Endolysosomal Cation Channels and Cancer—A Link with Great Potential


Abstract

{kind=link}
{kind=link}
1. Introduction
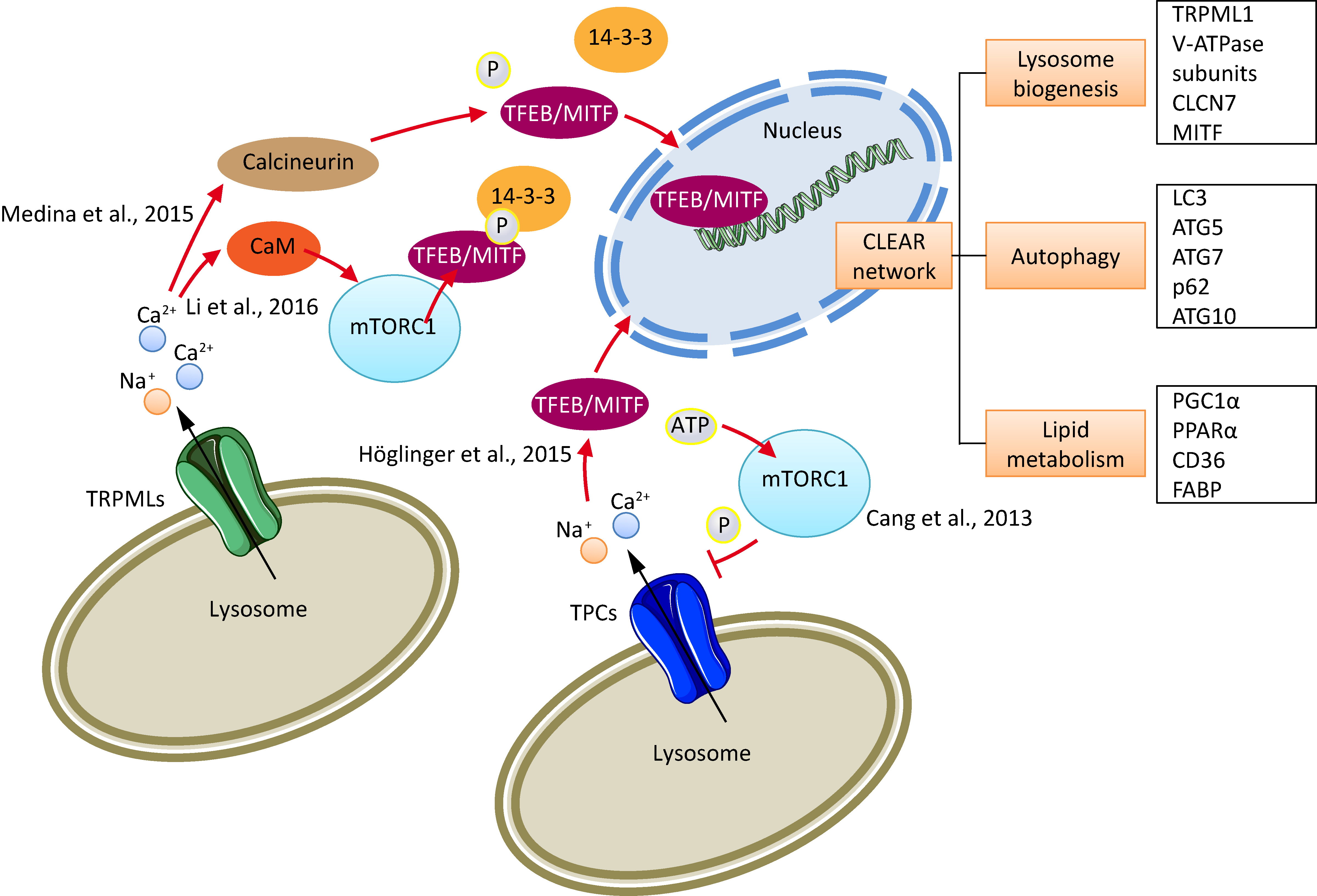
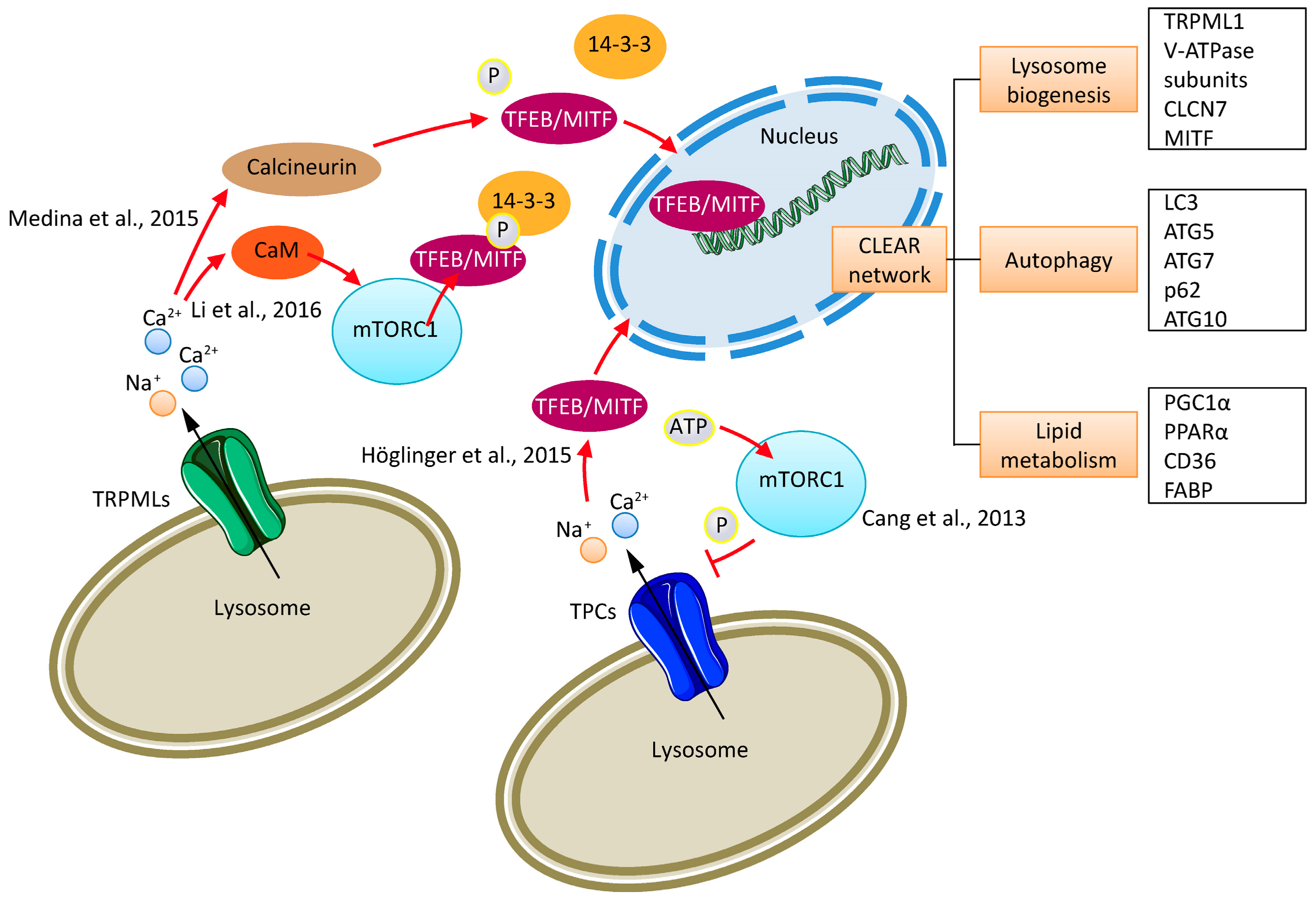
2. Cancer Cell Lysosomes
3. Cancer and Inflammation
4. TPCs and Cancer
5. TRPMLs and Cancer
Acknowledgments
Conflicts of Interest
References
- Davidson, S.M.; Vander Heiden, M.G. Critical Functions of the Lysosome in Cancer Biology. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 481–507. [Google Scholar] [CrossRef] [PubMed]
- Piao, S.; Amaravadi, R.K. Targeting the lysosome in cancer. Ann. N Y Acad. Sci. 2016, 1371, 45–54. [Google Scholar] [CrossRef] [PubMed]
- Saftig, P.; Sandhoff, K. Cancer: Killing from the inside. Nature 2013, 502, 312–313. [Google Scholar] [CrossRef] [PubMed]
- Allison, A.C. Lysosomes and cancer. In Lysosomes in Biology and Pathology; Dingle, J.T., Fell, H.B., Eds.; North-Holland: Amsterdam, The Netherlands, 1969; Volume 2, pp. 178–204. [Google Scholar]
- Allison, A.C. Lysosomes in cancer cells. J. Clin. Pathol. Suppl. 1974, 7, 43–50. [Google Scholar] [CrossRef]
- Poole, A.R. Tumour lysosomal enzymes and invasive growth. In Lysosomes in Biology and Pathology; Dingle, J.T., Ed.; North-Holland: Amsterdam, The Netherlands, 1973; Volume 3, p. 83. [Google Scholar]
- Fennelly, C.; Amaravadi, R.K. Lysosomal Biology in Cancer. Methods Mol. Biol. 2017, 1594, 293–308. [Google Scholar] [PubMed]
- Leanza, L.; Biasutto, L.; Managò, A.; Gulbins, E.; Zoratti, M.; Szabò, I. Intracellular ion channels and cancer. Front. Physiol. 2013, 4, 227. [Google Scholar] [CrossRef] [PubMed]
- Peruzzo, R.; Biasutto, L.; Szabò, I.; Leanza, L. Impact of intracellular ion channels on cancer development and progression. Eur. Biophys. J. 2016, 45, 685–707. [Google Scholar] [CrossRef] [PubMed]
- Gautier, M.; Dhennin-Duthille, I.; Ay, A.S.; Rybarczyk, P.; Korichneva, I.; Ouadid-Ahidouch, H. New insights into pharmacological tools to TR(i)P cancer up. Br. J. Pharmacol. 2014, 171, 2582–2592. [Google Scholar] [CrossRef] [PubMed]
- Shapovalov, G.; Ritaine, A.; Skryma, R.; Prevarskaya, N. Role of TRP ion channels in cancer and tumorigenesis. Semin. Immunopathol. 2016, 38, 357–369. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Butz, E.; Chen, C.-C.; Wahl-Schott, C.; Biel, M. From mucolipidosis type IV to Ebola: TRPML and two-pore channels at the crossroads of endo-lysosomal trafficking and disease. Cell Calcium 2017, 67, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Grimm, C.; Schneider, L.; Chao, Y.-K.; Watermann, A.; Ulrich, M.; Mayr, D.; Wahl-Schott, C.; Biel, M.; Vollmar, A.M. Two-pore channel function is crucial for migration of invasive cancer cells. Cancer Res. 2017, 77, 1427–1438. [Google Scholar] [CrossRef] [PubMed]
- Nomura, T.; Katunuma, N. Involvement of cathepsins in the invasion, metastasis and proliferation of cancer cells. J. Med. Investig. 2005, 52, 1–9. [Google Scholar] [CrossRef]
- Mohamed, M.M.; Sloane, B.F. Cysteine cathepsins: Multifunctional enzymes in cancer. Nat. Rev. Cancer 2006, 6, 764–775. [Google Scholar] [CrossRef] [PubMed]
- Tardy, C.; Codogno, P.; Autefage, H.; Levade, T.; Andrieu-Abadie, N. Lysosomes and lysosomal proteins in cancer cell death (new players of an old struggle). Biochim. Biophys. Acta 2006, 1765, 101–125. [Google Scholar] [CrossRef] [PubMed]
- Kallunki, T.; Olsen, O.D.; Jäättelä, M. Cancer-associated lysosomal changes: Friends or foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [PubMed]
- Kirkegaard, T.; Jäättelä, M. Lysosomal involvement in cell death and cancer. Biochim. Biophys. Acta 2009, 1793, 746–754. [Google Scholar] [CrossRef] [PubMed]
- Easton, J.B.; Houghton, P.J. mTOR and cancer therapy. Oncogene 2006, 25, 6436–6446. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Carreres, L.; Nasrallah, A.; Fajas, L. Cancer: Linking Powerhouses to Suicidal Bags. Front. Oncol. 2017, 7, 204. [Google Scholar] [CrossRef] [PubMed]
- Pópulo, H.; Lopes, J.M.; Soares, P. The mTOR signalling pathway in human cancer. Int. J. Mol. Sci. 2012, 13, 1886–1918. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.J.; DeWald, D.B.; Emr, S.D.; Plutner, H.; Balch, W.E. Role for phosphatidylinositol 3-kinase in the sorting and transport of newly synthesized lysosomal enzymes in mammalian cells. J. Cell Biol. 1995, 130, 781–796. [Google Scholar] [CrossRef] [PubMed]
- Mousavi, S.A.; Brech, A.; Berg, T.; Kjeken, R. Phosphoinositide 3-kinase regulates maturation of lysosomes in rat hepatocytes. Biochem. J. 2003, 372, 861–869. [Google Scholar] [CrossRef] [PubMed]
- Collins, D.; Chenard-Poirier, M.; Lopez, J. The PI3K pathway at the crossroads of cancer and the immune system: Strategies for next generation immunotherapy combinations. Curr. Cancer Drug Targets 2017, 26. [Google Scholar] [CrossRef] [PubMed]
- LoPiccolo, J.; Blumenthal, G.M.; Bernstein, W.B.; Dennis, P.A. Targeting the PI3K/Akt/mTOR pathway: Effective combinations and clinical considerations. Drug Resist. Updates 2008, 11, 32–50. [Google Scholar] [CrossRef] [PubMed]
- Wiedmann, R.M.; von Schwarzenberg, K.; Palamidessi, A.; Schreiner, L.; Kubisch, R.; Liebl, J.; Schempp, C.; Trauner, D.; Vereb, G.; Zahler, S.; et al. The V-ATPase-inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho-GTPase Rac1. Cancer Res. 2012, 72, 5976–5987. [Google Scholar] [CrossRef] [PubMed]
- Bartel, K.; Winzi, M.; Ulrich, M.; Koeberle, A.; Menche, D.; Werz, O.; Müller, R.; Guck, J.; Vollmar, A.M.; von Schwarzenberg, K. V-ATPase inhibition increases cancer cell stiffness and blocks membrane related Ras signaling—A new option for HCC therapy. Oncotarget 2017, 8, 9476–9487. [Google Scholar] [CrossRef] [PubMed]
- Hämälistö, S.; Jäätellä, M. Lysosomes in cancer-living on the edge (of the cell). Curr. Opin. Cell Biol. 2016, 39, 69–76. [Google Scholar] [CrossRef] [PubMed]
- Klinosky, D.J.; Abdelmohsen, K.; Abe, A.; Abedin, D.J.; Abeliovich, H.; Arozena, A.A. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 2016, 12, 1–222. [Google Scholar] [CrossRef] [PubMed]
- Landskron, G.; De la Fuente, M.; Thuwajit, P.; Thuwajit, C.; Hermoso, M.A. Chronic inflammation and cytokines in the tumor microenvironment. J. Immunol. Res. 2014, 2014, 149185. [Google Scholar] [CrossRef] [PubMed]
- Camoglio, L.; Te Velde, A.A.; Tigges, A.J.; Das, P.K.; Van Deventer, S.J. Altered expression of interferon-gamma and interleukin-4 in inflammatory bowel disease. Inflamm. Bowel Dis. 1998, 4, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Shacter, E.; Weitzman, S.A. Chronic inflammation and cancer. Oncology 2002, 16, 217–226. [Google Scholar] [PubMed]
- Coussens, L.M.; Werb, Z. Inflammation and cancer. Nature 2002, 420, 860–867. [Google Scholar] [CrossRef] [PubMed]
- Bretou, M.; Sáez, P.J.; Sanséau, D.; Maurin, M.; Lankar, D.; Chabaud, M.; Spampanato, C.; Malbec, O.; Barbier, L.; Muallem, S.; et al. Lysosome signaling controls the migration of dendritic cells. Sci. Immunol. 2017, 2, eaak9573. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-C.; Butz, E.; Chao, Y.-K.; Grishchuk, Y.; Becker, L.; Heller, S.; Slaugenhaupt, S.; Biel, M.; Wahl-Schott, C.; Grimm, C. Small molecules for early endosome specific patch-clamping. Cell Chem. Biol. 2017, 24, 907–916. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Hua, Y.; Vergarajauregui, S.; Diab, H.I.; Puertollano, R. Novel Role of TRPML2 in the Regulation of the Innate Immune Response. J. Immunol. 2015, 195, 4922–4932. [Google Scholar] [CrossRef] [PubMed]
- Parrington, J.; Lear, P.; Hachem, A. Calcium signals regulated by NAADP and two-pore channels—Their role in development, differentiation and cancer. Int. J. Dev. Biol. 2015, 59, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Huttenlocher, A.; Horwitz, A.R. Integrins in cell migration. Cold Spring Harb. Perspect. Biol. 2011, 3, a005074. [Google Scholar] [CrossRef] [PubMed]
- Paul, N.R.; Jacquemet, G.; Caswell, P.T. Endocytic Trafficking of Integrins in Cell Migration. Curr. Biol. 2015, 25, R1092–R1105. [Google Scholar] [CrossRef] [PubMed]
- Grimm, C.; Holdt, L.M.; Chen, C.-C.; Hassan, S.; Müller, C.; Jörs, S.; Cuny, H.; Kissing, S.; Schröder, B.; Butz, E.; et al. High susceptibility to fatty liver disease in two-pore channel 2-deficient mice. Nat. Commun. 2014, 5, 4699. [Google Scholar] [CrossRef] [PubMed]
- Pafumi, I.; Festa, M.; Papacci, F.; Lagostena, L.; Giunta, C.; Gutla, V.; Cornara, L.; Favia, A.; Palombi, F.; Gambale, F.; et al. Naringenin Impairs Two-Pore Channel 2 Activity and Inhibits VEGF-Induced Angiogenesis. Sci. Rep. 2017, 7, 5121. [Google Scholar] [CrossRef] [PubMed]
- Favia, A.; Desideri, M.; Gambara, G.; D’Alessio, A.; Ruas, M.; Esposito, B.; Del Bufalo, D.; Parrington, J.; Ziparo, E.; Palombi, F.; et al. VEGF-induced neoangiogenesis is mediated by NAADP and two-pore channel-2-dependent Ca2+ signaling. Proc. Natl. Acad. Sci. USA 2014, 111, E4706–E4715. [Google Scholar] [CrossRef] [PubMed]
- Cang, C.; Zhou, Y.; Navarro, B.; Seo, Y.J.; Aranda, K.; Shi, L.; Battaglia-Hsu, S.; Nissim, I.; Clapham, D.E.; Ren, D. mTOR regulates lysosomal ATP-sensitive two-pore Na+ channels to adapt to metabolic state. Cell 2013, 152, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed]
- Li, R.J.; Xu, J.; Fu, C.; Zhang, J.; Zheng, Y.G.; Jia, H.; Liu, J.O. Regulation of mTORC1 by lysosomal calcium and calmodulin. eLife 2016, 5, e19360. [Google Scholar] [CrossRef] [PubMed]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, M.; Impey, S.; Kang, H.; di Ronza, A.; Pelz, C.; Sardiello, M.; Ballabio, A. Characterization of the CLEAR network reveals an integrated control of cellular clearance pathways. Hum. Mol. Genet. 2011, 20, 3852–3866. [Google Scholar] [CrossRef] [PubMed]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, D.; Haberkant, P.; Aguilera-Romero, A.; Riezman, H.; Porter, F.D.; Platt, F.M.; Galione, A.; Schultz, C. Intracellular sphingosine releases calcium from lysosomes. eLife 2015, 4, e10616. [Google Scholar] [CrossRef] [PubMed]
- Di Paola, S.; Scotto-Rosato, A.; Medina, D.L. TRPML1: The Ca2+ retaker of the lysosome. Cell Calcium 2018, 69, 112–121. [Google Scholar] [CrossRef] [PubMed]
- Marchand, B.; Arsenault, D.; Raymond-Fleury, A.; Boisvert, F.M.; Boucher, M.J. Glycogen synthase kinase-3 (GSK3) inhibition induces prosurvival autophagic signals in human pancreatic cancer cells. J. Biol. Chem. 2015, 290, 5592–5605. [Google Scholar] [CrossRef] [PubMed]
- Giatromanolaki, A.; Kalamida, D.; Sivridis, E.; Karagounis, I.V.; Gatter, K.C.; Harris, A.L.; Koukourakis, M.I. Increased expression of transcription factor EB (TFEB) is associated with autophagy, migratory phenotype and poor prognosis in non-small cell lung cancer. Lung Cancer 2015, 90, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Calcagnì, A.; Kors, L.; Verschuren, E.; De Cegli, R.; Zampelli, N.; Nusco, E.; Confalonieri, S.; Bertalot, G.; Pece, S.; Settembre, C.; et al. Modelling TFE renal cell carcinoma in mice reveals a critical role of WNT signaling. eLife 2016, 5, e17047. [Google Scholar] [CrossRef] [PubMed]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [PubMed]
- Perera, R.M.; Stoykova, S.; Nicolay, B.N.; Ross, K.N.; Fitamant, J.; Boukhali, M.; Lengrand, J.; Deshpande, V.; Selig, M.K.; Ferrone, C.R.; et al. Transcriptional control of autophagy-lysosome function drives pancreatic cancer metabolism. Nature 2015, 524, 361–365. [Google Scholar] [CrossRef] [PubMed]
- Settembre, C.; De Cegli, R.; Mansueto, G.; Saha, P.K.; Vetrini, F.; Visvikis, O.; Huynh, T.; Carissimo, A.; Palmer, D.; Klisch, T.J. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat. Cell Biol. 2013, 15, 647–658. [Google Scholar] [CrossRef] [PubMed]
- Samie, M.; Wang, X.; Zhang, X.; Goschka, A.; Li, X.; Cheng, X.; Gregg, E.; Azar, M.; Zhuo, Y.; Garrity, A.G.; et al. A TRP channel in the lysosome regulates large particle phagocytosis via focal exocytosis. Dev. Cell 2013, 26, 511–524. [Google Scholar] [CrossRef] [PubMed]
- Park, S.; Ahuja, M.; Kim, M.S.; Brailoiu, G.C.; Jha, A.; Zeng, M.; Baydyuk, M.; Wu, L.G.; Wassif, C.A.; Porter, F.D.; et al. Fusion of lysosomes with secretory organelles leads to uncontrolled exocytosis in the lysosomal storage disease mucolipidosis type IV. EMBO Rep. 2016, 17, 266–278. [Google Scholar] [CrossRef] [PubMed]
- Ravi, S.; Peña, K.A.; Chu, C.T.; Kiselyov, K. Biphasic regulation of lysosomal exocytosis by oxidative stress. Cell Calcium 2016, 60, 356–362. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Rydzewski, N.; Hider, A.; Zhang, X.; Yang, J.; Wang, W.; Gao, Q.; Cheng, X.; Xu, H. A molecular mechanism to regulate lysosome motility for lysosome positioning and tubulation. Nat. Cell Biol. 2016, 18, 404–417. [Google Scholar] [CrossRef] [PubMed]
- Machado, E.; White-Gilbertson, S.; van de Vlekkert, D.; Janke, L.; Moshiach, S.; Campos, Y.; Finkelstein, D.; Gomero, E.; Mosca, R.; Qiu, X.; et al. Regulated lysosomal exocytosis mediates cancer progression. Sci. Adv. 2015, 1, e1500603. [Google Scholar] [CrossRef] [PubMed]
- Morelli, M.B.; Nabissi, M.; Amantini, C.; Tomassoni, D.; Rossi, F.; Cardinali, C.; Santoni, M.; Arcella, A.; Oliva, M.A.; Santoni, A.; et al. Overexpression of transient receptor potential mucolipin-2 ion channels in gliomas: Role in tumor growth and progression. Oncotarget 2016, 7, 43654–43668. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grimm, C.; Bartel, K.; Vollmar, A.M.; Biel, M. Endolysosomal Cation Channels and Cancer—A Link with Great Potential. Pharmaceuticals 2018, 11, 4. https://doi.org/10.3390/ph11010004
Grimm C, Bartel K, Vollmar AM, Biel M. Endolysosomal Cation Channels and Cancer—A Link with Great Potential. Pharmaceuticals. 2018; 11(1):4. https://doi.org/10.3390/ph11010004
Chicago/Turabian StyleGrimm, Christian, Karin Bartel, Angelika M. Vollmar, and Martin Biel. 2018. "Endolysosomal Cation Channels and Cancer—A Link with Great Potential" Pharmaceuticals 11, no. 1: 4. https://doi.org/10.3390/ph11010004
APA StyleGrimm, C., Bartel, K., Vollmar, A. M., & Biel, M. (2018). Endolysosomal Cation Channels and Cancer—A Link with Great Potential. Pharmaceuticals, 11(1), 4. https://doi.org/10.3390/ph11010004