In Silico Study, Synthesis, and Cytotoxic Activities of Porphyrin Derivatives

 ,
, 
Abstract
:1. Introduction
2. Results
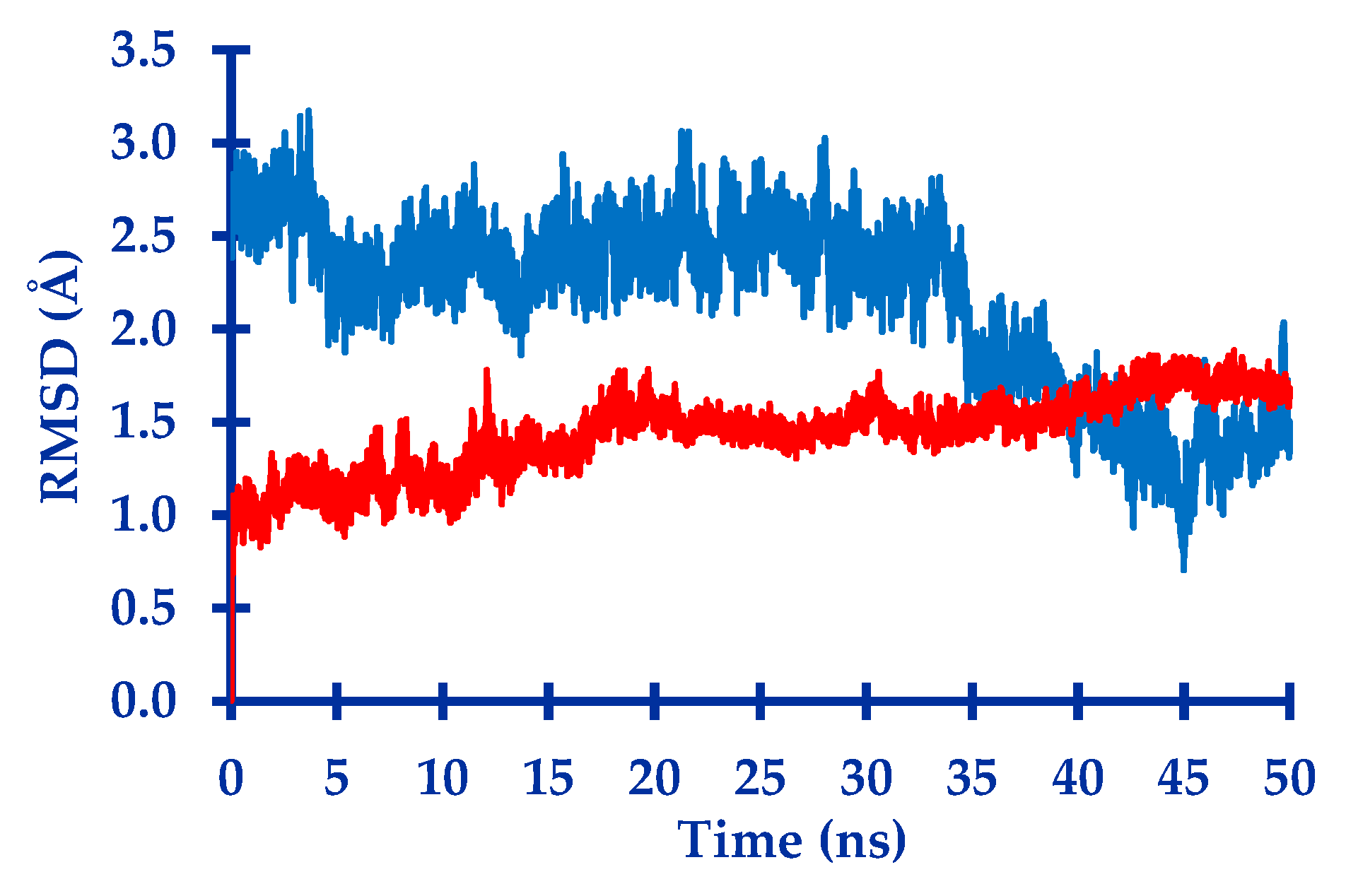
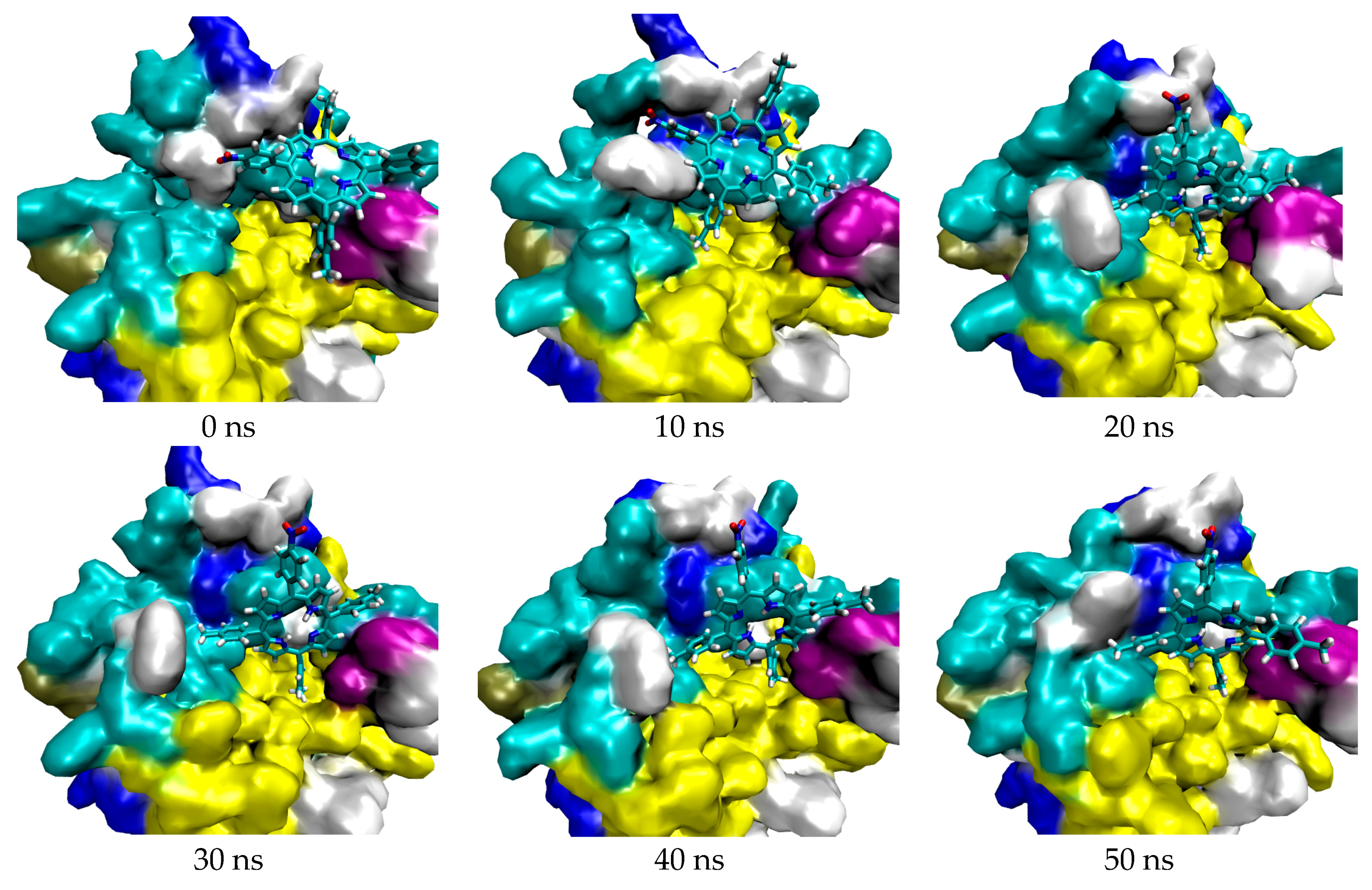
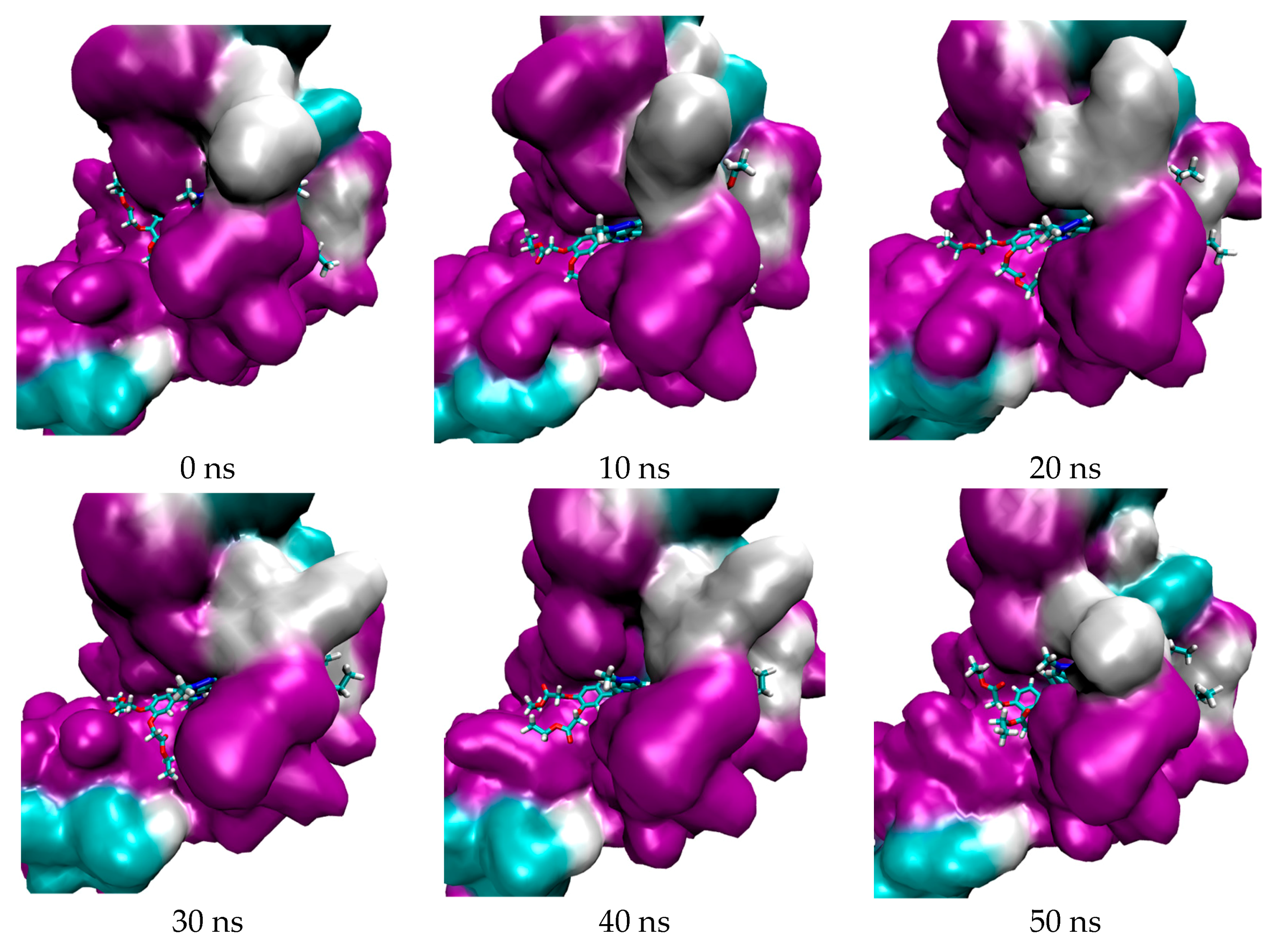
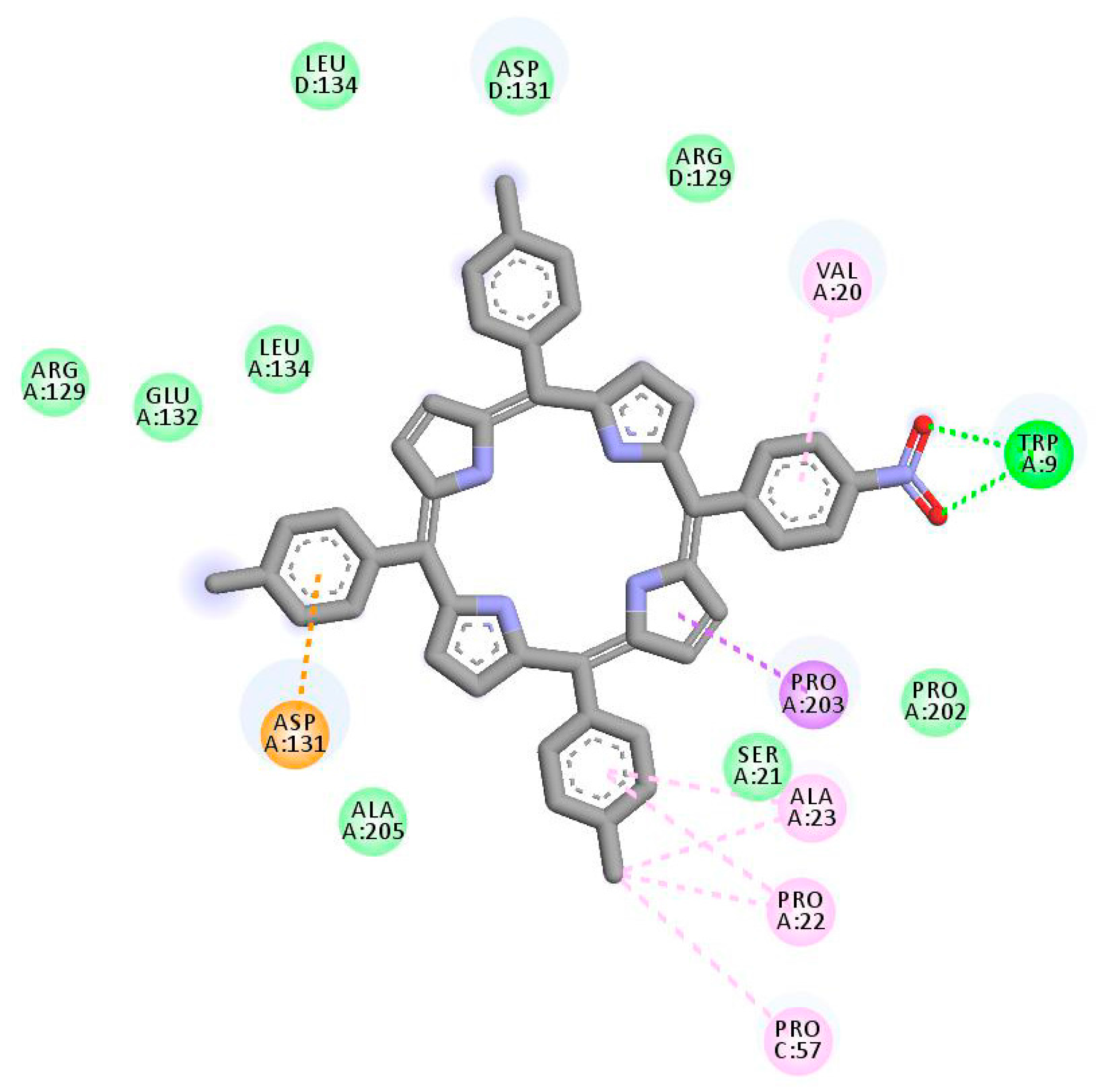
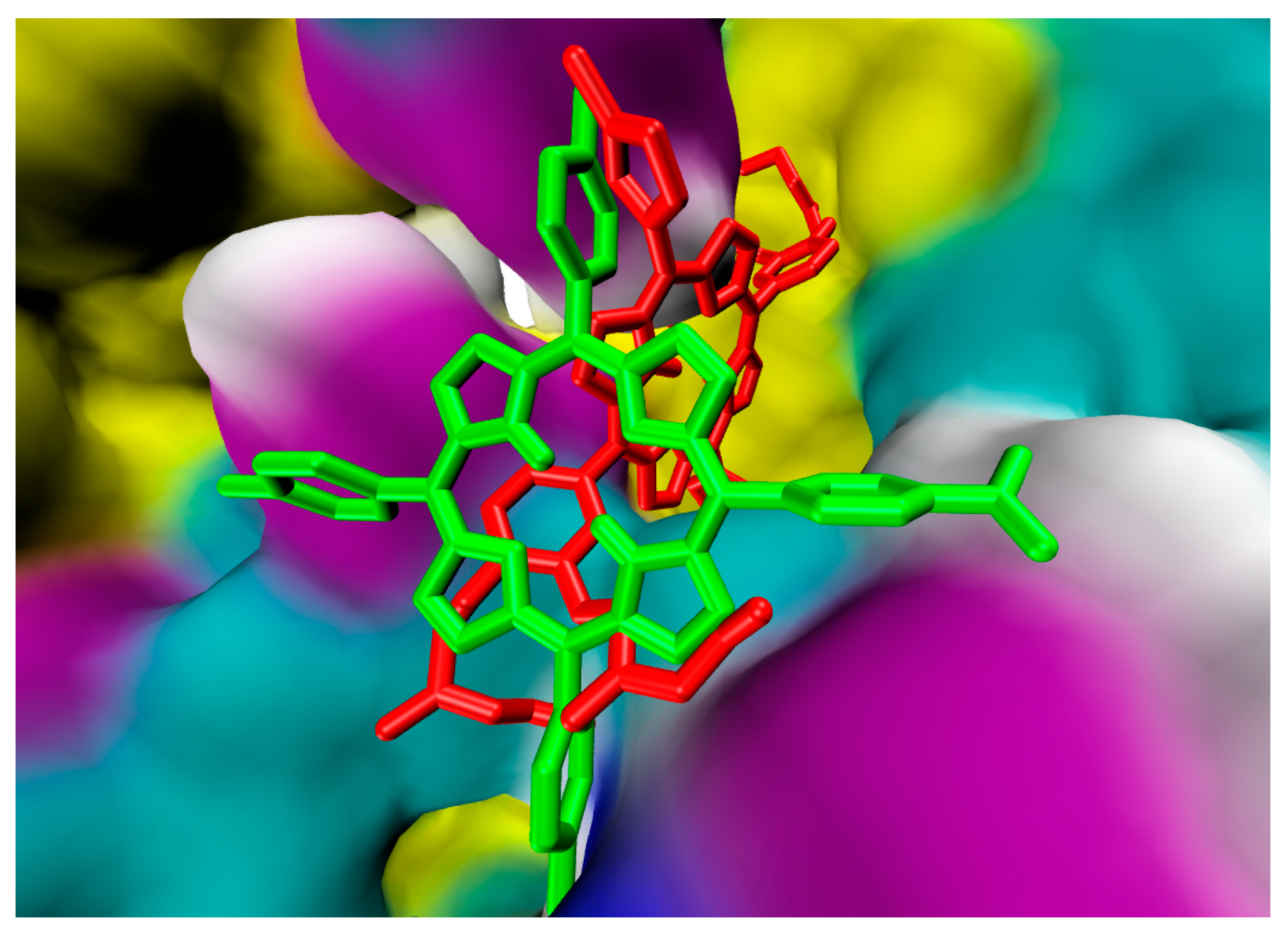
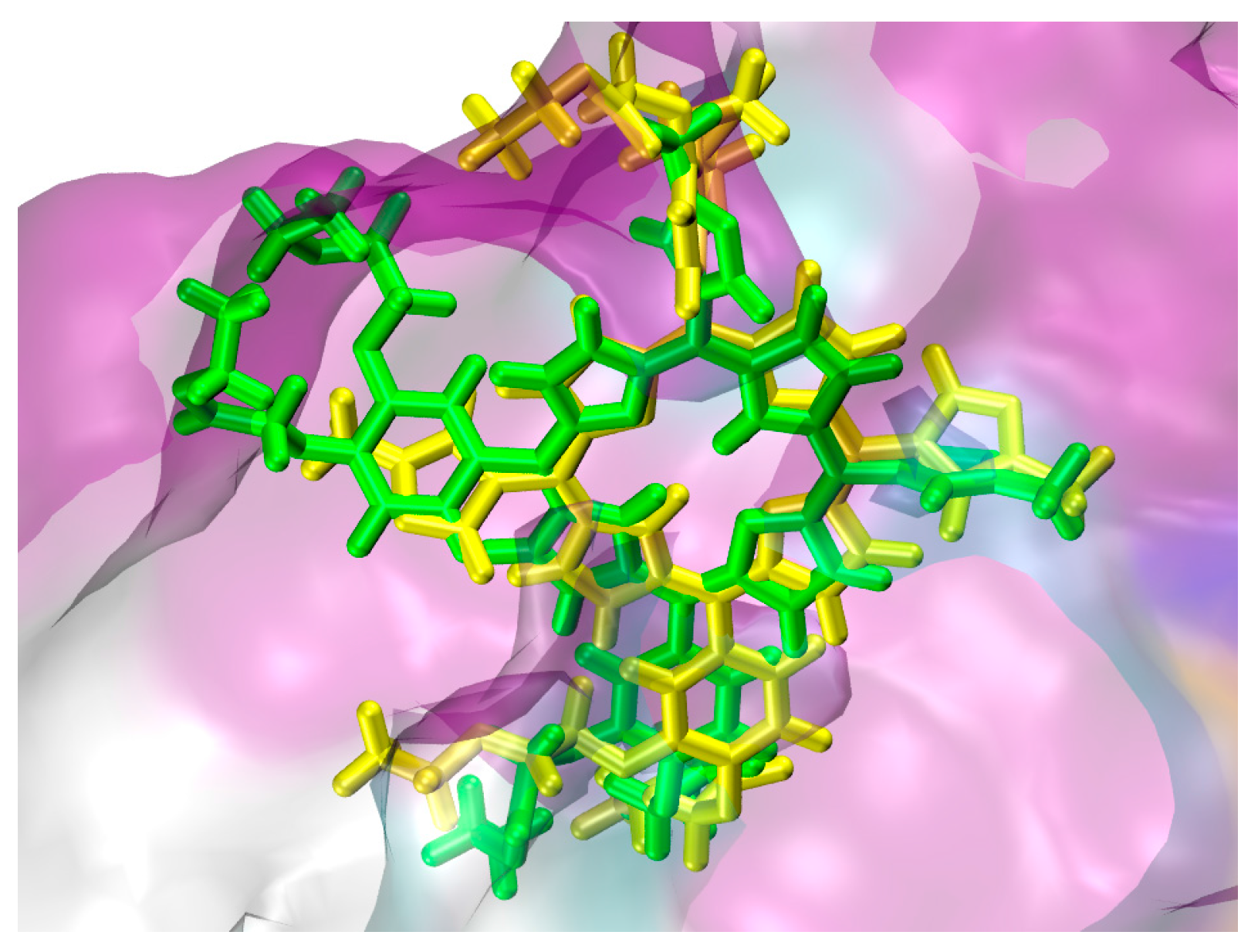
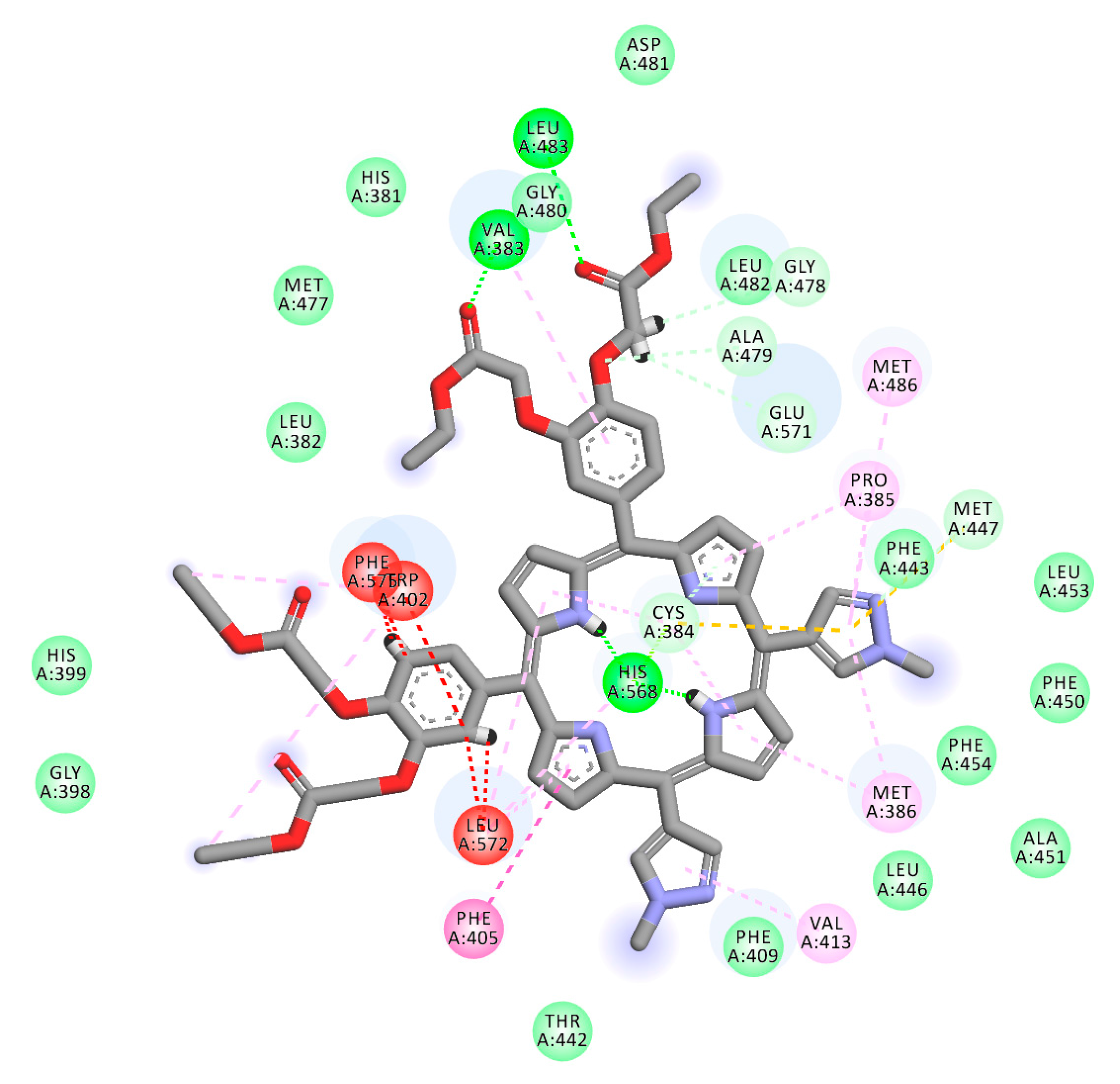
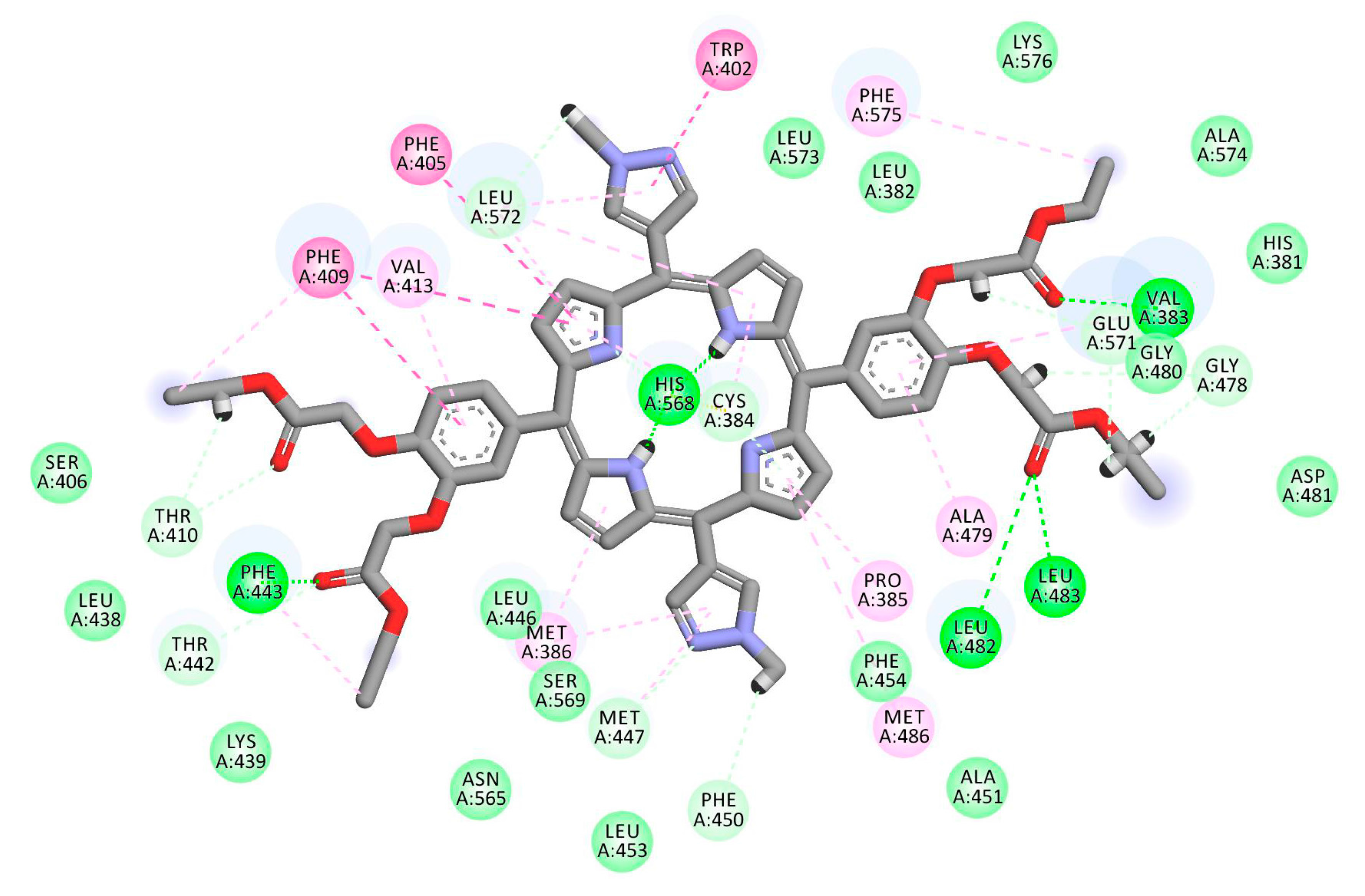
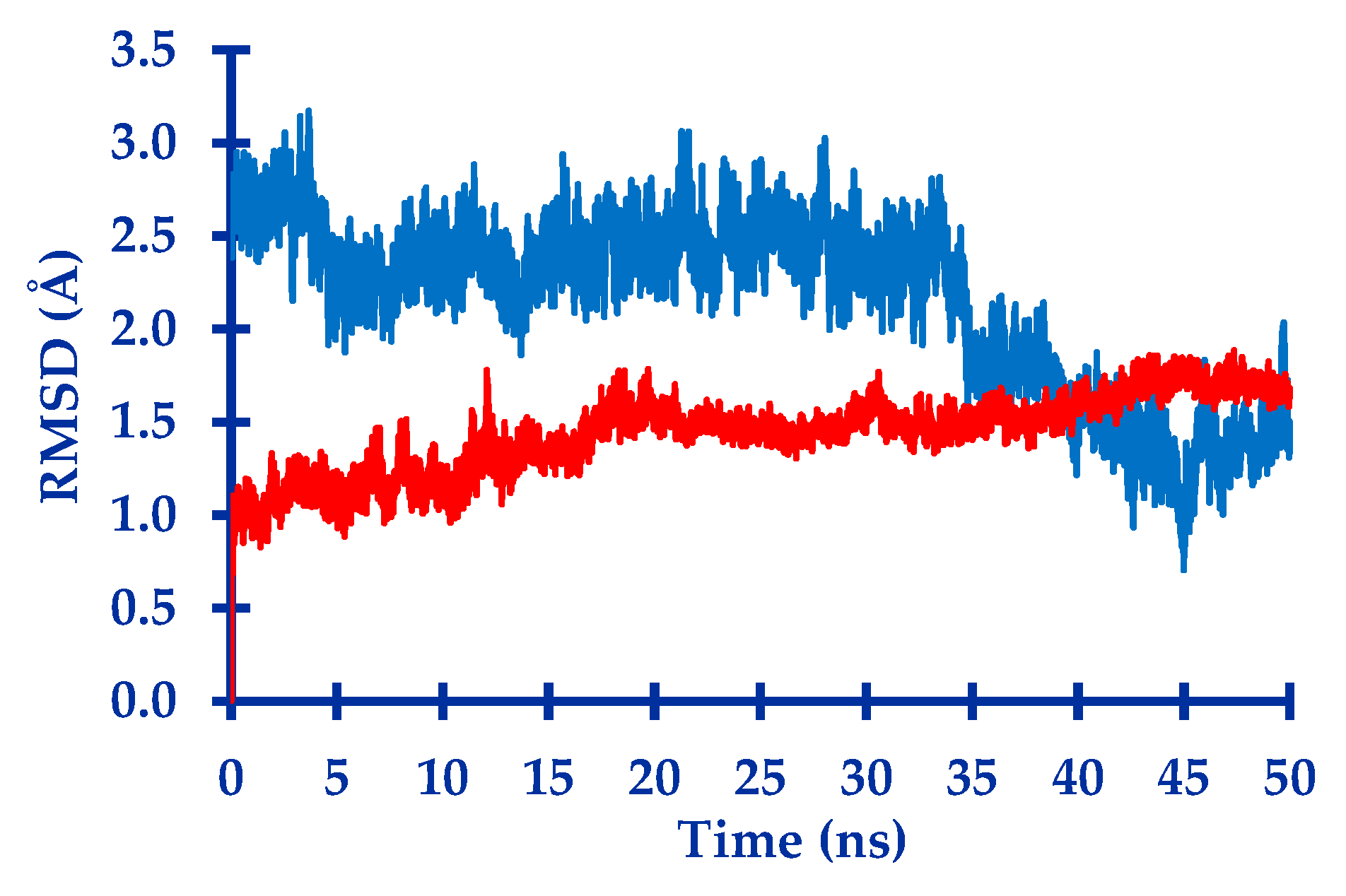
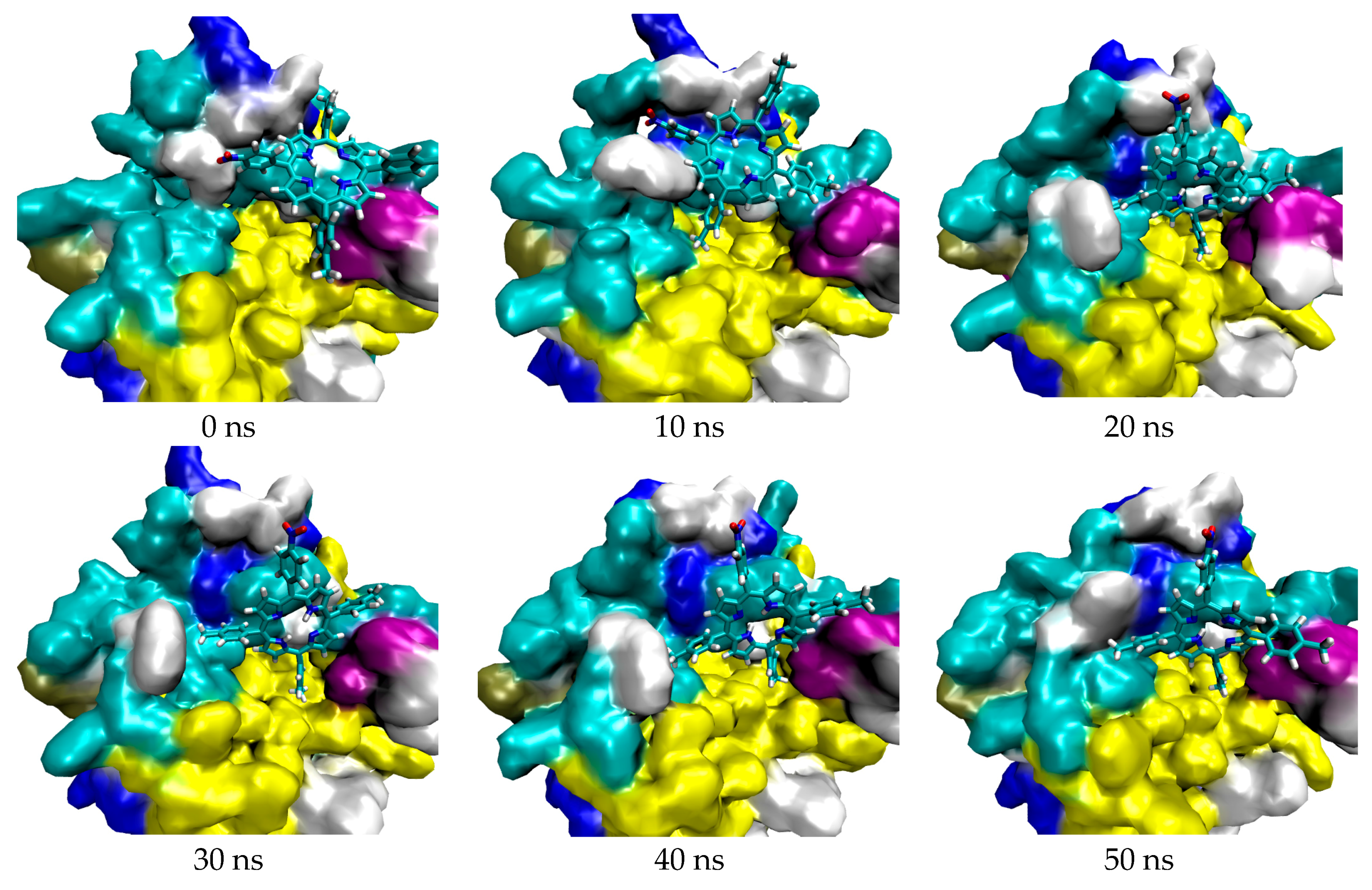
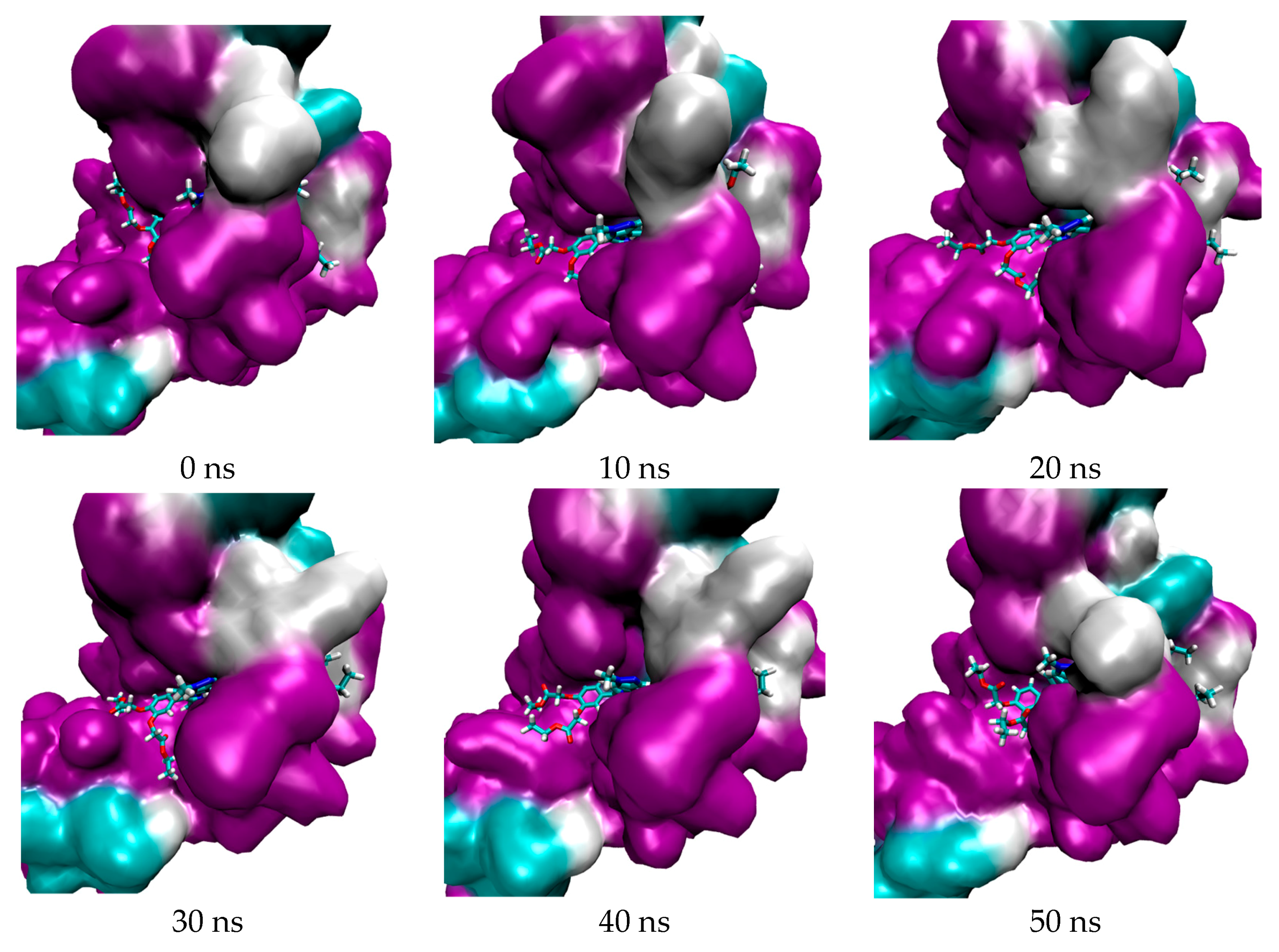
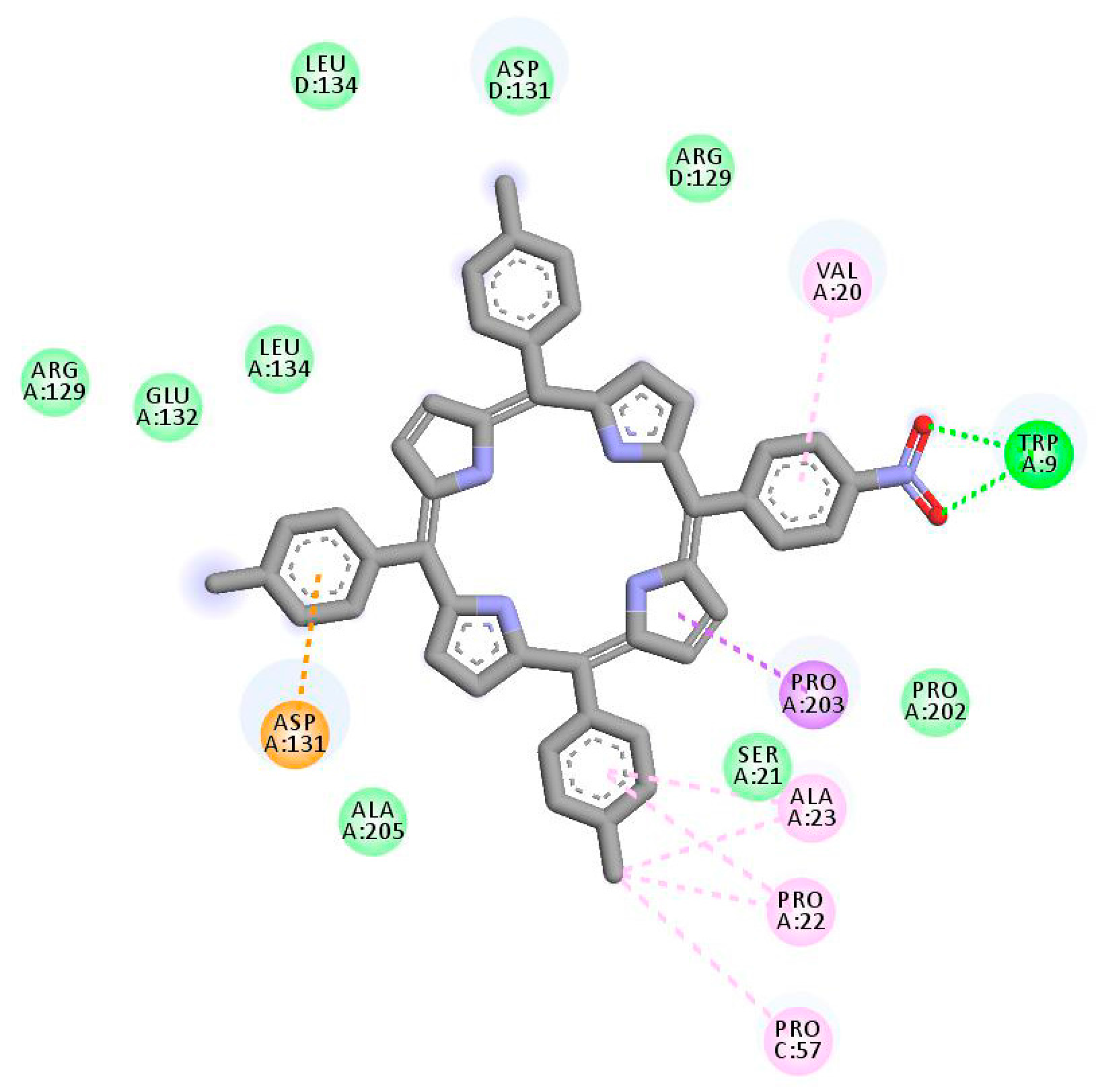
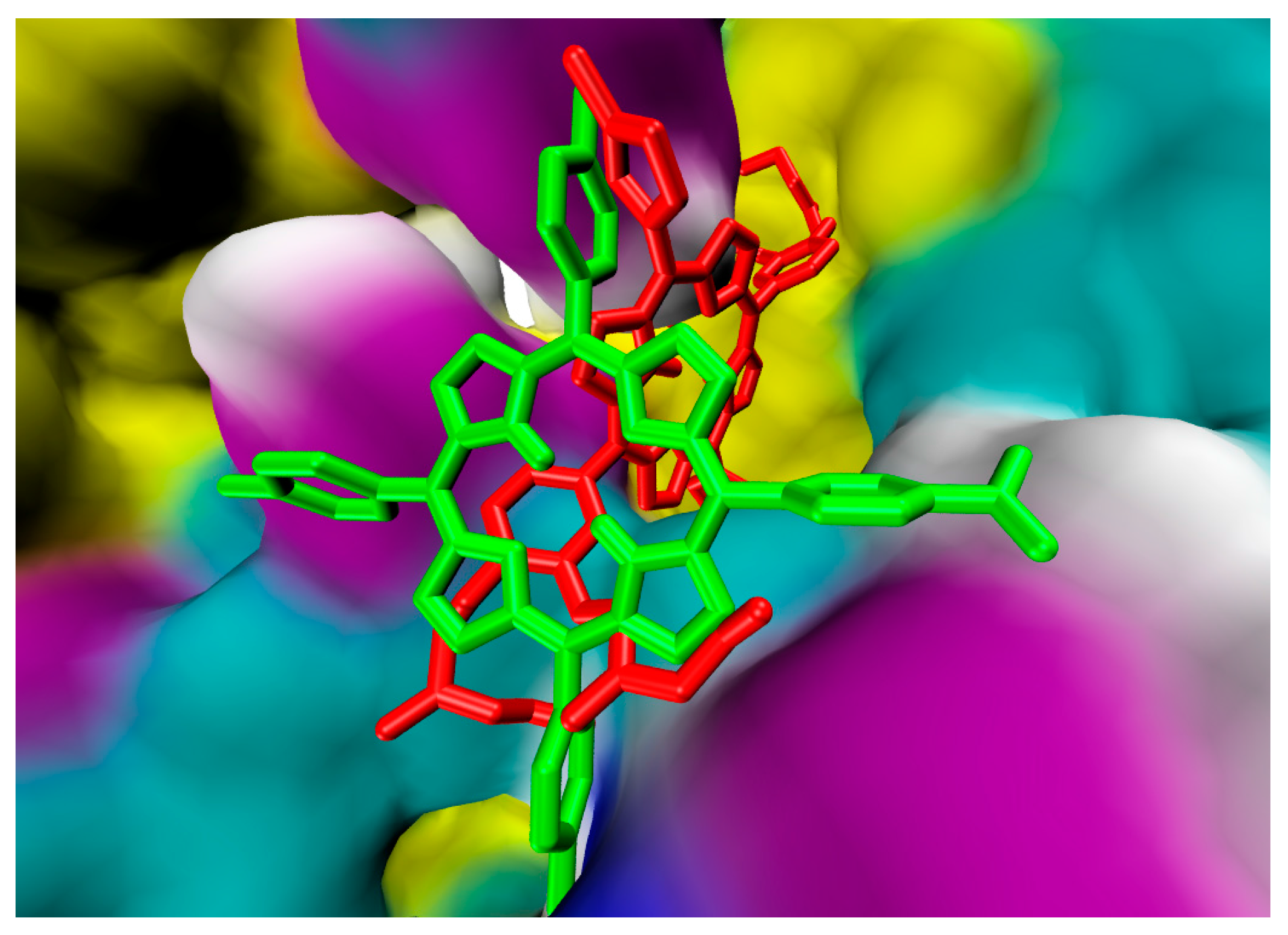
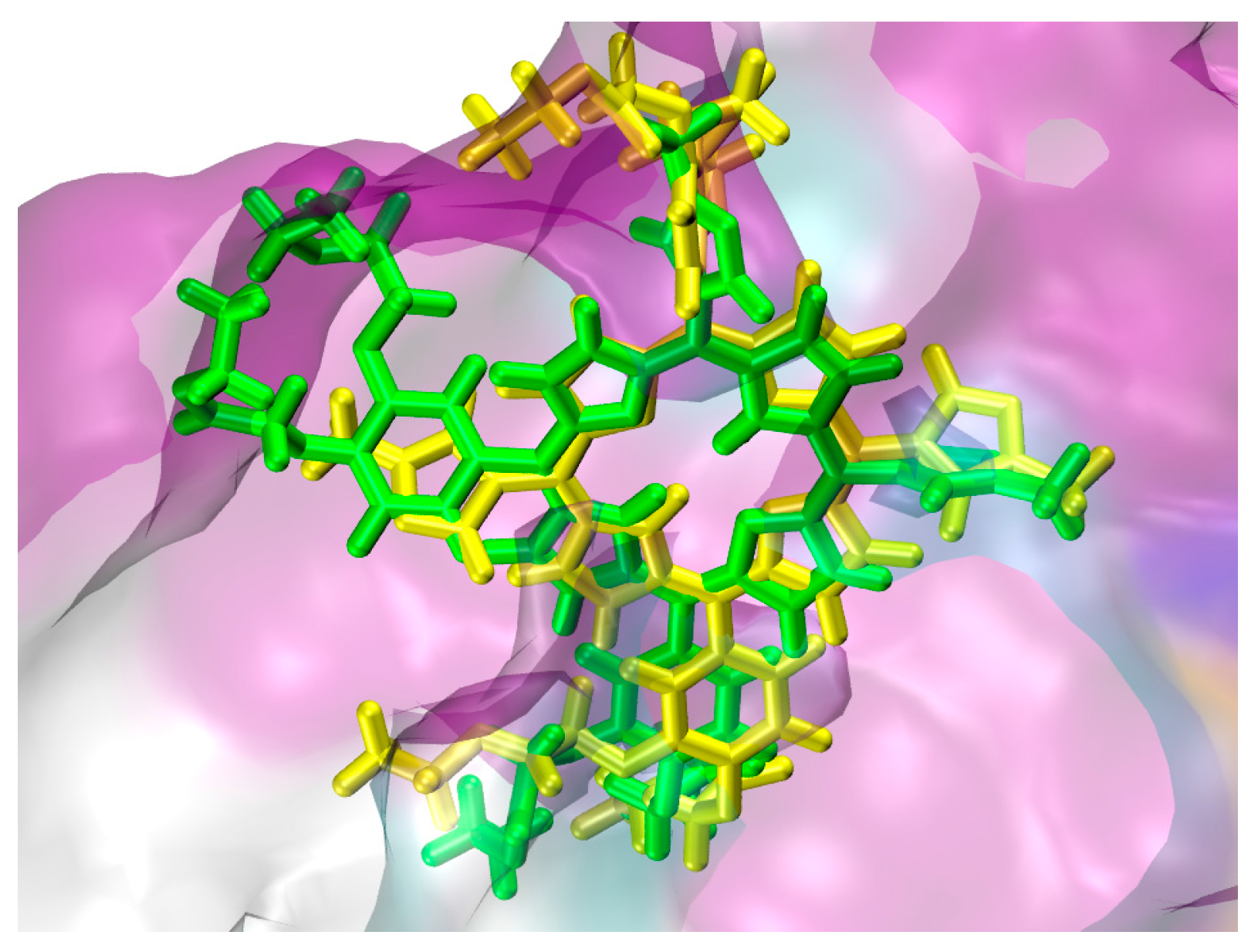
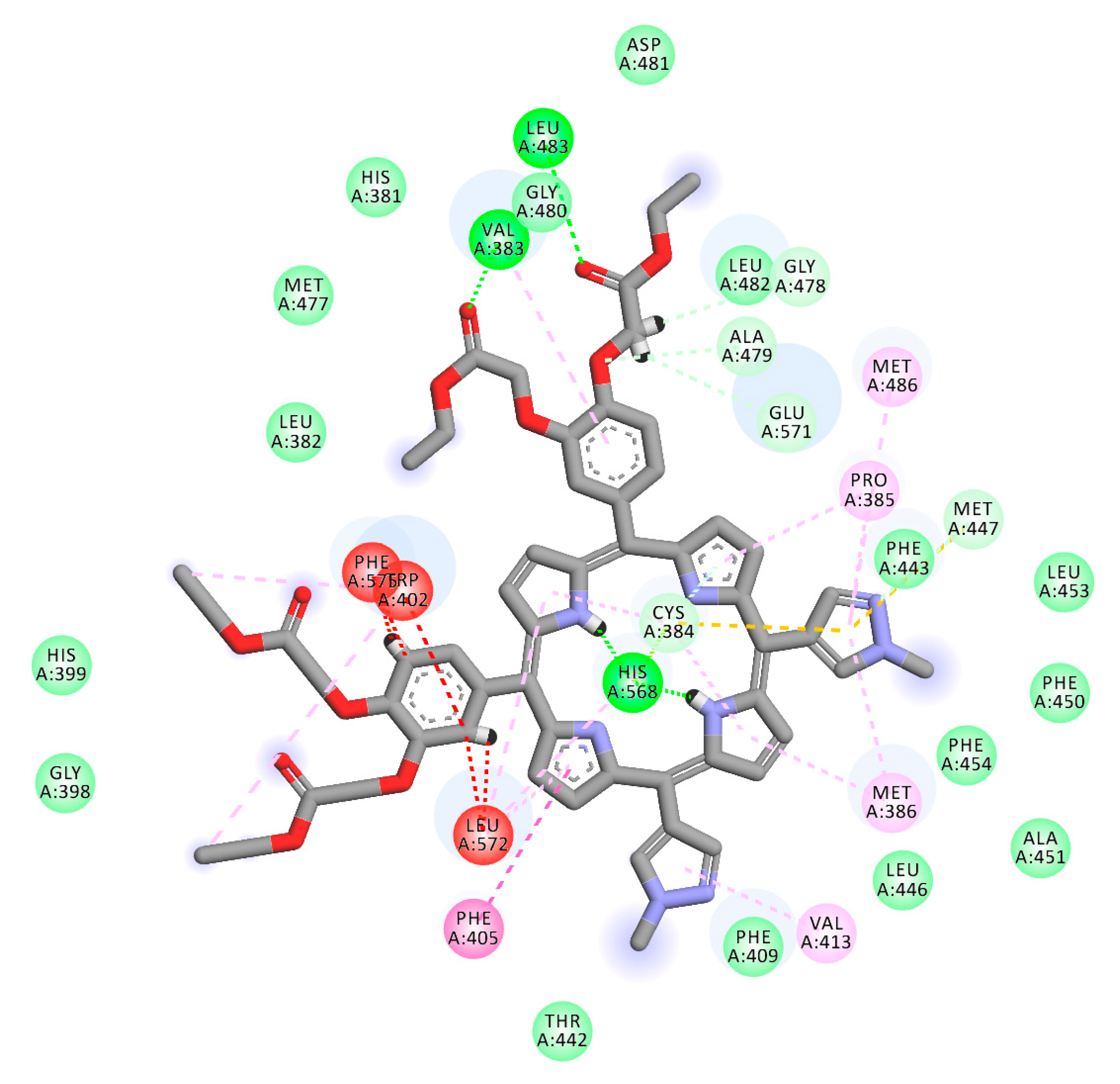
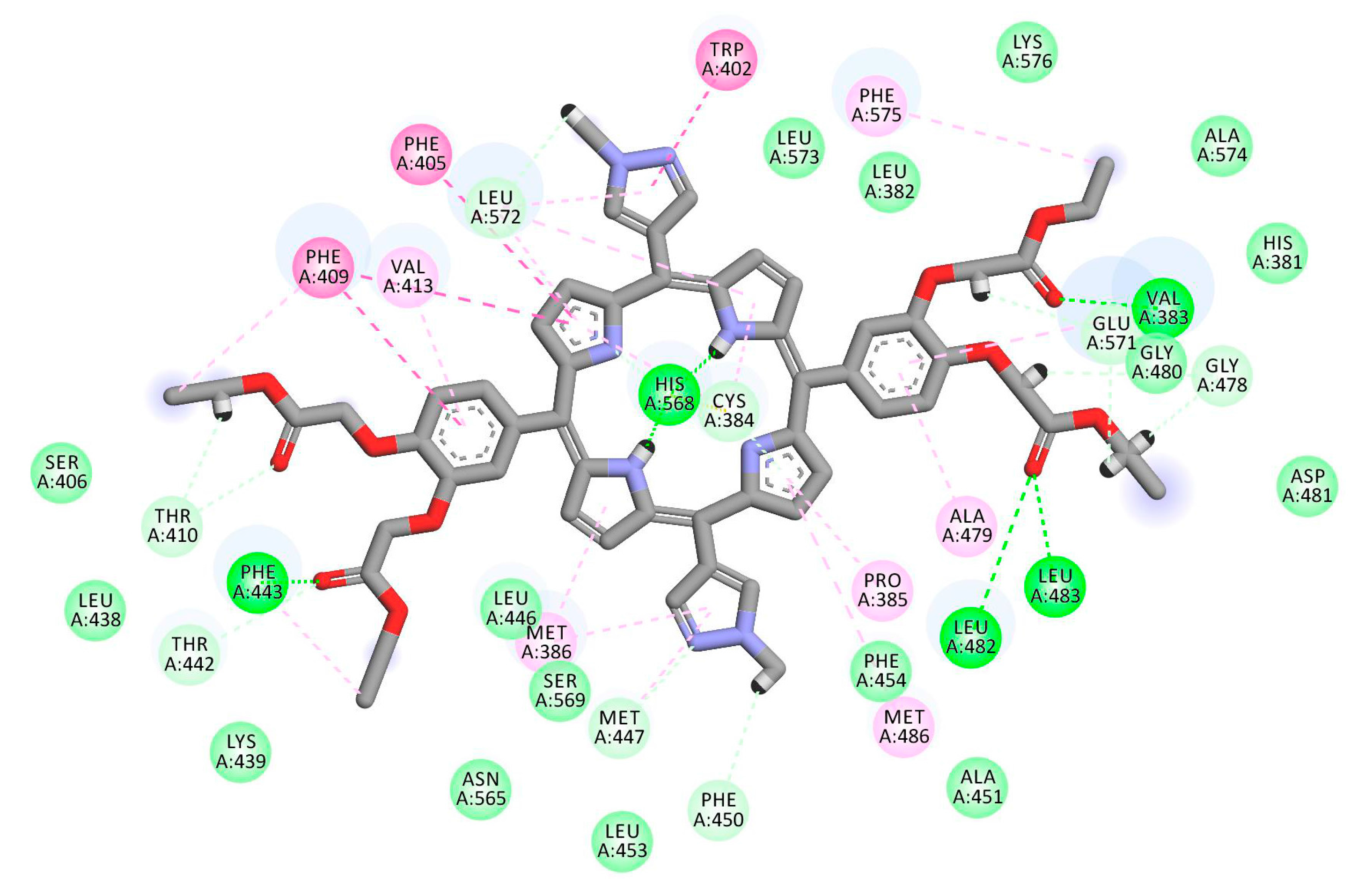
2.1. In Silico Study
2.2. Cytotoxicity Test
3. Discussion
3.1. In Silico Study
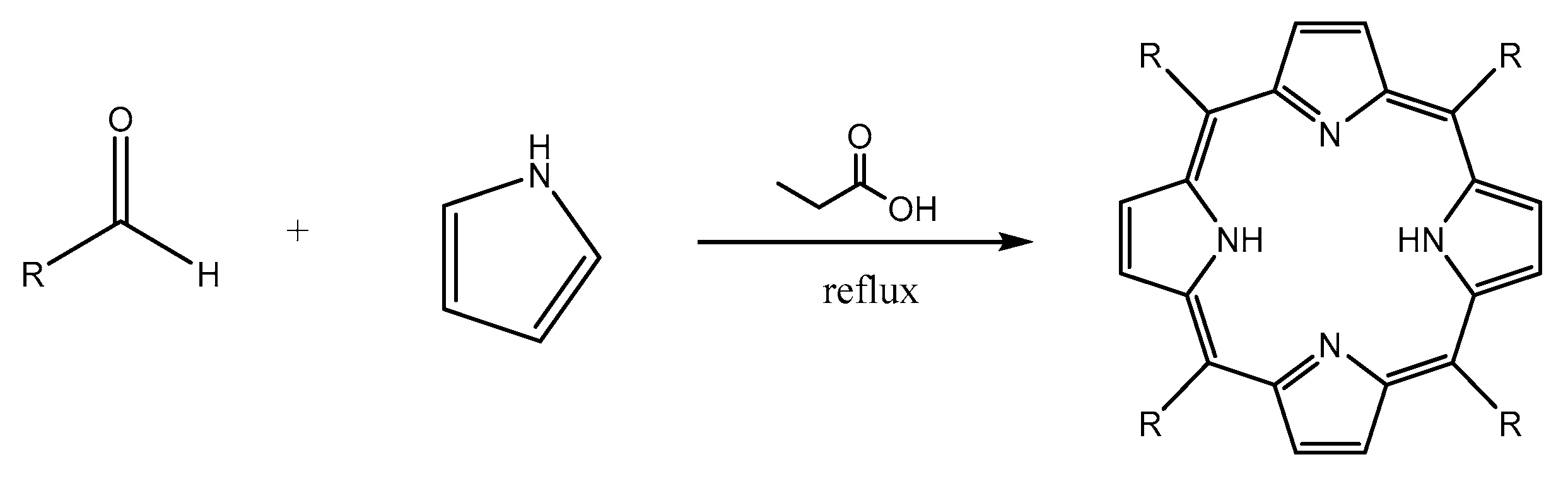
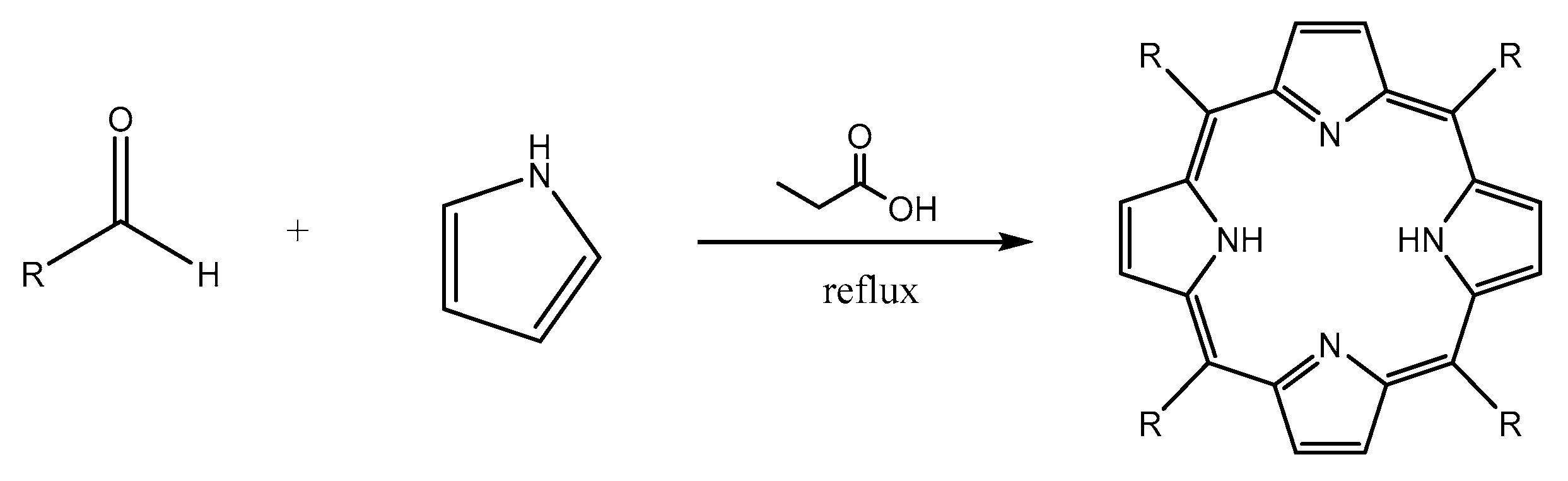
3.2. Synthesis of Porphyrin Derivatives
3.3. Cytotoxicity Test
4. Materials and Methods
4.1. In Silico Study
4.1.1. Macromolecule Preparation
4.1.2. Ligand Preparation
4.1.3. Molecular Docking Simulation
4.1.4. Molecular Dynamic Simulation
4.1.5. MM/PBSA Calculation
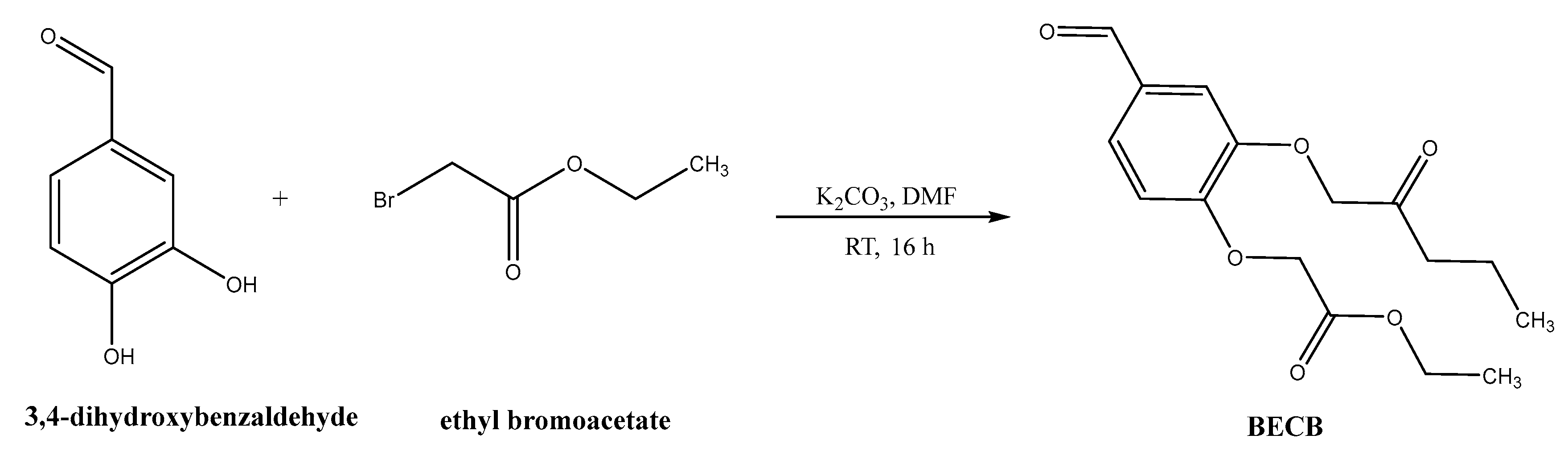
4.2. Synthesis of Porphyrin Derivatives
4.2.1. Materials
4.2.2. Procedure of Synthesis
Synthesis of 5,10,15,20-tetrakis(p-tolyl)porphyrin (TTP)
Synthesis of 5,10,15,20-tetrakis(p-bromophenyl)porphyrin(TBrPP)
Synthesis of 5,10,15,20-tetrakis(p-aminophenyl)porphyrin (TAPP)
Synthesis of 5,10,15-tris(tolyl)-20-mono(p-nitrophenyl)porphyrin (TrTMNP)
Synthesis of 5,10,15-tris(tolyl)-20-mono(p-aminophenyl)porphyrin (TrTMAP)
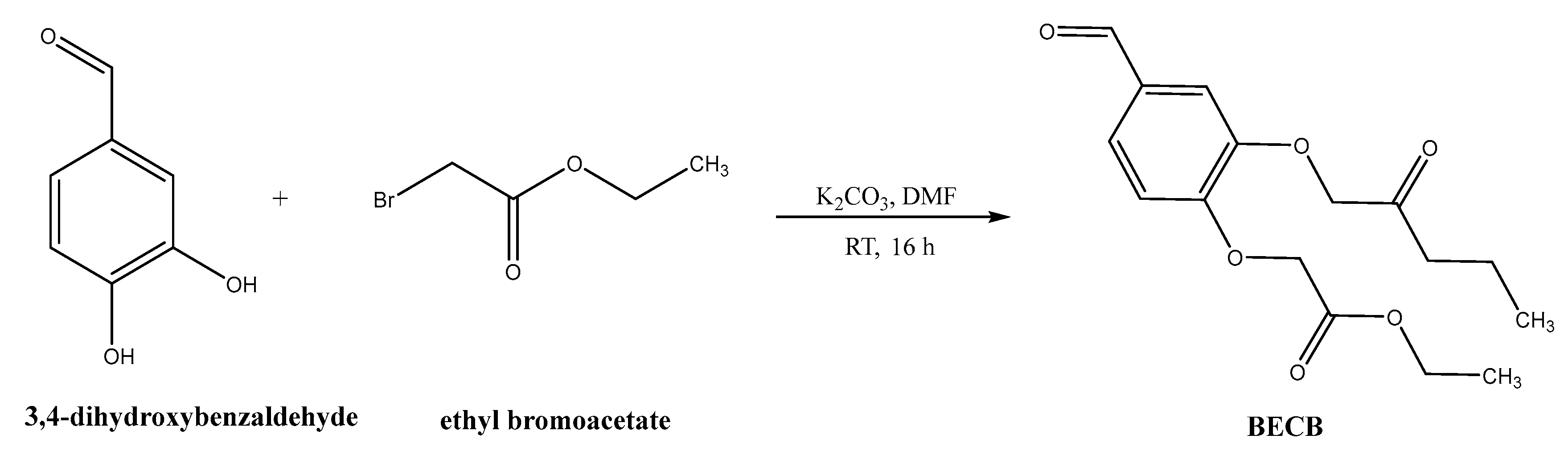
Synthesis of bis(3,4-ethylcarboxymethylenoxy) Benzaldehyde (BECB)
Synthesis of 1-Methylpyrazole-4-carbaldehyde
Synthesis of 5,15-di-[bis(3,4-ethylcarboxymethylenoxy)phenyl]-10,20-di(p-tolyl)porphyrin (DBECPDTP)
Synthesis of 5,10-di-[bis(3,4-ethylcarboxymethylenoxy)phenyl]-15,20-di-(methylpyrazole-4-yl)porphyrin (cDBECPDPzP)
Synthesis of 5,15-di-[bis(3,4-Ethylcarboxymethylenoxy)phenyl]-10,20-di-(methylpyrazole-4-yl)porphyrin (DBECPDPzP)
4.3. Cytotoxicity Test
4.3.1. Materials
4.3.2. Procedure
5. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seyfried, T.N.; Flores, R.E.; Poff, A.M.; Agostino, D.P.D. Cancer as a metabolic disease: Implications for novel therapeutics. Carcinogenesis 2014, 35, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer Statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Wu, L.; Qu, X. Cancer biomarker detection: Recent achievements and challenges. Chem. Soc. Rev. 2015, 44, 2963–2997. [Google Scholar] [CrossRef] [PubMed]
- Richter, M.; Zhang, H. Receptor-Targeted Cancer Therapy. DNA Cell Biol. 2005, 24, 271–282. [Google Scholar] [CrossRef] [PubMed]
- Mei, C.D.; Ercolani, L.; Parodi, C.; Veronesi, M.; Vecchio, C.L.; Bottegoni, G.; Torrente, E.; Scarpelli, R.; Marotta, R.; Ruffili, R.; et al. Dual inhibition of REV-ERBβ and autophagy as a novel pharmacological approach to induce cytotoxicity in cancer cells. Oncogene 2015, 34, 2597–2608. [Google Scholar] [CrossRef] [PubMed]
- Nógrádi, A. The Role of Carbonic Anhydrases in Tumors. Am. J. Pathol. 1998, 153, 1–4. [Google Scholar] [CrossRef]
- Maren, T.H. Carbonic anhydrase: Chemistry, physiology and inhibition. Physiol. Rev. 1967, 47, 595–743. [Google Scholar] [CrossRef] [PubMed]
- Tashian, R.E. The carbonic anhydrases: Widening perspectives on their evolution, expression and function. BioEssays 1989, 10, 186–192. [Google Scholar] [CrossRef] [PubMed]
- Hewett-Emmett, D.; Tashian, R.E. Functional diversity, conservation, and convergence in the evolution of the a-, b-, and g-carbonic anhydrase gene family. Mol. Phylogenet. Evol. 1996, 5, 50–77. [Google Scholar] [CrossRef] [PubMed]
- Sly, W.S.; Hu, P.Y. Human carbonic anhydrases and carbonic anhydrase deficiencies. Annu. Rev. Biochem. 1995, 64, 375–401. [Google Scholar] [CrossRef] [PubMed]
- Závada, J.; Závadová, Z.; Pastoreková, S.; Čiampor, F.; Pastorek, J.; Zelnik, V. Expression of MaTu-MN protein in human tumor cultures and in clinical specimens. Int. J. Cancer 1993, 54, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Stanbridge, E.J.; Der, C.J.; Doersen, C.-J.; Nishimi, R.Y.; Peehl, D.M.; Weissman, B.E.; Wilkinson, J.E. Human cell hybrids: Analysis of transformation and tumorigenicity. Science 1982, 215, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Liao, S.Y.; Brewer, C.; Závada, J.; Pastorek, J.; Pastoreková, S.; Manetta, A.; Berman, M.L.; DiSaia, P.J.; Stanbridge, E.J. Identification of the MN antigen as a diagnostic biomarker of cervical intraepithal squamous and glandular neoplasia and cervical carcinomas. Am. J. Pathol. 1994, 145, 598–609. [Google Scholar] [PubMed]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Colditz, G.A. Rotating night shifts and risk of breast cancer in women participating in the nurses’ health study. J. Natl. Cancer Inst. 2001, 93, 1563–1568. [Google Scholar] [CrossRef] [PubMed]
- Schernhammer, E.S.; Laden, F.; Speizer, F.E.; Willett, W.C.; Hunter, D.J.; Kawachi, I.; Fuchs, C.S.; Colditz, G.A. Night-shift work and risk of colorectal cancer in the Nurses’ Health Study. J. Natl. Cancer Inst. 2003, 95, 825–828. [Google Scholar] [CrossRef] [PubMed]
- Megdal, S.P.; Kroenke, C.H.; Laden, F.; Pukkala, E.; Schernhammer, E.S. Night work and breast cancer risk: A systematic review and meta-analysis. Eur. J. Cancer 2005, 41, 2023–2032. [Google Scholar] [CrossRef] [PubMed]
- Hansen, J. Increased breast cancer risk among women who work predominantly at night. Epidemiology 2001, 12, 74–77. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Kojetin, D.; Burris, T.P. Anti-Proliferative Actions of a Synthetic REV-ERBα/β Agonist in Breast Cancer Cell. Biochem. Pharmacol. 2015, 96, 315–322. [Google Scholar] [CrossRef] [PubMed]
- Straif, K.; Baan, R.; Grosse, Y.; Secretan, B.; El Ghissassi, F.; Bouvard, V.; Altieri, A.; Benbrahim-Tallaa, L.; Coglianno, V. Monograpgh WHOIARC, Carcinogenicity of shift-work, painting, and fire-fighting. Lancet Oncol. 2007, 8, 1065–1066. [Google Scholar] [CrossRef]
- Urruticoechea, A.; Alemany, R.; Balart, J.; Villanueva, A.; Vinals, F.; Capella, G. Recent Advances in Cancer Therapy: An Overview. Curr. Pharm. Des. 2010, 16, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Sebastian, R. Nanomedicine-the Future of Cancer Treatment: A review. J. Cancer Prev. Curr. Res. 2017, 8, 00265. [Google Scholar] [CrossRef]
- Tjahjono, D.H.; Akutsu, T.; Yoshioka, N.; Inoue, H. Cationic porphyrins bearing diazolium rings: Synthesis and their interaction with calf thymus DNA. Biochim. Biophys. Acta 1999, 1472, 333–343. [Google Scholar] [CrossRef]
- Tjahjono, D.H.; Yamamoto, T.; Ichimoto, S.; Yoshioka, N.; Inoue, H. Synthesis and DNA-binding properties of bisdiazoliumylporphyrins. J. Chem. Soc. Perkin Trans. 2000, 1, 3077–3081. [Google Scholar] [CrossRef]
- Subbarayan, M.; Shetty, S.J.; Srivastava, T.S.; Noronha, O.P.D.; Samuel, A.M. Evaluation studies of technetium-99m-porphyrin (T3,4BCPP) for tumor imaging. J. Porphyr. Phthalocyanines 2001, 5, 824–828. [Google Scholar] [CrossRef]
- Muttaqin, F.Z. Sintesis, Pelabelan, dan Uji Biodistribusi Meso-5,15-di [3,4-bis (karboksimetilenoksi)fenil] Porfirin dan Meso-5,15-di [3,4-bis (karboksimetilenoksi) fenil], 10,20-Difenil Porfirin Sebagai Ligan Radiofarmaka Teranostik. Ph.D. Thesis, Sekolah Farmasi Institut Teknologi Bandung, Bandung, Indonesia, 2014. [Google Scholar]
- Jia, Z.; Deng, H.; Luo, S. Rhenium-labelled meso-tetrakis [3,4-bis (carboxymethyleneoxy)phenyl] porphyrin for Targeted Radiotheraphy : Preliminary Biological Evaluation in Mice. Eur. J. Nucl. Med. Mol. Imaging 2008, 35, 734–742. [Google Scholar] [CrossRef] [PubMed]
- Izbicka, E.; Wheelhouse, R.T.; Raymond, E.; Davidson, K.K.; Lawrence, R.A.; Sun, D.Y.; Windle, B.E.; Hurley, L.H.; von Hoff, D.D. Effect of Cationic Porphyrins as G-quadruplex Interactive Agents in Human Tumor Cells. Cancer Res. 1999, 59, 639–644. [Google Scholar] [PubMed]
- Hurley, L.H.; Wheelhouse, R.T.; Sun, D.; Kerwin, S.M.; Salazar, M.; Fedoroff, O.Y.; Han, F.X.; Izbicka, E.; von Hoff, D.D. G-quadruplex as Targets for Drug Design. Pharmacol. Ther. 2000, 85, 141–158. [Google Scholar] [CrossRef]
- Kuhn, B.; Kollman, P.A. A Binding of diverse set of ligands to avidin and streptavidin: An accurate quantitative prediction of their relative affinities by a combination of molecular mechanics and continuum solvent models. J. Med. Chem. 2000, 43, 3786–3791. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, A.M.; Degliesposti, G.; Sgobba, M.; Rastelli, G. Validation of an automated procedure for the prediction of relative free energies of binding on a set of aldose reductase inhibitors. Bioorg. Med. Chem. 2007, 15, 7865–7877. [Google Scholar] [CrossRef] [PubMed]
- Arba, M.; Tjahjono, D.H. The binding modes of cationic porphyrin-anthraquinone hybrids to DNA duplexes: In silico study. J. Biomol. Struct. Dyn. 2015, 33, 657–665. [Google Scholar] [CrossRef] [PubMed]
- Adler, A.D.; Longo, F.R.; Finarelli, J.D.; Assour, J.; Korsakoff, L. A simplified synthesis for meso-tetraphenylporphine. J. Org. Chem. 1967, 32, 476. [Google Scholar] [CrossRef]
- Ni, J.; Auston, D.A.; Freilich, D.A.; Muralidharan, S.; Sobie, E.A.; Kao, J.P.Y. Photochemical Gating of Intracellular Ca2+ Release Channel. J. Am. Chem. Soc. 2007, 129, 5316–5317. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of Useful Decoys, Enhanced (DUD-E): Better Ligands and Decoys for Better Benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Dodoff, N.I.; Iordanov, I.; Tsoneva, I.; Grancharov, K.; Detcheva, R.; Pajpanova, T.; Berger, M.R. Cytotoxic Activity of Platinum(II) and Palladium(II) Complexes of N-3-Pyridinylmethanesulfonamide: The Influence of Electroporation. Z. Naturforsch. C 2009, 64, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Pastorekova, S.; Zavadova, Z.; Kostal, M.; Babusikova, O.; Zavada, J. A novel quasi-viral agent, MaTu, is a two-component system. Virology 1992, 187, 620–626. [Google Scholar] [CrossRef]
- McDonald, P.C.; Winum, J.; Supuran, C.T.; Dedhar, S. Recent Development in Targeting Carbonic Anhydrase IX for Cancer Therapeutics. Oncotarget 2012, 3, 84–97. [Google Scholar] [CrossRef] [PubMed]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Goodsell, D.S.; Morris, G.M.; Olson, A.J. Automated docking of flexible ligands: Applications of autodock. J. Mol. Recogn. 1996, 9, 1–5. [Google Scholar] [CrossRef]
- Morris, G.M.; Goodsell, D.S.; Halliday, R.S.; Huey, R.; Hart, W.E.; Belew, R.K.; Olson, A.J. Automated docking using a Lamarckian genetic algorithm and an empirical binding free energy function. J. Comput. Chem. 1998, 19, 1639–1662. [Google Scholar] [CrossRef]
- AutoDockTools, version 1.5.6; The Scripps Research Institute: La Jolla, CA, USA, 2015.
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Dassault Systemes BIOVIA, Discovery Studio Modeling Environment, Release 2017; Dassault Systemes: San Diego, CA, USA, 2016.
- Berendsen, H.J.C.; van der Spoel, D.; van Drunen, R. GROMACS: A message-passing parallel molecular dynamics implementation. Comput. Phys. Commun. 1995, 91, 43–56. [Google Scholar] [CrossRef]
- Lindahl, E.; Hess, B.; van der Spoel, D. GROMACS 3.0: A package for molecular simulation and trajectory analysis. J. Mol. Mod. 2001, 7, 306–317. [Google Scholar] [CrossRef]
- van der Spoel, D.; Lindahl, E.; Hess, B.; Groenhof, G.; Mark, A.E.; Berendsen, H.J.C. GROMACS: Fast, Flexible and Free. J. Comput. Chem. 2005, 26, 1701–1719. [Google Scholar] [CrossRef] [PubMed]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for highly efficient, load-balanced, and scalable molecular simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [PubMed]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; van der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef] [PubMed]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindahl, E. GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1, 19–25. [Google Scholar] [CrossRef]
- Páll, S.; Abraham, M.J.; Kutzner, C.; Hess, B.; Lindahl, E. Tackling Exascale Software Challenges in Molecular Dynamics Simulations with GROMACS. Solving Softw. Chall. Exascale 2015, 8759, 3–27. [Google Scholar]
- Aliev, A.E.; Kulke, M.; Khaneja, H.S.; Chudasama, V.; Sheppard, T.D.; Lanigan, R.M. Motional timescale predictions by molecular dynamics simulations: Case study using proline and hydroxyproline sidechain dynamics. Proteins 2014, 82, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, A.W.S.; Vranken, W.F. ACPYPE—AnteChamber Python Parser interface. BMC Res. Notes 2012, 5, 367. [Google Scholar] [CrossRef] [PubMed]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald—An NlogN method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- Baker, N.A.; Sept, D.; Joseph, S.; Holst, M.J.; McCammon, J.A. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc. Natl. Acad. Sci. USA 2001, 98, 10037–10041. [Google Scholar] [CrossRef] [PubMed]
- Kumari, R.; Kumar, R.; Open Source Drug Discovery Consortium; Lynn, A. g_mmpbsa—A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Hou, J.Q.; Chen, S.B.; Tan, J.H.; Ou, T.M.; Luo, H.B.; Li, D.; Xu, J.; Gu, L.Q.; Huang, Z.S. New Insights into the structures of ligand-quadruplex complexes from molecular dynamics simulations. J. Phys. Chem. B 2010, 114, 15301–15310. [Google Scholar] [CrossRef] [PubMed]
- Špačková, N.; Cheatham, T.E., III; Ryjáček, F.; Lankaš, F.; van Meervelt, L.; Hobza, P.; Šponer, J. Molecular dynamics simulations and thermodynamics analysis of DNA−drug complexes. Minor groove binding between 4′,6-diamidino-2-phenylindole and DNA duplexes in solution. J. Am. Chem. Soc. 2003, 125, 1759–1769. [Google Scholar]
- Sun, Z.; She, Y.; Zhou, Y.; Song, X.; Li, K. Synthesis, Characterization and Spectral Properties of Substituted Tetraphenylporphyrin Iron Chloride Complexes. Molecules 2011, 16, 2960–2970. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Xu, Y.; Guo, Z.; Nagai, A.; Jiang, D. Super absorbent conjugated microporous polymers: A synergistic structural effect on the exceptional uptake of amines. Chem. Commun. 2013, 49, 3233–3235. [Google Scholar] [CrossRef] [PubMed]
- Ormond, A.B.; Freemand, H.S. Effects of substituents on the photophysical properties of symmetrical porphyrins. Dyes Pigments 2013, 96, 440–448. [Google Scholar] [CrossRef]
- Sol, V.; Blais, J.C.; Carre, V.; Granet, R.; Guilloton, M.; Spiro, M.; Krausz, P. Synthesis, Spectroscopy, and Photocytotoxicity of Glycosylated Amino Acid Porphyrin Derivatives as Promising Molecules for Cancer Phototherapy. J. Org. Chem. 1999, 64, 4431–4444. [Google Scholar] [CrossRef]
- Finar, I.L.; Lord, G.H. The formylation of the pyrazole nucleus. J. Chem. Soc. 1957, 0, 3314–3315. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Porphyrin Ligand | Binding Energy (kJ/mol) | |
|---|---|---|
| 5FL6 | 4N73 | |
| TTP | −26.82 | −22.84 |
| TBrPP | −29.00 | −16.11 |
| TAPP | −21.30 | 179.37 |
| TrTMNP | −30.08 | −15.69 |
| TrTMAP | −28.66 | −23.81 |
| cDBECPDPzP | −22.34 | 36.15 |
| DBECPDPzP | −16.90 | −29.75 |
| DBECPDTP | −18.87 | 224.26 |
| Complex | ΔEvdw (kJ/mol) | ΔEele (kJ/mol) | ΔGPB (kJ/mol) | ΔGNP (kJ/mol) | ΔGBind (kJ/mol) |
|---|---|---|---|---|---|
| 5FL6-TrTMNP | −53.49 | −9.39 | 53.51 | −6.13 | −15.50 |
| 4N73-DBECPDPzP | −477.91 | −42.45 | 282.05 | −38.15 | −276.46 |
| Porphyrins | IC50 (μM) ± SD | |||||
|---|---|---|---|---|---|---|
| HeLa | WIDR | HepG2 | T47D | MCF-7 | Vero | |
| TTP | 714.6 ± 1.1 | 969.8 ± 1.2 | 1120.0 ± 1.3 | 1241.0 ± 1.2 | 1305.0 ± 1.3 | 15,612.0 ± 1.6 |
| TBrPP | 595.6 ± 1.3 | 772.7 ± 1.3 | 882.7 ± 1.2 | 443.9 ± 1.1 | 870.6 ± 1.3 | 1176.0 ± 1.1 |
| TAPP | 1007.0 ± 1.1 | 205.1 ± 1.2 | 36.0 ± 1.2 | 332.6 ± 1.2 | 262.2 ± 1.3 | 2264.0 ± 1.2 |
| TrTMNP | 226.1 ± 1.1 | 897.5 ± 1.1 | 146.6 ± 1.2 | 5372.0 ± 1.4 | 505.2 ± 1.2 | 10,079.0 ± 1.2 |
| TrTMAP | 593.1 ± 1.2 | 835.4 ± 1.3 | 106.7 ± 1.3 | 1099.0 ± 1.3 | 328.4 ± 1.4 | 4784.0 ± 1.2 |
| cDBECPDPzP | 1083.0 ± 1.2 | 28.9 ± 1.1 | 28.6 ± 1.2 | 164.5 ± 1.2 | 47.9 ± 1.1 | 548.8 ± 1.2 |
| DBECPDPzP | 941.9 ± 1.2 | 34.1 ± 1.1 | 31.7 ± 1.2 | 177.8 ± 1.2 | 64.2 ± 1.1 | 363.4 ± 1.1 |
| DBECPDTP | 201.7 ± 1.3 | 33.5 ± 1.1 | 37.2 ± 1.1 | 166.7 ± 1.3 | 42.1 ± 1.1 | 372.2 ± 1.1 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurniawan, F.; Miura, Y.; Kartasasmita, R.E.; Yoshioka, N.; Mutalib, A.; Tjahjono, D.H. In Silico Study, Synthesis, and Cytotoxic Activities of Porphyrin Derivatives. Pharmaceuticals 2018, 11, 8. https://doi.org/10.3390/ph11010008
Kurniawan F, Miura Y, Kartasasmita RE, Yoshioka N, Mutalib A, Tjahjono DH. In Silico Study, Synthesis, and Cytotoxic Activities of Porphyrin Derivatives. Pharmaceuticals. 2018; 11(1):8. https://doi.org/10.3390/ph11010008
Chicago/Turabian StyleKurniawan, Fransiska, Youhei Miura, Rahmana Emran Kartasasmita, Naoki Yoshioka, Abdul Mutalib, and Daryono Hadi Tjahjono. 2018. "In Silico Study, Synthesis, and Cytotoxic Activities of Porphyrin Derivatives" Pharmaceuticals 11, no. 1: 8. https://doi.org/10.3390/ph11010008
APA StyleKurniawan, F., Miura, Y., Kartasasmita, R. E., Yoshioka, N., Mutalib, A., & Tjahjono, D. H. (2018). In Silico Study, Synthesis, and Cytotoxic Activities of Porphyrin Derivatives. Pharmaceuticals, 11(1), 8. https://doi.org/10.3390/ph11010008