Mitochondrial Targeting in Neurodegeneration: A Heme Perspective


Abstract
1. Implication of Heme in Neurodegeneration
2. Role of Mitochondria in Neurodegenerative Diseases
2.1. Mutations on Mitochondrial DNA (mtDNA) Genes
2.2. Mutations on Nuclear DNA Genes Encoding Proteins Crucial for Mitochondrial Functionality
2.3. Alterations of Mitochondrial Dynamics (Fusion, Fission, Motility)
2.4. Inappropriate Activation of Cell Apoptosis by Mitochondria
2.5. Alteration of Mitochondria-Dependent Ca2+ Homeostasis
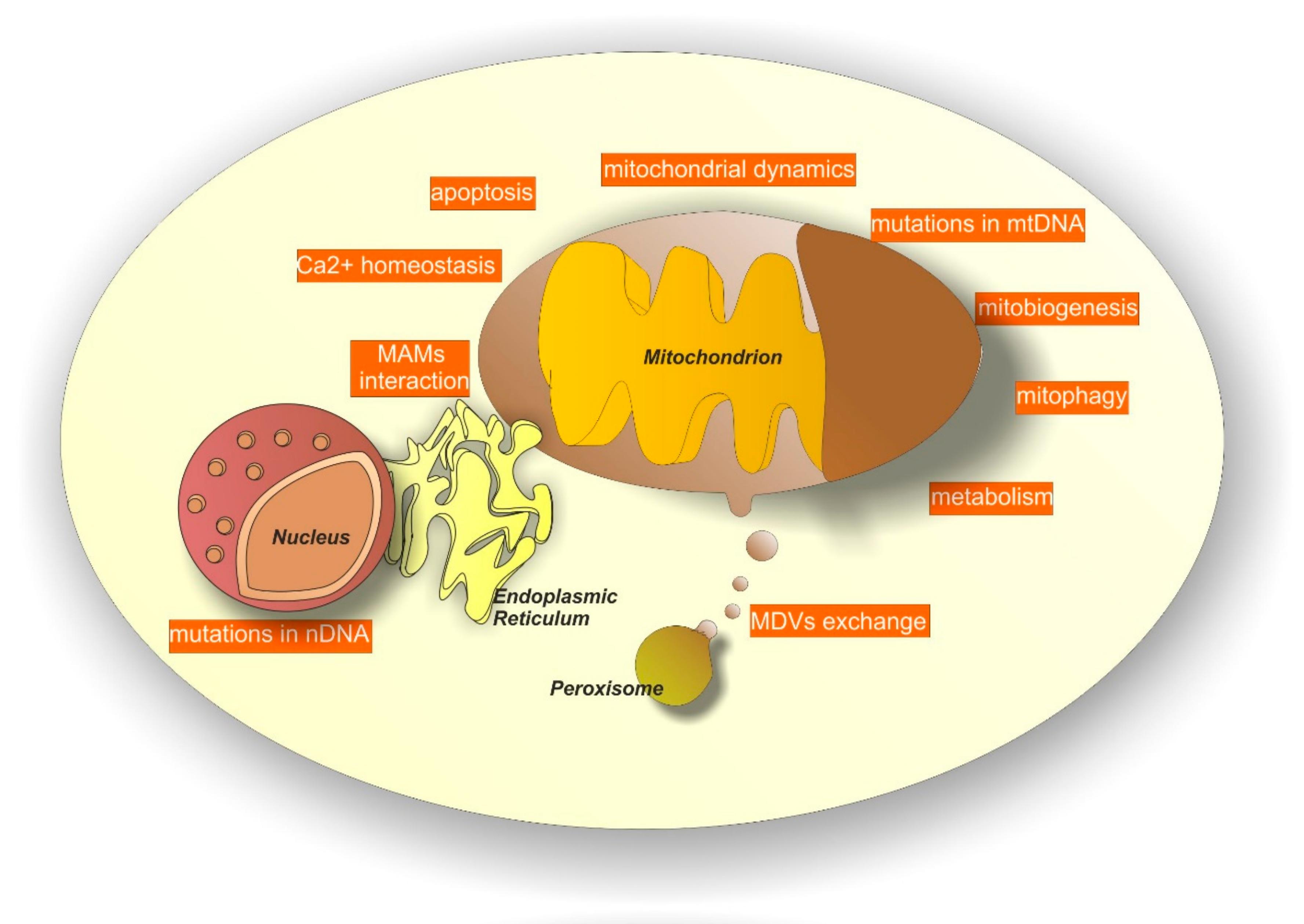
2.6. Additional Alterations of Mitochondrial-Related Processes: Biogenesis, Mitophagy, Mdvs Exchange, Interaction with Mams, Control of Cellular Metabolism
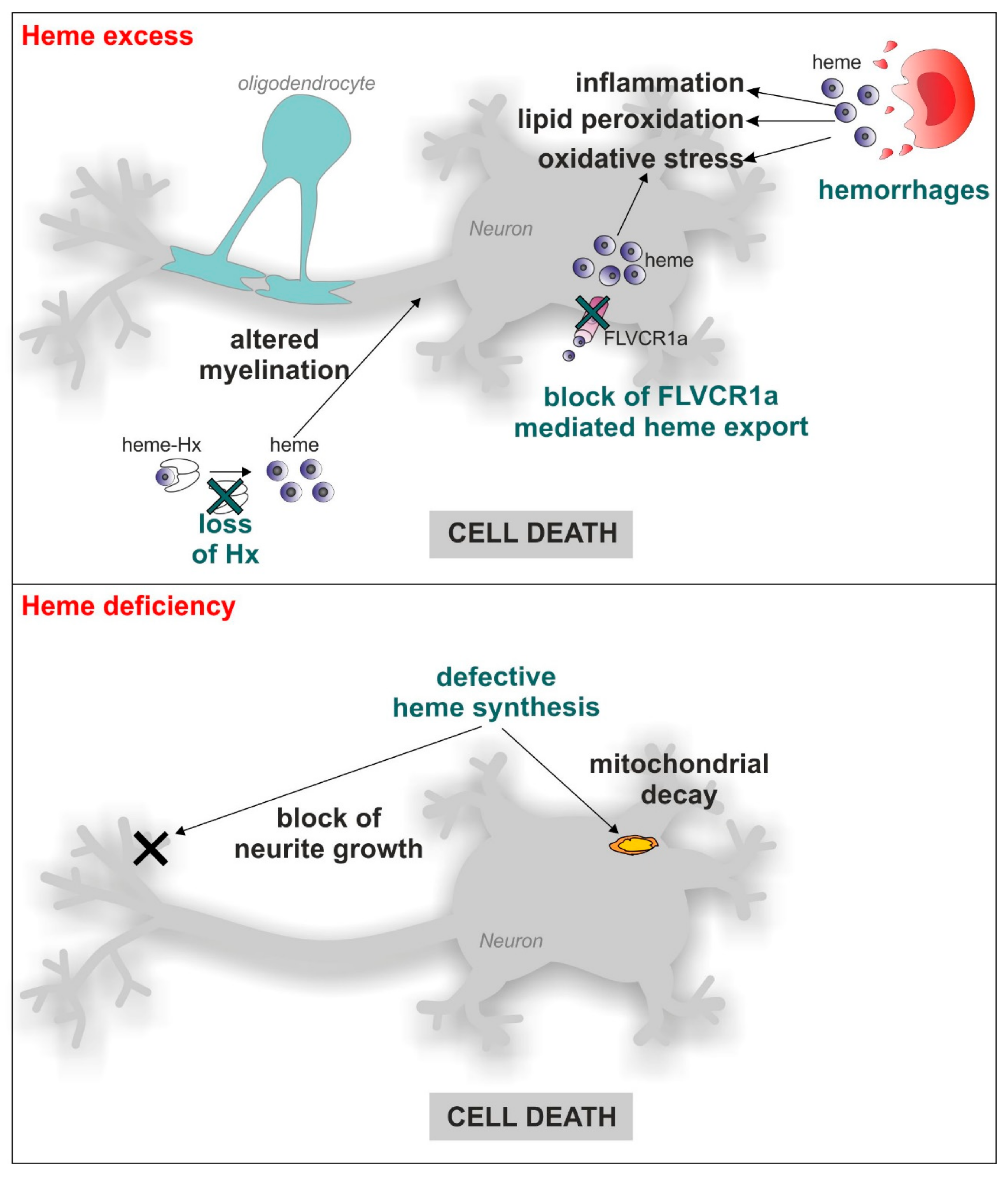
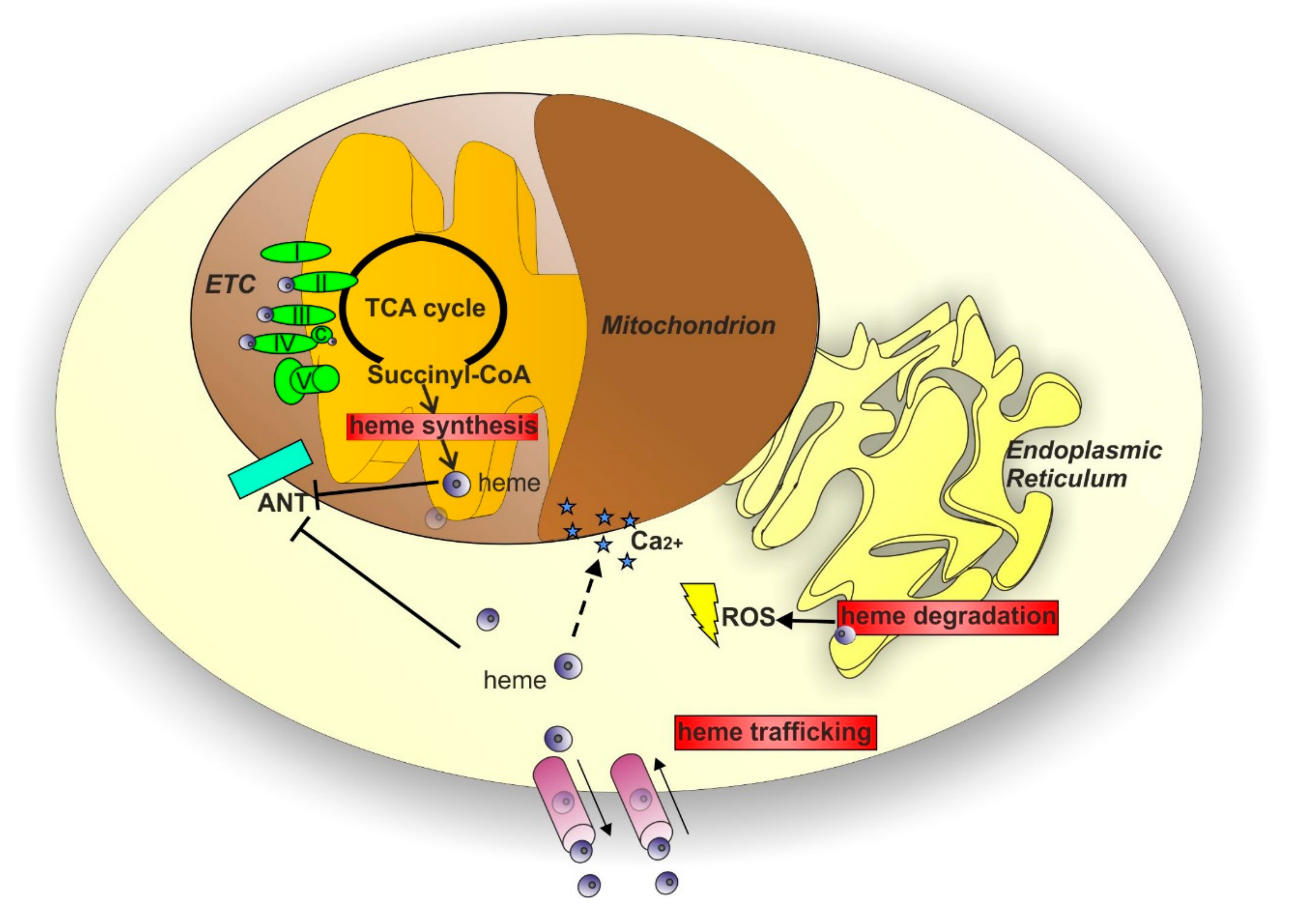
3. Heme and Mitochondrial Dysfunction Related to Neurodegenerative Diseases
4. Current Therapies and Potential Future Approaches to Face Mitochondrial Dysfunction in Neurodegenerative Diseases
5. Conclusions
Funding
Conflicts of Interest
References
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Mercurio, S.; Tolosano, E. Heme in pathophysiology: A matter of scavenging, metabolism and trafficking across cell membranes. Front. Pharmacol. 2014, 5, 61. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Hon, T.; Ye, W.; Zhang, L. Heme deficiency interferes with the Ras-mitogen-activated protein kinase signaling pathway and expression of a subset of neuronal genes. Cell Growth Differ. 2002, 13, 431–439. [Google Scholar] [PubMed]
- Sengupta, A.; Hon, T.; Zhang, L. Heme deficiency suppresses the expression of key neuronal genes and causes neuronal cell death. Brain Res. Mol. Brain Res. 2005, 137, 23–30. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Killilea, D.W.; Killilea, A.N.; Ames, B.N. Heme deficiency may be a factor in the mitochondrial and neuronal decay of aging. Proc. Natl. Acad. Sci. USA 2002, 99, 14807–14812. [Google Scholar] [CrossRef] [PubMed]
- Righy, C.; Bozza, M.T.; Oliveira, M.F.; Bozza, F.A. Molecular, Cellular and Clinical Aspects of Intracerebral Hemorrhage: Are the Enemies Within? Curr. Neuropharmacol. 2016, 14, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Ma, B.; Day, J.P.; Phillips, H.; Slootsky, B.; Tolosano, E.; Doré, S. Deletion of the hemopexin or heme oxygenase-2 gene aggravates brain injury following stroma-free hemoglobin-induced intracerebral hemorrhage. J. Neuroinflamm. 2016, 13, 26. [Google Scholar] [CrossRef] [PubMed]
- Hahl, P.; Davis, T.; Washburn, C.; Rogers, J.T.; Smith, A. Mechanisms of neuroprotection by hemopexin: Modeling the control of heme and iron homeostasis in brain neurons in inflammatory states. J. Neurochem. 2013, 125, 89–101. [Google Scholar] [CrossRef] [PubMed]
- Morello, N.; Tonoli, E.; Logrand, F.; Fiorito, V.; Fagoonee, S.; Turco, E.; Silengo, L.; Vercelli, A.; Altruda, F.; Tolosano, E. Haemopexin affects iron distribution and ferritin expression in mouse brain. J. Cell. Mol. Med. 2009, 13, 4192–4204. [Google Scholar] [CrossRef] [PubMed]
- Morello, N.; Bianchi, F.T.; Marmiroli, P.; Tonoli, E.; Rodriguez Menendez, V.; Silengo, L.; Cavaletti, G.; Vercelli, A.; Altruda, F.; Tolosano, E. A role for hemopexin in oligodendrocyte differentiation and myelin formation. PLoS ONE 2011, 6, e20173. [Google Scholar] [CrossRef] [PubMed]
- Tolosano, E.; Fagoonee, S.; Morello, N.; Vinchi, F.; Fiorito, V. Heme scavenging and the other facets of hemopexin. Antioxid. Redox Signal. 2010, 12, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Castori, M.; di Rocco, M.; Ungelenk, M.; Gießelmann, S.; Di Capua, M.; Madeo, A.; Grammatico, P.; Bartsch, S.; Hübner, C.A.; et al. Mutations in the Heme Exporter FLVCR1 Cause Sensory Neurodegeneration with Loss of Pain Perception. PLoS Genet. 2016, 12, e1006461. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Vinchi, F.; Fiorito, V.; Tolosano, E. Haptoglobin and Hemopexin in Heme Detoxification and Iron Recycling. In Acute Phase Proteins Francisco Veas; IntechOpen: London, UK, 2011; pp. 262–288. [Google Scholar]
- Blackburn, S.L.; Kumar, P.T.; McBride, D.; Zeineddine, H.A.; Leclerc, J.; Choi, H.A.; Dash, P.K.; Grotta, J.; Aronowski, J.; Cardenas, J.C.; et al. Unique Contribution of Haptoglobin and Haptoglobin Genotype in Aneurysmal Subarachnoid Hemorrhage. Front. Physiol. 2018, 9, 592. [Google Scholar] [CrossRef] [PubMed]
- Marro, S.; Barisani, D.; Chiabrando, D.; Fagoonee, S.; Muckenthaler, M.U.; Stolte, J.; Meneveri, R.; Haile, D.; Silengo, L.; Altruda, F.; et al. Lack of haptoglobin affects iron transport across duodenum by modulating ferroportin expression. Gastroenterology 2007, 133, 1261–1271. [Google Scholar] [CrossRef] [PubMed]
- Reddi, A.R.; Hamza, I. Heme Mobilization in Animals: A Metallolipid’s Journey. Acc. Chem. Res. 2016, 49, 1104–1110. [Google Scholar] [CrossRef] [PubMed]
- Gozzelino, R. The Pathophysiology of Heme in the Brain. Curr. Alzheimer Res. 2016, 13, 174–184. [Google Scholar] [CrossRef] [PubMed]
- Erkkinen, M.G.; Kim, M.O.; Geschwind, M.D. Clinical Neurology and Epidemiology of the Major Neurodegenerative Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10, a033118. [Google Scholar] [CrossRef] [PubMed]
- Tracy, J.A.; Dyck, P.J. Porphyria and its neurologic manifestations. Handb. Clin. Neurol. 2014, 120, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Albers, J.W.; Fink, J.K. Porphyric neuropathy. Muscle Nerve 2004, 30, 410–422. [Google Scholar] [CrossRef] [PubMed]
- Simon, N.G.; Herkes, G.K. The neurologic manifestations of the acute porphyrias. J. Clin. Neurosci. 2011, 18, 1147–1153. [Google Scholar] [CrossRef] [PubMed]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Yoon, T.; Cowan, J.A. Frataxin-mediated iron delivery to ferrochelatase in the final step of heme biosynthesis. J. Biol. Chem. 2004, 279, 25943–25946. [Google Scholar] [CrossRef] [PubMed]
- Lange, H.; Mühlenhoff, U.; Denzel, M.; Kispal, G.; Lill, R. The heme synthesis defect of mutants impaired in mitochondrial iron-sulfur protein biogenesis is caused by reversible inhibition of ferrochelatase. J. Biol. Chem. 2004, 279, 29101–29108. [Google Scholar] [CrossRef] [PubMed]
- Schoenfeld, R.A.; Napoli, E.; Wong, A.; Zhan, S.; Reutenauer, L.; Morin, D.; Buckpitt, A.R.; Taroni, F.; Lonnerdal, B.; Ristow, M.; et al. Frataxin deficiency alters heme pathway transcripts and decreases mitochondrial heme metabolites in mammalian cells. Hum. Mol. Genet. 2005, 14, 3787–3799. [Google Scholar] [CrossRef] [PubMed]
- Chernova, T.; Nicotera, P.; Smith, A.G. Heme deficiency is associated with senescence and causes suppression of N-methyl-d-aspartate receptor subunits expression in primary cortical neurons. Mol. Pharmacol. 2006, 69, 697–705. [Google Scholar] [CrossRef] [PubMed]
- Petrillo, S.; Chiabrando, D.; Genova, T.; Fiorito, V.; Ingoglia, G.; Vinchi, F.; Mussano, F.; Carossa, S.; Silengo, L.; Altruda, F.; et al. Heme accumulation in endothelial cells impairs angiogenesis by triggering paraptosis. Cell Death Differ. 2018, 25, 573–588. [Google Scholar] [CrossRef] [PubMed]
- Vinchi, F.; Ingoglia, G.; Chiabrando, D.; Mercurio, S.; Turco, E.; Silengo, L.; Altruda, F.; Tolosano, E. Heme exporter FLVCR1a regulates heme synthesis and degradation and controls activity of cytochromes P450. Gastroenterology 2014, 146, 1325–1338. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, V.; Neri, F.; Pala, V.; Silengo, L.; Oliviero, S.; Altruda, F.; Tolosano, E. Hypoxia controls Flvcr1 gene expression in Caco2 cells through HIF2α and ETS1. Biochim. Biophys. Acta 2014, 1839, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Fiorito, V.; Forni, M.; Silengo, L.; Altruda, F.; Tolosano, E. Crucial Role of FLVCR1a in the Maintenance of Intestinal Heme Homeostasis. Antioxid. Redox Signal. 2015, 23, 1410–1423. [Google Scholar] [CrossRef] [PubMed]
- Duffy, S.P.; Shing, J.; Saraon, P.; Berger, L.C.; Eiden, M.V.; Wilde, A.; Tailor, C.S. The Fowler syndrome-associated protein FLVCR2 is an importer of heme. Mol. Cell. Biol. 2010, 30, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Quigley, J.G.; Yang, Z.; Worthington, M.T.; Phillips, J.D.; Sabo, K.M.; Sabath, D.E.; Berg, C.L.; Sassa, S.; Wood, B.L.; Abkowitz, J.L. Identification of a human heme exporter that is essential for erythropoiesis. Cell 2004, 118, 757–766. [Google Scholar] [CrossRef] [PubMed]
- Rajadhyaksha, A.M.; Elemento, O.; Puffenberger, E.G.; Schierberl, K.C.; Xiang, J.Z.; Putorti, M.L.; Berciano, J.; Poulin, C.; Brais, B.; Michaelides, M.; et al. Mutations in FLVCR1 cause posterior column ataxia and retinitis pigmentosa. Am. J. Hum. Genet. 2010, 87, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Ishiura, H.; Fukuda, Y.; Mitsui, J.; Nakahara, Y.; Ahsan, B.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; Sakai, T.; Tsuji, S. Posterior column ataxia with retinitis pigmentosa in a Japanese family with a novel mutation in FLVCR1. Neurogenetics 2011, 12, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Shaibani, A.; Wong, L.J.; Wei Zhang, V.; Lewis, R.A.; Shinawi, M. Autosomal recessive posterior column ataxia with retinitis pigmentosa caused by novel mutations in the FLVCR1 gene. Int. J. Neurosci. 2015, 125, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, A.; Bahr, A.; Bähr, L.; Fleischhauer, J.; Zinkernagel, M.S.; Winkler, N.; Barthelmes, D.; Berger, L.; Gerth-Kahlert, C.; Neidhardt, J.; et al. Next generation sequencing based identification of disease-associated mutations in Swiss patients with retinal dystrophies. Sci. Rep. 2016, 6, 28755. [Google Scholar] [CrossRef] [PubMed]
- Yusuf, I.H.; Shanks, M.E.; Clouston, P.; MacLaren, R.E. A splice-site variant in FLVCR1 produces retinitis pigmentosa without posterior column ataxia. Ophthalmic Genet. 2018, 39, 263–267. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Morlino, S.; Ungelenk, M.; Pareyson, D.; Salsano, E.; Grammatico, P.; Tolosano, E.; Kurth, I.; Chiabrando, D. Posterior column ataxia with retinitis pigmentosa coexisting with sensory-autonomic neuropathy and leukemia due to the homozygous p.Pro221Ser FLVCR1 mutation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2017, 174, 732–739. [Google Scholar] [CrossRef] [PubMed]
- Meyer, E.; Ricketts, C.; Morgan, N.V.; Morris, M.R.; Pasha, S.; Tee, L.J.; Rahman, F.; Bazin, A.; Bessières, B.; Déchelotte, P.; et al. Mutations in FLVCR2 are associated with proliferative vasculopathy and hydranencephaly-hydrocephaly syndrome (Fowler syndrome). Am. J. Hum. Genet. 2010, 86, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Kvarnung, M.; Taylan, F.; Nilsson, D.; Albåge, M.; Nordenskjöld, M.; Anderlid, B.M.; Nordgren, A.; Syk Lundberg, E. Mutations in FLVCR2 associated with Fowler syndrome and survival beyond infancy. Clin. Genet. 2016, 89, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Dwyer, B.E.; Smith, M.A.; Richardson, S.L.; Perry, G.; Zhu, X. Down-regulation of aminolevulinate synthase, the rate-limiting enzyme for heme biosynthesis in Alzheimer’s disease. Neurosci. Lett. 2009, 460, 180–184. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Frey, W.H. A role for heme in Alzheimer’s disease: Heme binds amyloid beta and has altered metabolism. Proc. Natl. Acad. Sci. USA 2004, 101, 11153–11158. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H. Heme binding to Amyloid-beta peptide: Mechanistic role in Alzheimer’s disease. J. Alzheimers Dis. 2006, 10, 255–266. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.G.; Raven, E.L.; Chernova, T. The regulatory role of heme in neurons. Metallomics 2011, 3, 955–962. [Google Scholar] [CrossRef] [PubMed]
- Hayden, E.Y.; Kaur, P.; Williams, T.L.; Matsui, H.; Yeh, S.R.; Rousseau, D.L. Heme Stabilization of α-Synuclein Oligomers during Amyloid Fibril Formation. Biochemistry 2015, 54, 4599–4610. [Google Scholar] [CrossRef] [PubMed]
- Santiago, J.A.; Potashkin, J.A. Blood Transcriptomic Meta-analysis Identifies Dysregulation of Hemoglobin and Iron Metabolism in Parkinson’ Disease. Front. Aging Neurosci. 2017, 9, 73. [Google Scholar] [CrossRef] [PubMed]
- Scherzer, C.R.; Grass, J.A.; Liao, Z.; Pepivani, I.; Zheng, B.; Eklund, A.C.; Ney, P.A.; Ng, J.; McGoldrick, M.; Mollenhauer, B.; et al. GATA transcription factors directly regulate the Parkinson’s disease-linked gene alpha-synuclein. Proc. Natl. Acad. Sci. USA 2008, 105, 10907–10912. [Google Scholar] [CrossRef] [PubMed]
- Sampaio, T.B.; Marcondes Sari, M.H.; Pesarico, A.P.; Nogueira, C.W. δ-Aminolevulinate Dehydratase Activity is Stimulated in a MPTP Mouse Model of Parkinson’s Disease: Correlation with Myeloperoxidase Activity. Cell. Mol. Neurobiol. 2017, 37, 911–917. [Google Scholar] [CrossRef] [PubMed]
- Ringman, J.M.; Schulman, H.; Becker, C.; Jones, T.; Bai, Y.; Immermann, F.; Cole, G.; Sokolow, S.; Gylys, K.; Geschwind, D.H.; et al. Proteomic changes in cerebrospinal fluid of presymptomatic and affected persons carrying familial Alzheimer disease mutations. Arch. Neurol. 2012, 69, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Roher, A.E.; Maarouf, C.L.; Sue, L.I.; Hu, Y.; Wilson, J.; Beach, T.G. Proteomics-derived cerebrospinal fluid markers of autopsy-confirmed Alzheimer’s disease. Biomarkers 2009, 14, 493–501. [Google Scholar] [CrossRef] [PubMed]
- Castaño, E.M.; Roher, A.E.; Esh, C.L.; Kokjohn, T.A.; Beach, T. Comparative proteomics of cerebrospinal fluid in neuropathologically-confirmed Alzheimer’s disease and non-demented elderly subjects. Neurol. Res. 2006, 28, 155–163. [Google Scholar] [CrossRef] [PubMed]
- Song, I.U.; Kim, Y.D.; Chung, S.W.; Cho, H.J. Association between serum haptoglobin and the pathogenesis of Alzheimer’s disease. Intern. Med. 2015, 54, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Cocciolo, A.; Di Domenico, F.; Coccia, R.; Fiorini, A.; Cai, J.; Pierce, W.M.; Mecocci, P.; Butterfield, D.A.; Perluigi, M. Decreased expression and increased oxidation of plasma haptoglobin in Alzheimer disease: Insights from redox proteomics. Free Radic. Biol. Med. 2012, 53, 1868–1876. [Google Scholar] [CrossRef] [PubMed]
- de Farias, C.C.; Maes, M.; Bonifacio, K.L.; Matsumoto, A.K.; Bortolasci, C.C.; Nogueira, A.S.; Brinholi, F.F.; Morimoto, H.K.; de Melo, L.B.; Moreira, E.G.; et al. Parkinson’s Disease is Accompanied by Intertwined Alterations in Iron Metabolism and Activated Immune-inflammatory and Oxidative Stress Pathways. CNS Neurol. Disord. Drug Targets 2017, 16, 484–491. [Google Scholar] [CrossRef] [PubMed]
- Argüelles, S.; Venero, J.L.; García-Rodriguez, S.; Tomas-Camardiel, M.; Ayala, A.; Cano, J.; Machado, A. Use of haptoglobin and transthyretin as potential biomarkers for the preclinical diagnosis of Parkinson’s disease. Neurochem. Int. 2010, 57, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.C.; Wu, Y.R.; Tseng, M.Y.; Chen, Y.C.; Hsieh, S.Y.; Chen, C.M. Increased prothrombin, apolipoprotein A-IV, and haptoglobin in the cerebrospinal fluid of patients with Huntington’s disease. PLoS ONE 2011, 6, e15809. [Google Scholar] [CrossRef] [PubMed]
- Gajowiak, A.; Styś, A.; Starzyński, R.R.; Bednarz, A.; Lenartowicz, M.; Staroń, R.; Lipiński, P. Mice Overexpressing Both Non-Mutated Human SOD1 and Mutated SOD1(G93A) Genes: A Competent Experimental Model for Studying Iron Metabolism in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci. 2015, 8, 82. [Google Scholar] [CrossRef] [PubMed]
- Calingasan, N.Y.; Chen, J.; Kiaei, M.; Beal, M.F. Beta-amyloid 42 accumulation in the lumbar spinal cord motor neurons of amyotrophic lateral sclerosis patients. Neurobiol. Dis. 2005, 19, 340–347. [Google Scholar] [CrossRef] [PubMed]
- Arun, S.; Liu, L.; Donmez, G. Mitochondrial Biology and Neurological Diseases. Curr. Neuropharmacol. 2016, 14, 143–154. [Google Scholar] [CrossRef] [PubMed]
- Bose, A.; Beal, M.F. Mitochondrial dysfunction in Parkinson’s disease. J. Neurochem. 2016, 139 (Suppl. 1), 216–231. [Google Scholar] [CrossRef]
- Shi, P.; Gal, J.; Kwinter, D.M.; Liu, X.; Zhu, H. Mitochondrial dysfunction in amyotrophic lateral sclerosis. Biochim. Biophys. Acta 2010, 1802, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cabezas-Opazo, F.A.; Vergara-Pulgar, K.; Pérez, M.J.; Jara, C.; Osorio-Fuentealba, C.; Quintanilla, R.A. Mitochondrial Dysfunction Contributes to the Pathogenesis of Alzheimer’s Disease. Oxid. Med. Cell. Longev. 2015, 2015, 509654. [Google Scholar] [CrossRef] [PubMed]
- Costa, V.; Scorrano, L. Shaping the role of mitochondria in the pathogenesis of Huntington’s disease. EMBO J. 2012, 31, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Bennett, G.J.; Doyle, T.; Salvemini, D. Mitotoxicity in distal symmetrical sensory peripheral neuropathies. Nat. Rev. Neurol. 2014, 10, 326–336. [Google Scholar] [CrossRef] [PubMed]
- Larsson, N.G. Somatic mitochondrial DNA mutations in mammalian aging. Annu. Rev. Biochem. 2010, 79, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Chinnery, P.F.; Hudson, G. Mitochondrial genetics. Br. Med. Bull. 2013, 106, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Keogh, M.J.; Chinnery, P.F. Mitochondrial DNA mutations in neurodegeneration. Biochim. Biophys. Acta 2015, 1847, 1401–1411. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Kim, D.K.; Mook-Jung, I. The role of mitochondrial DNA mutation on neurodegenerative diseases. Exp. Mol. Med. 2015, 47, e150. [Google Scholar] [CrossRef] [PubMed]
- Nissanka, N.; Moraes, C.T. Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 2018, 592, 728–742. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.; Moraes, C.T. Mitochondrial genome changes and neurodegenerative diseases. Biochim. Biophys. Acta 2014, 1842, 1198–1207. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Ikebe, S.; Tanaka, M.; Ozawa, T. Point mutations of mitochondrial genome in Parkinson’s disease. Brain Res. Mol. Brain Res. 1995, 28, 281–295. [Google Scholar] [CrossRef]
- Perier, C.; Bender, A.; García-Arumí, E.; Melià, M.J.; Bové, J.; Laub, C.; Klopstock, T.; Elstner, M.; Mounsey, R.B.; Teismann, P.; et al. Accumulation of mitochondrial DNA deletions within dopaminergic neurons triggers neuroprotective mechanisms. Brain 2013, 136, 2369–2378. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Clark, J.; Zheng, K.; Kujoth, G.C.; Prolla, T.A.; Simon, D.K. Somatic mitochondrial DNA mutations do not increase neuronal vulnerability to MPTP in young POLG mutator mice. Neurotoxicol. Teratol. 2014, 46, 62–67. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, G.M.; Dayas, C.V.; Smith, D.W. Increased mitochondrial DNA deletions in substantia nigra dopamine neurons of the aged rat. Curr. Aging Sci. 2014, 7, 155–160. [Google Scholar] [CrossRef] [PubMed]
- Siddiqui, A.; Rivera-Sánchez, S.; Castro, M.E.R.; Acevedo-Torres, K.; Rane, A.; Torres-Ramos, C.A.; Nicholls, D.G.; Andersen, J.K.; Ayala-Torres, S. Mitochondrial DNA damage is associated with reduced mitochondrial bioenergetics in Huntington’s disease. Free Radic. Biol. Med. 2012, 53, 1478–1488. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.R.; Simpkins, J.W.; Roby, R.K. Mitochondrial DNA deletions in Alzheimer’s brains: A review. Alzheimers Dement. 2014, 10, 393–400. [Google Scholar] [CrossRef] [PubMed]
- Gerschütz, A.; Heinsen, H.; Grünblatt, E.; Wagner, A.K.; Bartl, J.; Meissner, C.; Fallgatter, A.J.; Al-Sarraj, S.; Troakes, C.; Ferrer, I.; et al. Neuron-specific mitochondrial DNA deletion levels in sporadic Alzheimer’s disease. Curr. Alzheimer Res. 2013, 10, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Smith, E.F.; Shaw, P.J.; De Vos, K.J. The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 2017. [Google Scholar] [CrossRef] [PubMed]
- Bekris, L.M.; Mata, I.F.; Zabetian, C.P. The genetics of Parkinson disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 228–242. [Google Scholar] [CrossRef] [PubMed]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Greene, J.C.; Whitworth, A.J.; Kuo, I.; Andrews, L.A.; Feany, M.B.; Pallanck, L.J. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. USA 2003, 100, 4078–4083. [Google Scholar] [CrossRef] [PubMed]
- Palacino, J.J.; Sagi, D.; Goldberg, M.S.; Krauss, S.; Motz, C.; Wacker, M.; Klose, J.; Shen, J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J. Biol. Chem. 2004, 279, 18614–18622. [Google Scholar] [CrossRef] [PubMed]
- Gautier, C.A.; Kitada, T.; Shen, J. Loss of PINK1 causes mitochondrial functional defects and increased sensitivity to oxidative stress. Proc. Natl. Acad. Sci. USA 2008, 105, 11364–11369. [Google Scholar] [CrossRef] [PubMed]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.; Willems, P.H.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [PubMed]
- Clark, I.E.; Dodson, M.W.; Jiang, C.; Cao, J.H.; Huh, J.R.; Seol, J.H.; Yoo, S.J.; Hay, B.A.; Guo, M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 2006, 441, 1162–1166. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Lee, S.B.; Lee, S.; Kim, Y.; Song, S.; Kim, S.; Bae, E.; Kim, J.; Shong, M.; Kim, J.M.; et al. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 2006, 441, 1157–1161. [Google Scholar] [CrossRef] [PubMed]
- Truban, D.; Hou, X.; Caulfield, T.R.; Fiesel, F.C.; Springer, W. PINK1, Parkin, and Mitochondrial Quality Control: What can we Learn about Parkinson’s Disease Pathobiology? J. Parkinsons Dis. 2017, 7, 13–29. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Shimoji, M.; Thomas, B.; Moore, D.J.; Yu, S.W.; Marupudi, N.I.; Torp, R.; Torgner, I.A.; Ottersen, O.P.; Dawson, T.M.; et al. Mitochondrial localization of the Parkinson’s disease related protein DJ-1: Implications for pathogenesis. Hum. Mol. Genet. 2005, 14, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Taira, T.; Saito, Y.; Niki, T.; Iguchi-Ariga, S.M.; Takahashi, K.; Ariga, H. DJ-1 has a role in antioxidative stress to prevent cell death. EMBO Rep. 2004, 5, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Zhou, W.; Freed, C.R. DJ-1 up-regulates glutathione synthesis during oxidative stress and inhibits A53T alpha-synuclein toxicity. J. Biol. Chem. 2005, 280, 43150–43158. [Google Scholar] [CrossRef] [PubMed]
- Yokota, T.; Sugawara, K.; Ito, K.; Takahashi, R.; Ariga, H.; Mizusawa, H. Down regulation of DJ-1 enhances cell death by oxidative stress, ER stress, and proteasome inhibition. Biochem. Biophys. Res. Commun. 2003, 312, 1342–1348. [Google Scholar] [CrossRef] [PubMed]
- Krebiehl, G.; Ruckerbauer, S.; Burbulla, L.F.; Kieper, N.; Maurer, B.; Waak, J.; Wolburg, H.; Gizatullina, Z.; Gellerich, F.N.; Woitalla, D.; et al. Reduced basal autophagy and impaired mitochondrial dynamics due to loss of Parkinson’s disease-associated protein DJ-1. PLoS ONE 2010, 5, e9367. [Google Scholar] [CrossRef] [PubMed]
- Ankarcrona, M.; Hultenby, K. Presenilin-1 is located in rat mitochondria. Biochem. Biophys. Res. Commun. 2002, 295, 766–770. [Google Scholar] [CrossRef]
- Area-Gomez, E.; de Groof, A.J.; Boldogh, I.; Bird, T.D.; Gibson, G.E.; Koehler, C.M.; Yu, W.H.; Duff, K.E.; Yaffe, M.P.; Pon, L.A.; et al. Presenilins are enriched in endoplasmic reticulum membranes associated with mitochondria. Am. J. Pathol. 2009, 175, 1810–1816. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, T.L. Mitochondrial trafficking in neurons. Cold Spring Harb. Perspect. Biol. 2013, 5, a011304. [Google Scholar] [CrossRef] [PubMed]
- Burté, F.; Carelli, V.; Chinnery, P.F.; Yu-Wai-Man, P. Disturbed mitochondrial dynamics and neurodegenerative disorders. Nat. Rev. Neurol. 2015, 11, 11–24. [Google Scholar] [CrossRef] [PubMed]
- Dupuis, L. Mitochondrial quality control in neurodegenerative diseases. Biochimie 2014, 100, 177–183. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.J.; Hoek, S.; Fon, E.A.; Wade-Martins, R. Mitochondrial dysfunction and mitophagy in Parkinson’s: From familial to sporadic disease. Trends Biochem. Sci. 2015, 40, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Franco-Iborra, S.; Vila, M.; Perier, C. Mitochondrial Quality Control in Neurodegenerative Diseases: Focus on Parkinson’s Disease and Huntington’s Disease. Front. Neurosci. 2018, 12, 342. [Google Scholar] [CrossRef] [PubMed]
- Van Laar, V.S.; Berman, S.B. The interplay of neuronal mitochondrial dynamics and bioenergetics: Implications for Parkinson’s disease. Neurobiol. Dis. 2013, 51, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Yan, M.H.; Wang, X.; Zhu, X. Mitochondrial defects and oxidative stress in Alzheimer disease and Parkinson disease. Free Radic. Biol. Med. 2013, 62, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Carmo, C.; Naia, L.; Lopes, C.; Rego, A.C. Mitochondrial Dysfunction in Huntington’s Disease. Adv. Exp. Med. Biol. 2018, 1049, 59–83. [Google Scholar] [CrossRef] [PubMed]
- Shirendeb, U.; Reddy, A.P.; Manczak, M.; Calkins, M.J.; Mao, P.; Tagle, D.A.; Reddy, P.H. Abnormal mitochondrial dynamics, mitochondrial loss and mutant huntingtin oligomers in Huntington’s disease: Implications for selective neuronal damage. Hum. Mol. Genet. 2011, 20, 1438–1455. [Google Scholar] [CrossRef] [PubMed]
- De Vos, K.J.; Chapman, A.L.; Tennant, M.E.; Manser, C.; Tudor, E.L.; Lau, K.F.; Brownlees, J.; Ackerley, S.; Shaw, P.J.; McLoughlin, D.M.; et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007, 16, 2720–2728. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.; Wang, L.; Liu, J.; Xie, F.; Su, B.; Wang, X. Abnormalities of Mitochondrial Dynamics in Neurodegenerative Diseases. Antioxidants 2017, 6, 25. [Google Scholar] [CrossRef] [PubMed]
- Carrì, M.T.; Cozzolino, M. SOD1 and mitochondria in ALS: A dangerous liaison. J. Bioenerg. Biomembr. 2011, 43, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Wood-Kaczmar, A.; Gandhi, S.; Yao, Z.; Abramov, A.Y.; Abramov, A.S.; Miljan, E.A.; Keen, G.; Stanyer, L.; Hargreaves, I.; Klupsch, K.; et al. PINK1 is necessary for long term survival and mitochondrial function in human dopaminergic neurons. PLoS ONE 2008, 3, e2455. [Google Scholar] [CrossRef]
- Luo, D.J.; Feng, Q.; Wang, Z.H.; Sun, D.S.; Wang, Q.; Wang, J.Z.; Liu, G.P. Knockdown of phosphotyrosyl phosphatase activator induces apoptosis via mitochondrial pathway and the attenuation by simultaneous tau hyperphosphorylation. J. Neurochem. 2014, 130, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.J.; Cao, Q.; Zhang, Y.; Su, X.D. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [PubMed]
- Takeuchi, H.; Kobayashi, Y.; Ishigaki, S.; Doyu, M.; Sobue, G. Mitochondrial localization of mutant superoxide dismutase 1 triggers caspase-dependent cell death in a cellular model of familial amyotrophic lateral sclerosis. J. Biol. Chem. 2002, 277, 50966–50972. [Google Scholar] [CrossRef] [PubMed]
- Calì, T.; Ottolini, D.; Brini, M. Mitochondrial Ca2+ and neurodegeneration. Cell Calcium 2012, 52, 73–85. [Google Scholar] [CrossRef] [PubMed]
- Pelizzoni, I.; Macco, R.; Zacchetti, D.; Grohovaz, F.; Codazzi, F. Iron and calcium in the central nervous system: A close relationship in health and sickness. Biochem. Soc. Trans. 2008, 36, 1309–1312. [Google Scholar] [CrossRef] [PubMed]
- Fernyhough, P.; Roy Chowdhury, S.K.; Schmidt, R.E. Mitochondrial stress and the pathogenesis of diabetic neuropathy. Expert Rev. Endocrinol. Metab. 2010, 5, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Jones, A.W.; Yao, Z.; Vicencio, J.M.; Karkucinska-Wieckowska, A.; Szabadkai, G. PGC-1 family coactivators and cell fate: Roles in cancer, neurodegeneration, cardiovascular disease and retrograde mitochondria-nucleus signalling. Mitochondrion 2012, 12, 86–99. [Google Scholar] [CrossRef] [PubMed]
- Weydt, P.; Pineda, V.V.; Torrence, A.E.; Libby, R.T.; Satterfield, T.F.; Lazarowski, E.R.; Gilbert, M.L.; Morton, G.J.; Bammler, T.K.; Strand, A.D.; et al. Thermoregulatory and metabolic defects in Huntington’s disease transgenic mice implicate PGC-1alpha in Huntington’s disease neurodegeneration. Cell Metab. 2006, 4, 349–362. [Google Scholar] [CrossRef] [PubMed]
- Cui, L.; Jeong, H.; Borovecki, F.; Parkhurst, C.N.; Tanese, N.; Krainc, D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell 2006, 127, 59–69. [Google Scholar] [CrossRef] [PubMed]
- Qin, W.; Haroutunian, V.; Katsel, P.; Cardozo, C.P.; Ho, L.; Buxbaum, J.D.; Pasinetti, G.M. PGC-1alpha expression decreases in the Alzheimer disease brain as a function of dementia. Arch. Neurol. 2009, 66, 352–361. [Google Scholar] [CrossRef] [PubMed]
- St-Pierre, J.; Drori, S.; Uldry, M.; Silvaggi, J.M.; Rhee, J.; Jäger, S.; Handschin, C.; Zheng, K.; Lin, J.; Yang, W.; et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell 2006, 127, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Zheng, B.; Liao, Z.; Locascio, J.J.; Lesniak, K.A.; Roderick, S.S.; Watt, M.L.; Eklund, A.C.; Zhang-James, Y.; Kim, P.D.; Hauser, M.A.; et al. PGC-1α, a potential therapeutic target for early intervention in Parkinson’s disease. Sci. Transl. Med. 2010, 2, 52ra73. [Google Scholar] [CrossRef] [PubMed]
- Chu, C.T. Mechanisms of selective autophagy and mitophagy: Implications for neurodegenerative diseases. Neurobiol. Dis. 2018. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M. Neuronal Mitophagy in Neurodegenerative Diseases. Front. Mol. Neurosci. 2017, 10, 64. [Google Scholar] [CrossRef] [PubMed]
- Rodolfo, C.; Campello, S.; Cecconi, F. Mitophagy in neurodegenerative diseases. Neurochem. Int. 2018, 117, 156–166. [Google Scholar] [CrossRef] [PubMed]
- Kondapalli, C.; Kazlauskaite, A.; Zhang, N.; Woodroof, H.I.; Campbell, D.G.; Gourlay, R.; Burchell, L.; Walden, H.; Macartney, T.J.; Deak, M.; et al. PINK1 is activated by mitochondrial membrane potential depolarization and stimulates Parkin E3 ligase activity by phosphorylating Serine 65. Open Biol. 2012, 2, 120080. [Google Scholar] [CrossRef] [PubMed]
- Moon, H.E.; Paek, S.H. Mitochondrial Dysfunction in Parkinson’s Disease. Exp. Neurobiol. 2015, 24, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Vilariño-Güell, C.; Wider, C.; Ross, O.A.; Dachsel, J.C.; Kachergus, J.M.; Lincoln, S.J.; Soto-Ortolaza, A.I.; Cobb, S.A.; Wilhoite, G.J.; Bacon, J.A.; et al. VPS35 mutations in Parkinson disease. Am. J. Hum. Genet. 2011, 89, 162–167. [Google Scholar] [CrossRef] [PubMed]
- Zimprich, A.; Benet-Pagès, A.; Struhal, W.; Graf, E.; Eck, S.H.; Offman, M.N.; Haubenberger, D.; Spielberger, S.; Schulte, E.C.; Lichtner, P.; et al. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am. J. Hum. Genet. 2011, 89, 168–175. [Google Scholar] [CrossRef] [PubMed]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef] [PubMed]
- Shlevkov, E.; Schwarz, T.L. Have you seen? For parkin, it’s not all or nothing. EMBO J. 2014, 33, 277–279. [Google Scholar] [CrossRef] [PubMed]
- Friedman, J.R.; Lackner, L.L.; West, M.; DiBenedetto, J.R.; Nunnari, J.; Voeltz, G.K. ER tubules mark sites of mitochondrial division. Science 2011, 334, 358–362. [Google Scholar] [CrossRef] [PubMed]
- de Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Zhu, X. Endoplasmic reticulum-mitochondria tethering in neurodegenerative diseases. Transl. Neurodegener. 2017, 6, 21. [Google Scholar] [CrossRef] [PubMed]
- Camandola, S.; Mattson, M.P. Brain metabolism in health, aging, and neurodegeneration. EMBO J. 2017, 36, 1474–1492. [Google Scholar] [CrossRef] [PubMed]
- Requejo-Aguilar, R.; Lopez-Fabuel, I.; Fernandez, E.; Martins, L.M.; Almeida, A.; Bolaños, J.P. PINK1 deficiency sustains cell proliferation by reprogramming glucose metabolism through HIF1. Nat. Commun. 2014, 5, 4514. [Google Scholar] [CrossRef] [PubMed]
- Herrero-Mendez, A.; Almeida, A.; Fernández, E.; Maestre, C.; Moncada, S.; Bolaños, J.P. The bioenergetic and antioxidant status of neurons is controlled by continuous degradation of a key glycolytic enzyme by APC/C-Cdh1. Nat. Cell Biol. 2009, 11, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Mercurio, S.; Tolosano, E. Heme and erythropoieis: More than a structural role. Haematologica 2014, 99, 973–983. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Khalimonchuk, O.; Smith, P.M.; Winge, D.R. Structure, function, and assembly of heme centers in mitochondrial respiratory complexes. Biochim. Biophys. Acta 2012, 1823, 1604–1616. [Google Scholar] [CrossRef] [PubMed]
- Sabová, L.; Zeman, I.; Supek, F.; Kolarov, J. Transcriptional control of AAC3 gene encoding mitochondrial ADP/ATP translocator in Saccharomyces cerevisiae by oxygen, heme and ROX1 factor. Eur. J. Biochem. 1993, 213, 547–553. [Google Scholar] [CrossRef] [PubMed]
- Giraud, S.; Bonod-Bidaud, C.; Wesolowski-Louvel, M.; Stepien, G. Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J. Mol. Biol. 1998, 281, 409–418. [Google Scholar] [CrossRef] [PubMed]
- Azuma, M.; Kabe, Y.; Kuramori, C.; Kondo, M.; Yamaguchi, Y.; Handa, H. Adenine nucleotide translocator transports haem precursors into mitochondria. PLoS ONE 2008, 3, e3070. [Google Scholar] [CrossRef] [PubMed]
- Frezza, C.; Zheng, L.; Folger, O.; Rajagopalan, K.N.; MacKenzie, E.D.; Jerby, L.; Micaroni, M.; Chaneton, B.; Adam, J.; Hedley, A.; et al. Haem oxygenase is synthetically lethal with the tumour suppressor fumarate hydratase. Nature 2011, 477, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, Y.; Wang, Y.; Lian, S.; Lynch, J.; Nagai, S.; Fanshawe, B.; Kandilci, A.; Janke, L.J.; Neale, G.; Fan, Y.; et al. Upregulated heme biosynthesis, an exploitable vulnerability in MYCN-driven leukemogenesis. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [PubMed]
- Horowitz, M.P.; Greenamyre, J.T. Mitochondrial iron metabolism and its role in neurodegeneration. J. Alzheimers Dis. 2010, 20 (Suppl. 2), S551–S568. [Google Scholar] [CrossRef] [PubMed]
- Walter, P.B.; Knutson, M.D.; Paler-Martinez, A.; Lee, S.; Xu, Y.; Viteri, F.E.; Ames, B.N. Iron deficiency and iron excess damage mitochondria and mitochondrial DNA in rats. Proc. Natl. Acad. Sci. USA 2002, 99, 2264–2269. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Campian, J.L.; Qian, M.; Sun, X.F.; Eaton, J.W. Mitochondrial DNA damage in iron overload. J. Biol. Chem. 2009, 284, 4767–4775. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Newberry, J.; Erlitzki, R.; Schultz, C.S.; Ames, B.N. Biotin deficiency inhibits heme synthesis and impairs mitochondria in human lung fibroblasts. J. Nutr. 2007, 137, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Doré, S.; Ferris, C.D.; Tomita, T.; Sawa, A.; Wolosker, H.; Borchelt, D.R.; Iwatsubo, T.; Kim, S.H.; Thinakaran, G.; et al. Amyloid precursor proteins inhibit heme oxygenase activity and augment neurotoxicity in Alzheimer’s disease. Neuron 2000, 28, 461–473. [Google Scholar] [CrossRef]
- Sheng, X.J.; Tu, H.J.; Chien, W.L.; Kang, K.H.; Lu, D.H.; Liou, H.H.; Lee, M.J.; Fu, W.M. Antagonism of proteasome inhibitor-induced heme oxygenase-1 expression by PINK1 mutation. PLoS ONE 2017, 12, e0183076. [Google Scholar] [CrossRef] [PubMed]
- Clements, C.M.; McNally, R.S.; Conti, B.J.; Mak, T.W.; Ting, J.P. DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA 2006, 103, 15091–15096. [Google Scholar] [CrossRef] [PubMed]
- Alam, J.; Stewart, D.; Touchard, C.; Boinapally, S.; Choi, A.M.; Cook, J.L. Nrf2, a Cap’n’Collar transcription factor, regulates induction of the heme oxygenase-1 gene. J. Biol. Chem. 1999, 274, 26071–26078. [Google Scholar] [CrossRef] [PubMed]
- Hull, T.D.; Boddu, R.; Guo, L.; Tisher, C.C.; Traylor, A.M.; Patel, B.; Joseph, R.; Prabhu, S.D.; Suliman, H.B.; Piantadosi, C.A.; et al. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 2016, 1, e85817. [Google Scholar] [CrossRef] [PubMed]
- Atamna, H.; Walter, P.B.; Ames, B.N. The role of heme and iron-sulfur clusters in mitochondrial biogenesis, maintenance, and decay with age. Arch. Biochem. Biophys. 2002, 397, 345–353. [Google Scholar] [CrossRef] [PubMed]
- Suliman, H.B.; Keenan, J.E.; Piantadosi, C.A. Mitochondrial quality-control dysregulation in conditional HO-1. JCI Insight 2017, 2, e89676. [Google Scholar] [CrossRef] [PubMed]
- Higdon, A.N.; Benavides, G.A.; Chacko, B.K.; Ouyang, X.; Johnson, M.S.; Landar, A.; Zhang, J.; Darley-Usmar, V.M. Hemin causes mitochondrial dysfunction in endothelial cells through promoting lipid peroxidation: The protective role of autophagy. Am. J. Physiol. Heart Circ. Physiol. 2012, 302, H1394–H1409. [Google Scholar] [CrossRef] [PubMed]
- Chiabrando, D.; Marro, S.; Mercurio, S.; Giorgi, C.; Petrillo, S.; Vinchi, F.; Fiorito, V.; Fagoonee, S.; Camporeale, A.; Turco, E.; et al. The mitochondrial heme exporter FLVCR1b mediates erythroid differentiation. J. Clin. Investig. 2012, 122, 4569–4579. [Google Scholar] [CrossRef] [PubMed]
- Homedan, C.; Laafi, J.; Schmitt, C.; Gueguen, N.; Lefebvre, T.; Karim, Z.; Desquiret-Dumas, V.; Wetterwald, C.; Deybach, J.C.; Gouya, L.; et al. Acute intermittent porphyria causes hepatic mitochondrial energetic failure in a mouse model. Int. J. Biochem. Cell Biol. 2014, 51, 93–101. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gago, M.; Alonso, A.; Pintos-Martínez, E.; Beiras-Iglesias, A.; Campos, Y.; Arenas, J.; Novo-Rodríguez, M.I.; Eirís-Puñal, J. Congenital hydranencephalic-hydrocephalic syndrome associated with mitochondrial dysfunction. J. Child Neurol. 1999, 14, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Castro-Gago, M.; Pintos-Martínez, E.; Forteza-Vila, J.; Iglesias-Diz, M.; Ucieda-Somoza, R.; Silva-Villar, I.; Codesido-López, J.; Viso-Lorenzo, A.; Campos, Y.; Arenas, J.; et al. Congenital hydranencephalic-hydrocephalic syndrome with proliferative vasculopathy: A possible relation with mitochondrial dysfunction. J. Child Neurol. 2001, 16, 858–862. [Google Scholar] [CrossRef] [PubMed]
- Kiaei, M. New hopes and challenges for treatment of neurodegenerative disorders: Great opportunities for young neuroscientists. Basic Clin. Neurosci. 2013, 4, 3–4. [Google Scholar] [PubMed]
- Andreux, P.A.; Houtkooper, R.H.; Auwerx, J. Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 2013, 12, 465–483. [Google Scholar] [CrossRef] [PubMed]
- Timmers, S.; Konings, E.; Bilet, L.; Houtkooper, R.H.; van de Weijer, T.; Goossens, G.H.; Hoeks, J.; van der Krieken, S.; Ryu, D.; Kersten, S.; et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab. 2011, 14, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Douglas, P.M.; Dillin, A. Protein homeostasis and aging in neurodegeneration. J. Cell Biol. 2010, 190, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Lutz, A.K.; Exner, N.; Fett, M.E.; Schlehe, J.S.; Kloos, K.; Lämmermann, K.; Brunner, B.; Kurz-Drexler, A.; Vogel, F.; Reichert, A.S.; et al. Loss of parkin or PINK1 function increases Drp1-dependent mitochondrial fragmentation. J. Biol. Chem. 2009, 284, 22938–22951. [Google Scholar] [CrossRef] [PubMed]
- Rappold, P.M.; Cui, M.; Grima, J.C.; Fan, R.Z.; de Mesy-Bentley, K.L.; Chen, L.; Zhuang, X.; Bowers, W.J.; Tieu, K. Drp1 inhibition attenuates neurotoxicity and dopamine release deficits in vivo. Nat. Commun. 2014, 5, 5244. [Google Scholar] [CrossRef] [PubMed]
- Lackner, L.L.; Nunnari, J. Small molecule inhibitors of mitochondrial division: Tools that translate basic biological research into medicine. Chem. Biol. 2010, 17, 578–583. [Google Scholar] [CrossRef] [PubMed]
- Wang, D.; Wang, J.; Bonamy, G.M.; Meeusen, S.; Brusch, R.G.; Turk, C.; Yang, P.; Schultz, P.G. A small molecule promotes mitochondrial fusion in mammalian cells. Angew. Chem. Int. Ed. Engl. 2012, 51, 9302–9305. [Google Scholar] [CrossRef] [PubMed]
- Yue, W.; Chen, Z.; Liu, H.; Yan, C.; Chen, M.; Feng, D.; Wu, H.; Du, L.; Wang, Y.; Liu, J.; et al. A small natural molecule promotes mitochondrial fusion through inhibition of the deubiquitinase USP30. Cell Res. 2014, 24, 482–496. [Google Scholar] [CrossRef] [PubMed]
- Cassidy-Stone, A.; Chipuk, J.E.; Ingerman, E.; Song, C.; Yoo, C.; Kuwana, T.; Kurth, M.J.; Shaw, J.T.; Hinshaw, J.E.; Green, D.R.; et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev. Cell 2008, 14, 193–204. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; Tang, X.; Christian, W.V.; Yoon, Y.; Tieu, K. Perturbations in mitochondrial dynamics induced by human mutant PINK1 can be rescued by the mitochondrial division inhibitor mdivi-1. J. Biol. Chem. 2010, 285, 11740–11752. [Google Scholar] [CrossRef] [PubMed]
- Bordt, E.A.; Clerc, P.; Roelofs, B.A.; Saladino, A.J.; Tretter, L.; Adam-Vizi, V.; Cherok, E.; Khalil, A.; Yadava, N.; Ge, S.X.; et al. The Putative Drp1 Inhibitor mdivi-1 Is a Reversible Mitochondrial Complex I Inhibitor that Modulates Reactive Oxygen Species. Dev. Cell 2017, 40, 583–594. [Google Scholar] [CrossRef] [PubMed]
- Sherer, T.B.; Betarbet, R.; Testa, C.M.; Seo, B.B.; Richardson, J.R.; Kim, J.H.; Miller, G.W.; Yagi, T.; Matsuno-Yagi, A.; Greenamyre, J.T. Mechanism of toxicity in rotenone models of Parkinson’s disease. J. Neurosci. 2003, 23, 10756–10764. [Google Scholar] [CrossRef] [PubMed]
- Lezi, E.; Swerdlow, R.H. Mitochondria in neurodegeneration. Adv. Exp. Med. Biol. 2012, 942, 269–286. [Google Scholar] [CrossRef] [PubMed]
- Witte, M.E.; Mahad, D.J.; Lassmann, H.; van Horssen, J. Mitochondrial dysfunction contributes to neurodegeneration in multiple sclerosis. Trends Mol. Med. 2014, 20, 179–187. [Google Scholar] [CrossRef] [PubMed]
- Green, D.R.; Galluzzi, L.; Kroemer, G. Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 2011, 333, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Koentjoro, B.; Park, J.S.; Sue, C.M. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci. Rep. 2017, 7, 44373. [Google Scholar] [CrossRef] [PubMed]
- Chen, J. Heme oxygenase in neuroprotection: From mechanisms to therapeutic implications. Rev. Neurosci. 2014, 25, 269–280. [Google Scholar] [CrossRef] [PubMed]
- Song, W.; Su, H.; Song, S.; Paudel, H.K.; Schipper, H.M. Over-expression of heme oxygenase-1 promotes oxidative mitochondrial damage in rat astroglia. J. Cell Physiol. 2006, 206, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Zukor, H.; Song, W.; Liberman, A.; Mui, J.; Vali, H.; Fillebeen, C.; Pantopoulos, K.; Wu, T.D.; Guerquin-Kern, J.L.; Schipper, H.M. HO-1-mediated macroautophagy: A mechanism for unregulated iron deposition in aging and degenerating neural tissues. J. Neurochem. 2009, 109, 776–791. [Google Scholar] [CrossRef] [PubMed]
- Nitti, M.; Piras, S.; Brondolo, L.; Marinari, U.M.; Pronzato, M.A.; Furfaro, A.L. Heme Oxygenase 1 in the Nervous System: Does It Favor Neuronal Cell Survival or Induce Neurodegeneration? Int. J. Mol. Sci. 2018, 19, 2260. [Google Scholar] [CrossRef] [PubMed]
- Schipper, H.M.; Gupta, A.; Szarek, W.A. Suppression of glial HO-1 activity as a potential neurotherapeutic intervention in AD. Curr. Alzheimer Res. 2009, 6, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Gupta, A.; Lacoste, B.; Pistell, P.J.; Pistel, P.J.; Ingram, D.K.; Hamel, E.; Alaoui-Jamali, M.A.; Szarek, W.A.; Vlahakis, J.Z.; Jie, S.; et al. Neurotherapeutic effects of novel HO-1 inhibitors in vitro and in a transgenic mouse model of Alzheimer’s disease. J. Neurochem. 2014, 131, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Omori, C.; Motodate, R.; Shiraki, Y.; Chiba, K.; Sobu, Y.; Kimura, A.; Nakaya, T.; Kondo, H.; Kurumiya, S.; Tanaka, T.; et al. Facilitation of brain mitochondrial activity by 5-aminolevulinic acid in a mouse model of Alzheimer’s disease. Nutr. Neurosci. 2017, 20, 538–546. [Google Scholar] [CrossRef] [PubMed]
- Gomes, A.T.P.C.; Neves, M.G.P.M.; Cavaleiro, J.A.S. Cancer, Photodynamic Therapy and Porphyrin-Type Derivatives. An. Acad. Bras. Cienc. 2018, 90, 993–1026. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Disease | Gene | Inheritance | Clinical Features | OMIM |
|---|---|---|---|---|
| ALAD deficiency | 5-aminolevulinate dehydratase (ALAD) | autosomal recessive | Neuropathic Porphyria: acute neurovisceral attacks involving severe abdominal pain, peripheral neuropathies and psychiatric disturbances | 612740 |
| Acute intermittent porphyria (AIP) | Hydroxymethylbilane synthase (HMBS) | autosomal dominant | 176000 | |
| Hereditary coproporphyria (HCP) | Coproporphyrinogen oxidase (CPOX) | autosomal dominant | 121300 | |
| Variegate porphyria (VP) | Protoporphyrinogen oxidase (PPOX) | autosomal dominant | 176200 | |
| Friederich Ataxia (FRDA) | Frataxin (FXN) | autosomal recessive | Progressive gait and limb ataxia associated with cardiomyopathy and diabetes | 229300 |
| Posterior Column Ataxia and Retinitis Pigmentosa (PCARP) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Sensory ataxia and retinitis pigmentosa | 609033 |
| Non syndromic Retinitis pigmentosa (RP) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Retinitis pigmentosa | 268000 |
| Hereditary Sensory and Autonomic Neuropathy (HSAN) | Feline Leukemia Virus Subgroup C Receptor 1 (FLVCR1) | autosomal recessive | Loss of pain perception | 201300 |
| Fowler syndrome (PVHH) | Feline Leukemia Virus Subgroup C Receptor 2 (FLVCR2) | autosomal recessive | Proliferative glomerular vasculopathy in the central nervous system associated with severe hydrocephaly, ventriculomegaly, cortical thinning and hypoplastic cerebellum. | 225790 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fiorito, V.; Chiabrando, D.; Tolosano, E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals 2018, 11, 87. https://doi.org/10.3390/ph11030087
Fiorito V, Chiabrando D, Tolosano E. Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals. 2018; 11(3):87. https://doi.org/10.3390/ph11030087
Chicago/Turabian StyleFiorito, Veronica, Deborah Chiabrando, and Emanuela Tolosano. 2018. "Mitochondrial Targeting in Neurodegeneration: A Heme Perspective" Pharmaceuticals 11, no. 3: 87. https://doi.org/10.3390/ph11030087
APA StyleFiorito, V., Chiabrando, D., & Tolosano, E. (2018). Mitochondrial Targeting in Neurodegeneration: A Heme Perspective. Pharmaceuticals, 11(3), 87. https://doi.org/10.3390/ph11030087