igaku Zasshi 1998, 73, 215–225. [Google Scholar] [PubMed]
- Fonderico, M.; Laudisi, M.; Andreasi, N.G.; Bigoni, S.; Lamperti, C.; Panteghini, C.; Garavaglia, B.; Carecchio, M.; Emanuele, E.A.; Forni, G.L.; et al. Patient Affected by Beta-Propeller Protein-Associated Neurodegeneration: A Therapeutic Attempt with Iron Chelation Therapy. Front. Neurol. 2017, 8, 385. [Google Scholar] [CrossRef] [PubMed]
- Faux, N.G.; Ritchie, C.W.; Gunn, A.; Rembach, A.; Tsatsanis, A.; Bedo, J.; Harrison, J.; Lannfelt, L.; Blennow, K.; Zetterberg, H.; et al. PBT2 rapidly improves cognition in Alzheimer’s Disease: Additional phase II analyses. J. Alzheimer’s Dis. 2010, 20, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, J.L. Current recommendations for chelation for transfusion-dependent thalassemia. Ann. N. Y. Acad. Sci. 2016, 1368, 107–114. [Google Scholar] [CrossRef] [PubMed]
- Porter, J.B. A risk-benefit assessment of iron-chelation therapy. Drug Saf. 1997, 17, 407–421. [Google Scholar] [CrossRef] [PubMed]
- Galanello, R. Deferiprone in the treatment of transfusion-dependent thalassemia: A review and perspective. Ther. Clin. Risk Manag. 2007, 3, 795–805. [Google Scholar] [PubMed]
- Cohen, A.R.; Galanello, R.; Piga, A.; De Sanctis, V.; Tricta, F. Safety and effectiveness of long-term therapy with the oral iron chelator deferiprone. Blood 2003, 102, 1583–1587. [Google Scholar] [CrossRef] [PubMed]
- Henter, J.I.; Karlen, J. Fatal agranulocytosis after deferiprone therapy in a child with Diamond-Blackfan anemia. Blood 2007, 109, 5157–5159. [Google Scholar] [CrossRef] [PubMed]
- Cappellini, M.D.; Cohen, A.; Piga, A.; Bejaoui, M.; Perrotta, S.; Agaoglu, L.; Aydinok, Y.; Kattamis, A.; Kilinc, Y.; Porter, J.; et al. A phase 3 study of deferasirox (ICL670), a once-daily oral iron chelator, in patients with beta-thalassemia. Blood 2006, 107, 3455–3462. [Google Scholar] [CrossRef] [PubMed]
- Botzenhardt, S.; Li, N.; Chan, E.W.; Sing, C.W.; Wong, I.C.; Neubert, A. Safety profiles of iron chelators in young patients with haemoglobinopathies. Eur. J. Haematol. 2017, 98, 198–217. [Google Scholar] [CrossRef] [PubMed]
- al-Refaie, F.N.; Wonke, B.; Wickens, D.G.; Aydinok, Y.; Fielding, A.; Hoffbrand, A.V. Zinc concentration in patients with iron overload receiving oral iron chelator 1,2-dimethyl-3-hydroxypyrid-4-one or desferrioxamine. J. Clin. Pathol. 1994, 47, 657–660. [Google Scholar] [CrossRef] [PubMed]
- Crisponi, G.; Nurchi, V.M.; Crespo-Alonso, M.; Sanna, G.; Zoroddu, M.A.; Alberti, G.; Biesuz, R. A Speciation Study on the Perturbing Effects of Iron Chelators on the Homeostasis of Essential Metal Ions. PLoS ONE 2015, 10, e0133050. [Google Scholar] [CrossRef] [PubMed]
- Lalioti, V.; Muruais, G.; Tsuchiya, Y.; Pulido, D.; Sandoval, I.V. Molecular mechanisms of copper homeostasis. Front. Biosci. 2009, 14, 4878–4903. [Google Scholar] [CrossRef]
- Rae, T.D.; Schmidt, P.J.; Pufahl, R.A.; Culotta, V.C.; O’Halloran, T.V. Undetectable intracellular free copper: The requirement of a copper chaperone for superoxide dismutase. Science 1999, 284, 805–808. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Singh, S.; Lillard, J.W., Jr. Past, present, and future technologies for oral delivery of therapeutic proteins. J. Pharm. Sci. 2008, 97, 2497–2523. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Boil. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed]
- Aoun, M.; Tiranti, V. Mitochondria: A crossroads for lipid metabolism defect in neurodegeneration with brain iron accumulation diseases. Int. J. Biochem. Cell Boil. 2015, 63, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Urrutia, P.J.; Mena, N.P.; Núñez, M.T. The interplay between iron accumulation, mitochondrial dysfunction, and inflammation during the execution step of neurodegenerative disorders. Front. Pharmacol. 2014, 5, 38. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.S.; Breuer, W.; Munnich, A.; Cabantchik, Z.I. Redistribution of accumulated cell iron: A modality of chelation with therapeutic implications. Blood 2008, 111, 1690–1699. [Google Scholar] [CrossRef] [PubMed]
- Levites, Y.; Weinreb, O.; Maor, G.; Youdim, M.B.; Mandel, S. Green tea polyphenol (−)-epigallocatechin-3- gallate prevents N-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced dopaminergic neurodegeneration. J. Neurochem. 2001, 78, 1073–1082. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Maor, G.; Youdim, M.B. Iron and alpha-synuclein in the substantia nigra of MPTP-treated mice: Effect of neuroprotective drugs R-apomorphine and green tea polyphenol (−)-epigallocatechin-3-gallate. J. Mol. Neurosci. 2004, 24, 401–416. [Google Scholar] [CrossRef]
- Pan, T.; Fei, J.; Zhou, X.; Jankovic, J.; Le, W. Effects of green tea polyphenols on dopamine uptake and on MPP+-induced dopamine neuron injury. Life Sci. 2003, 72, 1073–1083. [Google Scholar] [CrossRef]
- Li, R.; Peng, N.; Du, F.; Li, X.P.; Le, W.D. Epigallocatechin gallate protects dopaminergic neurons against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity by inhibiting microglial cell activation. Nan Fang Yi Ke Da Xue Xue Bao 2006, 26, 376–380. [Google Scholar] [PubMed]
- Chen, M.; Wang, T.; Yue, F.; Li, X.; Wang, P.; Li, Y.; Chan, P.; Yu, S. Tea polyphenols alleviate motor impairments, dopaminergic neuronal injury, and cerebral alpha-synuclein aggregation in MPTP-intoxicated parkinsonian monkeys. Neuroscience 2015, 286, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Zheng, H.; Weiner, L.M.; Bar-Am, O.; Epsztejn, S.; Cabantchik, Z.I.; Warshawsky, A.; Youdim, M.B.; Fridkin, M. Design, synthesis, and evaluation of novel bifunctional iron-chelators as potential agents for neuroprotection in Alzheimer’s, Parkinson’s, and other neurodegenerative diseases. Bioorg. Med. Chem. 2005, 13, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, L.; Weinreb, O.; Amit, T.; Mandel, S.; Bar-Am, O.; Youdim, M.B. Novel molecular targets of the neuroprotective/neurorescue multimodal iron chelating drug M30 in the mouse brain. Neuroscience 2011, 189, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Kupershmidt, L.; Amit, T.; Bar-Am, O.; Weinreb, O.; Youdim, M.B. Multi-target, neuroprotective and neurorestorative M30 improves cognitive impairment and reduces Alzheimer’s-like neuropathology and age-related alterations in mice. Mol. Neurobiol. 2012, 46, 217–220. [Google Scholar] [CrossRef] [PubMed]
- Bar-Am, O.; Amit, T.; Kupershmidt, L.; Aluf, Y.; Mechlovich, D.; Kabha, H.; Danovitch, L.; Zurawski, V.R.; Youdim, M.B.; Weinreb, O. Neuroprotective and neurorestorative activities of a novel iron chelator-brain selective monoamine oxidase-A/monoamine oxidase-B inhibitor in animal models of Parkinson’s disease and aging. Neurobiol. Aging 2015, 36, 1529–1542. [Google Scholar] [CrossRef] [PubMed]
- Storr, T.; Merkel, M.; Song-Zhao, G.X.; Scott, L.E.; Green, D.E.; Bowen, M.L.; Thompson, K.H.; Patrick, B.O.; Schugar, H.J.; Orvig, C. Synthesis, characterization, and metal coordinating ability of multifunctional carbohydrate-containing compounds for Alzheimer’s therapy. J. Am. Chem. Soc. 2007, 129, 7453–7463. [Google Scholar] [CrossRef] [PubMed]
- Bolognesi, M.L.; Cavalli, A.; Valgimigli, L.; Bartolini, M.; Rosini, M.; Andrisano, V.; Recanatini, M.; Melchiorre, C. Multi-target-directed drug design strategy: From a dual binding site acetylcholinesterase inhibitor to a trifunctional compound against Alzheimer’s disease. J. Med. Chem. 2007, 50, 6446–6449. [Google Scholar] [CrossRef] [PubMed]
- Fernandez-Bachiller, M.I.; Perez, C.; Gonzalez-Munoz, G.C.; Conde, S.; Lopez, M.G.; Villarroya, M.; Garcia, A.G.; Rodriguez-Franco, M.I. Novel tacrine-8-hydroxyquinoline hybrids as multifunctional agents for the treatment of Alzheimer’s disease, with neuroprotective, cholinergic, antioxidant, and copper-complexing properties. J. Med. Chem. 2010, 53, 4927–4937. [Google Scholar] [CrossRef] [PubMed]
- Mao, F.; Huang, L.; Luo, Z.; Liu, A.; Lu, C.; Xie, Z.; Li, X. O-Hydroxyl- or o-amino benzylamine-tacrine hybrids: Multifunctional biometals chelators, antioxidants, and inhibitors of cholinesterase activity and amyloid-beta aggregation. Bioorg. Med. Chem. 2012, 20, 5884–5892. [Google Scholar] [CrossRef] [PubMed]
- Hiremathad, A.; Keri, R.S.; Esteves, A.R.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Novel Tacrine-Hydroxyphenylbenzimidazole hybrids as potential multitarget drug candidates for Alzheimer’s disease. Eur. J. Med. Chem. 2018, 148, 255–267. [Google Scholar] [CrossRef] [PubMed]
- Hamulakova, S.; Poprac, P.; Jomova, K.; Brezova, V.; Lauro, P.; Drostinova, L.; Jun, D.; Sepsova, V.; Hrabinova, M.; Soukup, O.; et al. Targeting copper(II)-induced oxidative stress and the acetylcholinesterase system in Alzheimer’s disease using multifunctional tacrine-coumarin hybrid molecules. J. Inorg. Biochem. 2016, 161, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, B.; Antonio, T.; Reith, M.E.; Dutta, A.K. Discovery of 4-(4-(2-((5-Hydroxy-1,2,3,4-tetrahydronaphthalen-2-yl)(propyl)amino)ethyl)piperazin-1-yl)quinolin-8-ol and its analogues as highly potent dopamine D2/D3 agonists and as iron chelator: In vivo activity indicates potential application in symptomatic and neuroprotective therapy for Parkinson’s disease. J. Med. Chem. 2010, 53, 2114–2125. [Google Scholar] [PubMed]
- Das, B.; Kandegedara, A.; Xu, L.; Antonio, T.; Stemmler, T.; Reith, M.E.A.; Dutta, A.K. A Novel Iron(II) Preferring Dopamine Agonist Chelator as Potential Symptomatic and Neuroprotective Therapeutic Agent for Parkinson’s Disease. ACS Chem. Neurosci. 2017, 8, 723–730. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Rajagopalan, S.; Joshi, G.S.; Xu, L.; Luo, D.; Andersen, J.K.; Todi, S.V.; Dutta, A.K. A novel iron (II) preferring dopamine agonist chelator D-607 significantly suppresses alpha-syn- and MPTP-induced toxicities in vivo. Neuropharmacology 2017, 123, 88–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Y.; Chen, Y.; Li, Y.P.; Chen, S.H.; Tan, J.H.; Ou, T.M.; Gu, L.Q.; Huang, Z.S. Design, synthesis, and biological evaluation of curcumin analogues as multifunctional agents for the treatment of Alzheimer’s disease. Bioorg. Med. Chem. 2011, 19, 5596–5604. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Lu, C.; Sun, Y.; Mao, F.; Luo, Z.; Su, T.; Jiang, H.; Shan, W.; Li, X. Multitarget-directed benzylideneindanone derivatives: Anti-beta-amyloid (Abeta) aggregation, antioxidant, metal chelation, and monoamine oxidase B (MAO-B) inhibition properties against Alzheimer’s disease. J. Med. Chem. 2012, 55, 8483–8492. [Google Scholar] [CrossRef] [PubMed]
- Nunes, A.; Marques, S.M.; Quintanova, C.; Silva, D.F.; Cardoso, S.M.; Chaves, S.; Santos, M.A. Multifunctional iron-chelators with protective roles against neurodegenerative diseases. Dalton Trans. 2013, 42, 6058–6073. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Chen, J.; Li, X.; Su, T.; Wang, Y.; Wang, Z.; Huang, L.; Li, X. Synthesis and evaluation of selegiline derivatives as monoamine oxidase inhibitor, antioxidant and metal chelator against Alzheimer’s disease. Bioorg. Med. Chem. 2015, 23, 3722–3729. [Google Scholar] [CrossRef] [PubMed]
- Sheng, R.; Tang, L.; Jiang, L.; Hong, L.; Shi, Y.; Zhou, N.; Hu, Y. Novel 1-Phenyl-3-hydroxy-4-pyridinone Derivatives as Multifunctional Agents for the Therapy of Alzheimer’s Disease. ACS Chem. Neurosci. 2016, 7, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, P.; Garcia-Beltran, O.; Tapia, V.; Munoz, Y.; Cassels, B.K.; Nunez, M.T. Neuroprotective effect of a new 7,8-dihydroxycoumarin-based Fe2+/Cu2+ chelator in cell and animal models of Parkinson’s disease. ACS Chem. Neurosci. 2017, 12, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Beltran, O.; Mena, N.P.; Aguirre, P.; Barriga-Gonzalez, G.; Galdamez, A.; Nagles, E.; Adasme, T.; Hidalgo, C.; Nunez, M.T. Development of an iron-selective antioxidant probe with protective effects on neuronal function. PLoS ONE 2017, 12, e0189043. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.Q.; Wang, Z.S.; Ma, Y.X.; Zhang, W.; Lu, J.L.; Liang, Y.R.; Zheng, X.Q. Neuroprotective Effects and Mechanisms of Tea Bioactive Components in Neurodegenerative Diseases. Molecules 2018, 23, 512. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Reznichenko, L.; Amit, T.; Youdim, M.B. Green tea polyphenol (−)-epigallocatechin-3-gallate protects rat PC12 cells from apoptosis induced by serum withdrawal independent of P13-Akt pathway. Neurotox. Res. 2003, 5, 419–424. [Google Scholar] [CrossRef] [PubMed]
- Xu, Q.; Langley, M.; Kanthasamy, A.G.; Reddy, M.B. Epigallocatechin Gallate Has a Neurorescue Effect in a Mouse Model of Parkinson Disease. J. Nutr. 2017, 147, 1926–1931. [Google Scholar] [CrossRef] [PubMed]
- Li, R.; Peng, N.; Li, X.P.; Le, W.D. (−)-Epigallocatechin gallate regulates dopamine transporter internalization via protein kinase C-dependent pathway. Brain Res. 2006, 1097, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Miller, G.W.; Gainetdinov, R.R.; Levey, A.I.; Caron, M.G. Dopamine transporters and neuronal injury. Trends Pharmacol. Sci. 1999, 20, 424–429. [Google Scholar] [CrossRef]
- McKinley, E.T.; Baranowski, T.C.; Blavo, D.O.; Cato, C.; Doan, T.N.; Rubinstein, A.L. Neuroprotection of MPTP-induced toxicity in zebrafish dopaminergic neurons. Brain Res. Mol. Brain Res. 2005, 141, 128–137. [Google Scholar] [CrossRef] [PubMed]
- Gainetdinov, R.R.; Fumagalli, F.; Jones, S.R.; Caron, M.G. Dopamine transporter is required for in vivo MPTP neurotoxicity: Evidence from mice lacking the transporter. J. Neurochem. 1997, 69, 1322–1325. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Gross, C.E.; Fournier, M.C.; Dovero, S.; Bloch, B.; Jaber, M. Absence of MPTP-induced neuronal death in mice lacking the dopamine transporter. Exp. Neurol. 1999, 155, 268–273. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Zhang, X.; Zhao, J.F.; Wang, Z.Y.; Bickers, D.; Lebwohl, M. Scavenging of hydrogen peroxide and inhibition of ultraviolet light-induced oxidative DNA damage by aqueous extracts from green and black teas. Free. Radic. Boil. Med. 1999, 26, 1427–1435. [Google Scholar] [CrossRef]
- Nakagawa, T.; Yokozawa, T. Direct scavenging of nitric oxide and superoxide by green tea. Food Chem. Toxicol. 2002, 40, 1745–1750. [Google Scholar] [CrossRef]
- Frei, B.; Higdon, J.V. Antioxidant activity of tea polyphenols in vivo: Evidence from animal studies. J. Nutr. 2003, 133, 3275s–3284s. [Google Scholar] [CrossRef] [PubMed]
- Raza, H.; John, A. Green tea polyphenol epigallocatechin-3-gallate differentially modulates oxidative stress in PC12 cell compartments. Toxicol. Appl. Pharmacol. 2005, 207, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Noda, C.; He, J.; Takano, T.; Tanaka, C.; Kondo, T.; Tohyama, K.; Yamamura, H.; Tohyama, Y. Induction of apoptosis by epigallocatechin-3-gallate in human lymphoblastoid B cells. Biochem. Biophys. Res. Commun. 2007, 362, 951–957. [Google Scholar] [CrossRef] [PubMed]
- Hsuuw, Y.D.; Chan, W.H. Epigallocatechin gallate dose-dependently induces apoptosis or necrosis in human MCF-7 cells. Ann. N. Y. Acad. Sci. 2007, 1095, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Yin, S.T.; Tang, M.L.; Deng, H.M.; Xing, T.R.; Chen, J.T.; Wang, H.L.; Ruan, D.Y. Epigallocatechin-3-gallate induced primary cultures of rat hippocampal neurons death linked to calcium overload and oxidative stress. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2009, 379, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Suh, K.S.; Chon, S.; Oh, S.; Kim, S.W.; Kim, J.W.; Kim, Y.S.; Woo, J.T. Prooxidative effects of green tea polyphenol (−)-epigallocatechin-3-gallate on the HIT-T15 pancreatic beta cell line. Cell Boil. Toxicol. 2010, 26, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.H.; Cheng, J.; Li, C.R.; Ye, M.; Ma, Z.; Cai, F. Modulation of Ca(2)(+) signals by epigallocatechin-3-gallate(EGCG) in cultured rat hippocampal neurons. Int. J. Mol. Sci. 2011, 12, 742–754. [Google Scholar] [CrossRef] [PubMed]
- Qanungo, S.; Das, M.; Haldar, S.; Basu, A. Epigallocatechin-3-gallate induces mitochondrial membrane depolarization and caspase-dependent apoptosis in pancreatic cancer cells. Carcinogenesis 2005, 26, 958–967. [Google Scholar] [CrossRef] [PubMed]
- Cavet, M.E.; Harrington, K.L.; Vollmer, T.R.; Ward, K.W.; Zhang, J.Z. Anti-inflammatory and anti-oxidative effects of the green tea polyphenol epigallocatechin gallate in human corneal epithelial cells. Mol. Vis. 2011, 17, 533–542. [Google Scholar] [PubMed]
- Wang, L.; Tian, X. Epigallocatechin-3-Gallate Protects against Homocysteine-Induced Brain Damage in Rats. Planta Med. 2018, 84, 34–41. [Google Scholar] [CrossRef] [PubMed]
- Mandel, S.; Amit, T.; Bar-Am, O.; Youdim, M.B. Iron dysregulation in Alzheimer’s disease: Multimodal brain permeable iron chelating drugs, possessing neuroprotective-neurorescue and amyloid precursor protein-processing regulatory activities as therapeutic agents. Prog. Neurobiol. 2007, 82, 348–360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Liu, F.; Jin, H.; Li, R.; Wang, Y.; Zhang, W.; Wang, H.; Chen, W. Involvement of PKCalpha and ERK1/2 signaling pathways in EGCG’s protection against stress-induced neural injuries in Wistar rats. Neuroscience 2017, 346, 226–237. [Google Scholar] [CrossRef] [PubMed]
- Youdim, M.B.; Kupershmidt, L.; Amit, T.; Weinreb, O. Promises of novel multi-target neuroprotective and neurorestorative drugs for Parkinson’s disease. Parkinsonism Relat. Disord. 2014, 20 (Suppl. S1), S132–S136. [Google Scholar] [CrossRef]
- Amit, T.; Bar-Am, O.; Mechlovich, D.; Kupershmidt, L.; Youdim, M.B.H.; Weinreb, O. The novel multitarget iron chelating and propargylamine drug M30 affects APP regulation and processing activities in Alzheimer’s disease models. Neuropharmacology 2017, 123, 359–367. [Google Scholar] [CrossRef] [PubMed]
- Avramovich-Tirosh, Y.; Bar-Am, O.; Amit, T.; Youdim, M.B.; Weinreb, O. Up-regulation of hypoxia-inducible factor (HIF)-1α and HIF-target genes in cortical neurons by the novel multifunctional iron chelator anti-Alzheimer drug, M30. Curr. Alzheimer Res. 2010, 7, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Park, S.S.; Bae, I.; Lee, Y.J. Flavonoids-induced accumulation of hypoxia-inducible factor (HIF)-1alpha/2alpha is mediated through chelation of iron. J. Cell Biochem. 2008, 103, 1989–1998. [Google Scholar] [CrossRef] [PubMed]
- Golko-Perez, S.; Amit, T.; Bar-Am, O.; Youdim, M.B.; Weinreb, O. A novel iron chelator-radical scavenger ameliorates motor dysfunction and improves life span and mitochondrial biogenesis in SOD1 G93A ALS mice. Neurotox. Res. 2017, 31, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Golko-Perez, S.; Amit, T.; Youdim, M.B.; Weinreb, O. Beneficial Effects of Multitarget Iron Chelator on Central Nervous System and Gastrocnemius Muscle in SOD1 G93A Transgenic ALS Mice. J. Mol. Neurosci. 2016, 59, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Qutub, A.A.; Hunt, C.A. Glucose transport to the brain: A systems model. Brain Res. Rev. 2005, 49, 595–617. [Google Scholar] [CrossRef] [PubMed]
- Schugar, H.; Green, D.E.; Bowen, M.L.; Scott, L.E.; Storr, T.; Bohmerle, K.; Thomas, F.; Allen, D.D.; Lockman, P.R.; Merkel, M.; et al. Combating Alzheimer’s disease with multifunctional molecules designed for metal passivation. Angew. Chem. 2007, 46, 1716–1718. [Google Scholar] [CrossRef] [PubMed]
- Jakoby, P.; Schmidt, E.; Ruminot, I.; Gutierrez, R.; Barros, L.F.; Deitmer, J.W. Higher transport and metabolism of glucose in astrocytes compared with neurons: A multiphoton study of hippocampal and cerebellar tissue slices. Cereb. Cortex 2014, 24, 222–231. [Google Scholar] [CrossRef] [PubMed]
- Tumiatti, V.; Minarini, A.; Bolognesi, M.L.; Milelli, A.; Rosini, M.; Melchiorre, C. Tacrine derivatives and Alzheimer’s disease. Curr. Med. Chem. 2010, 17, 1825–1838. [Google Scholar] [CrossRef] [PubMed]
- Giacobini, E. Modulation of brain acetylcholine levels with cholinesterase inhibitors as a treatment of Alzheimer disease. Keio J. Med. 1987, 36, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Jogani, V.V.; Shah, P.J.; Mishra, P.; Mishra, A.K.; Misra, A.R. Intranasal mucoadhesive microemulsion of tacrine to improve brain targeting. Alzheimer Dis. Assoc. Disord. 2008, 22, 116–124. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Compound | Properties/Characteristics | Metal Specificity | In-Vivo Testing | Route of Administration | Brain Permeability | Disease Model | References |
|---|---|---|---|---|---|---|---|
| EGCG | Metal chelation; antioxidant; neuroprotective; activation of cell survival genes. | Cu2+; Fe3+; Al3+; Mn2+ | Yes | Intraperitoneal; Oral | Yes | PD, AD | [146,147,148,149,150] |
| Hydroxyquinoline‒propargyl hybrids M30, VAR10303 | Metal chelation; MAO-B inhibition; antiapoptotic; activation of cell survival genes; neuroprotective; neuritogenic. | Fe3+ > Cu2+ > Zn2+ | Yes | Oral | Yes | PD, AD, amyotrophic lateral sclerosis | [151,152,153,154] |
| Hydroxypyridinone glycoconjugates H2GL1, H2GL2 | Metal chelation; reduction of amyloid-beta aggregation | Cu2+ > Zn2+ | No | Not tested | Not tested; probably yes | AD | [155] |
| Bis-tacrine hybrids | Metal chelation; AChE inhibition; reduction of amyloid-beta aggregation | Cu2+ | No | Not tested | Not tested | AD | [156] |
| 8-OH-Quinoline‒tacrine hybrids | Metal chelation; AChE inhibition | Cu2+ | No | Not tested | Probably yes | AD | [157] |
| Benzylamine‒tacrine hybrids | Metal chelation; AChE inhibition; inhibition of amyloid-beta aggregation; moderate antioxidant activity | Cu2+; Fe2+; Zn2+ | No | Not tested | Not tested | AD | [158] |
| Phenyl–benzimidazole‒tacrine hybrid | AChE inhibition; metal chelation; inhibition of Cu-induced amyloid-beta aggregation; free radical scavenger | Cu2+; other metals not tested | No | Not tested | Not tested | AD | [159] |
| Coumarin‒tacrine hybrid | Metal chelation; AChE inhibition; inhibition of amyloid-beta aggregation; free radical scavenger | Cu2+; other metals not tested | No | Not tested | Not tested | AD | [160] |
| Piperazine–8-OH-quinolone hybrids | Metal chelation; dopamine D2/D3 receptor agonists; hydroxyl radical scavenger | Fe2+; Fe3+ | Yes | Subcutaneous | Yes | PD | [161] |
| Dipyridyl‒D2R/D3R agonist hybrids | Metal chelation; dopamine D2/D3 receptor agonist; antioxidant; neuroprotective | Fe2+ >>> Fe3+ | Yes | Intraperitoneal | Yes | PD | [162,163] |
| Curcumin hybrids | Metal chelation; antioxidant activity; reduction of amyloid-beta aggregation | Cu2+; Fe2+ | No | Not tested | Not tested | AD | [164] |
| Benzyl–indanone hybrid compound 41 | Metal chelation; antioxidant activity; AChE inhibition; inhibition of amyloid-beta aggregation | Cu2+ | No | Not tested | Not tested | AD | [165] |
| Benzothiazole–linker–pyridinone hybrids | Metal chelation; antioxidant activity; AChE inhibition; inhibition of amyloid-beta aggregation | Fe3+ | No | Not tested | Probably yes | AD | [166] |
| Clioquinol‒selegiline hybrids | MAO-B inhibition; metal chelation; antioxidant activity | Cu2+; Fe2+; Zn2+ | No | Not tested | Probably yes | PD | [167] |
| Deferiprone‒H3 receptor antagonist hybrid C5 | H3R inhibition; metal chelation; antioxidant activity; reduction of amyloid-beta aggregation | Cu2+∼ Fe2+ >>> Zn2+ | Yes | Intraperitoneal | Yes | AD | [168] |
| 7,8-Dihydroxycoumarin derivative DHC12 | Metal chelation; MAO-B inhibition; mitochondriotropic; free radical scavenger; neuroprotective | Cu2+∼ Fe2+ > Zn2+ > Fe3+ | Yes | Oral | Yes | PD | [169] |
| Coumarin–tris hybrid CT51 | Metal chelation; impedes Fe2+/Fe3+cycling; antioxidant; mitochondriotropic; calcium upsurge blocker | Fe2+ > Fe3+ | No | Not tested | Not tested | PD | [170] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nuñez, M.T.; Chana-Cuevas, P. New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases. Pharmaceuticals 2018, 11, 109. https://doi.org/10.3390/ph11040109
Nuñez MT, Chana-Cuevas P. New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases. Pharmaceuticals. 2018; 11(4):109. https://doi.org/10.3390/ph11040109
Chicago/Turabian StyleNuñez, Marco T., and Pedro Chana-Cuevas. 2018. "New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases" Pharmaceuticals 11, no. 4: 109. https://doi.org/10.3390/ph11040109
APA StyleNuñez, M. T., & Chana-Cuevas, P. (2018). New Perspectives in Iron Chelation Therapy for the Treatment of Neurodegenerative Diseases. Pharmaceuticals, 11(4), 109. https://doi.org/10.3390/ph11040109