Semi-Solid and Solid Dosage Forms for the Delivery of Phage Therapy to Epithelia



Abstract
1. Introduction
2. Formulations of Semi-Solid and Solid Dosage Forms for Delivery to Epithelia
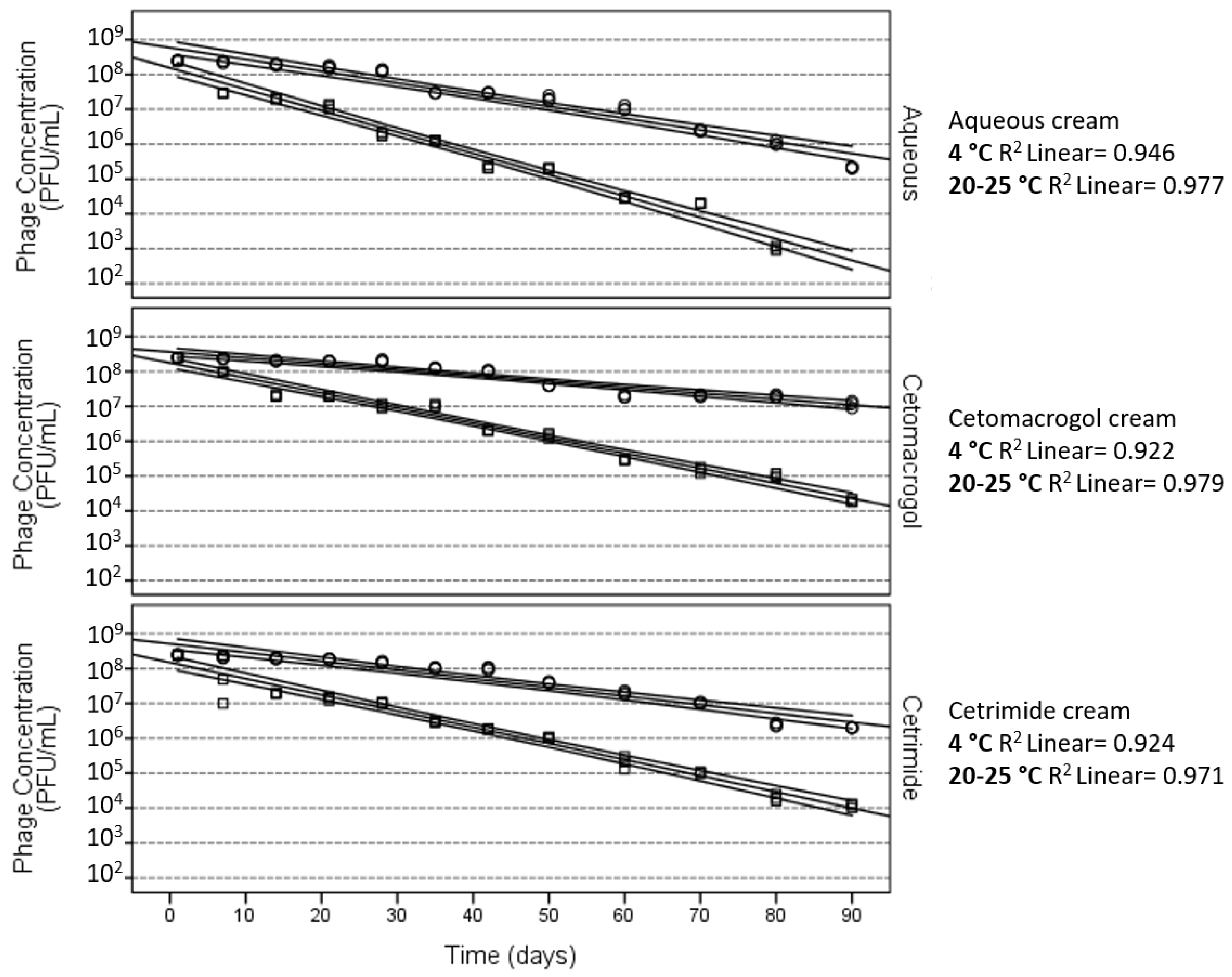
3. Stability of Phages in Semi-Solid Preparations
4. Effect of the Ionic Nature of the Semi-Solid Base
5. Lipophilicity of the Semi-Solid Base to Be Used for Phage Formulations
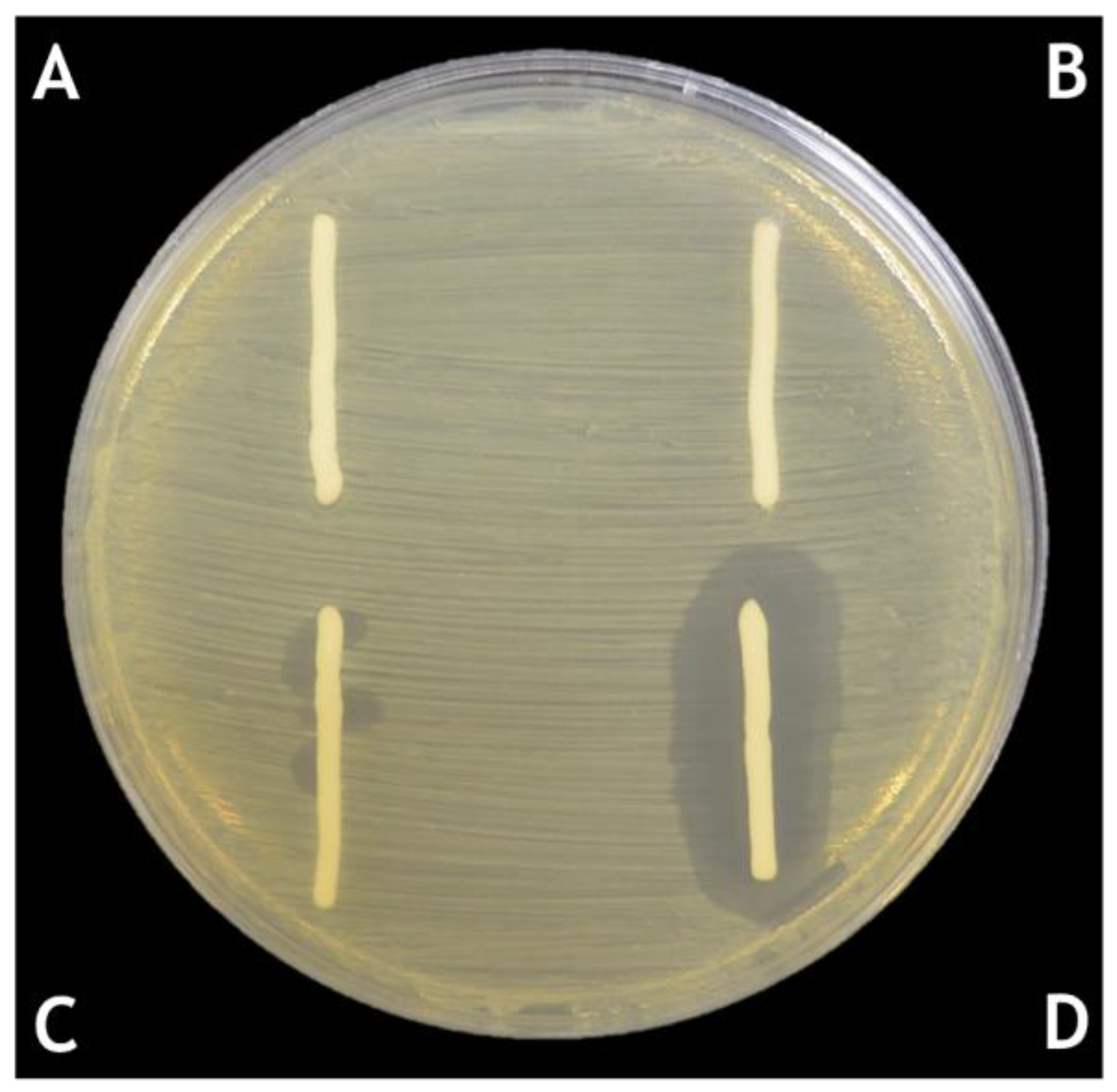
6. Solid Formulations for the Delivery of Phages
Author Contributions
Conflicts of Interest
References
- Spellberg, B.; Bartlett, J.G.; Gilbert, D.N. The future of antibiotics and resistance. N. Engl. J. Med. 2013, 368, 299–302. [Google Scholar] [CrossRef] [PubMed]
- Aryee, A.; Price, N. Antimicrobial stewardship—Can we afford to do without it? Br. J. Clin. Pharmacol. 2015, 79, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Cosgrove, S.E.; Carmeli, Y. The impact of antimicrobial resistance on health and economic outcomes. Clin. Infect. Dis. 2003, 36, 1433–1437. [Google Scholar] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef] [PubMed]
- Levy, S.B.; Marshall, B. Antibacterial resistance worldwide: Causes, challenges and responses. Nat. Med. 2004, 10, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Schuts, E.C.; Hulscher, M.E.; Mouton, J.W.; Verduin, C.M.; Stuart, J.W.; Overdiek, H.W.; van der Linden, P.D.; Natsch, S.; Hertogh, C.M.; Wolfs, T.F.; et al. Current evidence on hospital antimicrobial stewardship objectives: A systematic review and meta-analysis. Lancet Infect. Dis. 2016, 16, 847–856. [Google Scholar] [CrossRef]
- Sulakvelidze, A.; Alavidze, Z.; Morris, J.G., Jr. Bacteriophage therapy. Antimicrob. Agents Chemother. 2001, 45, 649–659. [Google Scholar] [CrossRef] [PubMed]
- Cisek, A.A.; Dabrowska, I.; Gregorczyk, K.P.; Wyzewski, Z. Phage therapy in bacterial infections treatment: One hundred years after the discovery of bacteriophages. Curr. Microbiol. 2017, 74, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Bruynoghe, R.; Maisin, J. Essais de therapeutique au moyen du bacteriophage du staphylocoque. J. Compt. Rend Soc. Biol. 1921, 85, 1020–1121. [Google Scholar]
- Chanishvili, N. Phage therapy-history from twort and d'herelle through soviet experience to current approaches. Adv. Virus Res. 2012, 83, 3–40. [Google Scholar] [PubMed]
- Debarbieux, L.; Pirnay, J.P.; Verbeken, G.; De Vos, D.; Merabishvili, M.; Huys, I.; Patey, O.; Schoonjans, D.; Vaneechoutte, M.; Zizi, M.; et al. A bacteriophage journey at the european medicines agency. Microbiol. Lett. 2016, 363. [Google Scholar] [CrossRef] [PubMed]
- Vandenheuvel, D.; Lavigne, R.; Brussow, H. Bacteriophage therapy: Advances in formulation strategies and human clinical trials. Annu. Rev. Virol. 2015, 2, 599–618. [Google Scholar] [CrossRef] [PubMed]
- Loc-Carrillo, C.; Abedon, S.T. Pros and cons of phage therapy. Bacteriophage 2011, 1, 111–114. [Google Scholar] [CrossRef] [PubMed]
- Kutter, E.; De Vos, D.; Gvasalia, G.; Alavidze, Z.; Gogokhia, L.; Kuhl, S.; Abedon, S.T. Phage therapy in clinical practice: Treatment of human infections. Curr. Pharm. Biotechnol. 2010, 11, 69–86. [Google Scholar] [CrossRef] [PubMed]
- NIH. New NIH Awards Will Support Development of Therapeutic Alternatives to Traditional Antibiotics. Available online: https://www.niaid.nih.gov/news-events/new-nih-awards-will-support-development-therapeutic-alternatives-traditional-antibiotics (accessed on 16 November 2017).
- Chaplin, S. Topical antibacterial and antiviral agents: Prescribing and resistance. Prescriber 2016, 27, 29–36. [Google Scholar] [CrossRef]
- Abedon, S.T.; Kuhl, S.J.; Blasdel, B.G.; Kutter, E.M. Phage treatment of human infections. Bacteriophage 2011, 1, 66–85. [Google Scholar] [CrossRef] [PubMed]
- Gu, J.; Liu, X.; Li, Y.; Han, W.; Lei, L.; Yang, Y.; Zhao, H.; Gao, Y.; Song, J.; Lu, R.; et al. A method for generation phage cocktail with great therapeutic potential. PLoS ONE 2012, 7, e31698. [Google Scholar] [CrossRef] [PubMed]
- Tothova, L.; Celec, P.; Babickova, J.; Gajdosova, J.; Al-Alami, H.; Kamodyova, N.; Drahovska, H.; Liptakova, A.; Turna, J.; Hodosy, J. Phage therapy of cronobacter-induced urinary tract infection in mice. Med. Sci. Monit. 2011, 17, BR173–BR178. [Google Scholar] [CrossRef] [PubMed]
- Dufour, N.; Clermont, O.; La Combe, B.; Messika, J.; Dion, S.; Khanna, V.; Denamur, E.; Ricard, J.D.; Debarbieux, L. Bacteriophage LM33_P1, a fast-acting weapon against the pandemic ST131-O25b:H4 Escherichia coli clonal complex. J. Antimicrob. Chemother. 2016, 71, 3072–3080. [Google Scholar] [CrossRef] [PubMed]
- Laanto, E.; Bamford, J.K.; Ravantti, J.J.; Sundberg, L.R. The use of phage FCL-2 as an alternative to chemotherapy against columnaris disease in aquaculture. Front. Microbiol. 2015, 6, 829. [Google Scholar] [CrossRef] [PubMed]
- Kishor, C.; Mishra, R.R.; Saraf, S.K.; Kumar, M.; Srivastav, A.K.; Nath, G. Phage therapy of staphylococcal chronic osteomyelitis in experimental animal model. Indian J. Med. Res. 2016, 143, 87–94. [Google Scholar] [PubMed]
- Gu, J.; Li, X.; Yang, M.; Du, C.; Cui, Z.; Gong, P.; Xia, F.; Song, J.; Zhang, L.; Li, J.; et al. Therapeutic effect of pseudomonas aeruginosa phage YH30 on mink hemorrhagic pneumonia. Vet. Microbiol. 2016, 190, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Keen, E.C. Phage therapy: Concept to cure. Front. Microbiol. 2012, 3, 238. [Google Scholar] [CrossRef] [PubMed]
- Cooper, C.J.; Khan Mirzaei, M.; Nilsson, A.S. Adapting drug approval pathways for bacteriophage-based therapeutics. Front. Microbiol. 2016, 7, 1209. [Google Scholar] [CrossRef] [PubMed]
- Brayfield, A. Martindale: The Complete Drug Reference; Pharmaceutical Press: London, UK, 2014. [Google Scholar]
- Allen, L.V. Remington: An. Introduction to Pharmacy; Pharmaceutical Press: London, UK, 2013. [Google Scholar]
- Aulton, M.E. Aulton's Pharmaceutics: The Design and Manufacture of Medicine; Churchill Livingstone: Edinburgh, IN, USA, 2007. [Google Scholar]
- British Pharmacopoeia Commission. British Pharmacopoeia; Stationery Office: London, UK, 2012. [Google Scholar]
- Alyami, H.; Dahmash, E.; Bowen, J.; Mohammed, A.R. An investigation into the effects of excipient particle size, blending techniques and processing parameters on the homogeneity and content uniformity of a blend containing low-dose model drug. PLoS ONE 2017, 12, e0178772. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.L.; Thomas, T.; Odgers, J.; Petrovski, S.; Spark, M.J.; Tucci, J. Bacteriophage formulated into a range of semisolid and solid dosage forms maintain lytic capacity against isolated cutaneous and opportunistic oral bacteria. J. Pharm. Pharmacol. 2017, 69, 244–253. [Google Scholar] [CrossRef] [PubMed]
- O'Flaherty, S.; Ross, R.; Meaney, W.; Fitzgerald, G.; Elbreki, M.; Coffey, A. Potential of the polyvalent anti-staphylococcus bacteriophage K for control of antibiotic-resistant staphylococci from hospitals. Appl. Environ. Microbiol. 2005, 71, 1836–1842. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-K.; Liu, Y.-L.; Hu, A.; Chang, K.-C.; Lin, N.-T.; Lai, M.-J.; Tseng, C.-C. Potential of bacteriophage ΦAB2 as an environmental biocontrol agent for the control of multidrug-resistant Acinetobacter baumannii. BMC Microbiol. 2013, 13, 154. [Google Scholar] [CrossRef] [PubMed]
- Merabishvili, M.; Monserez, R.; van Belleghem, J.; Rose, T.; Jennes, S.; De Vos, D.; Verbeken, G.; Vaneechoutte, M.; Pirnay, J.-P. Stability of bacteriophages in burn wound care products. PLoS ONE 2017, 12, e0182121. [Google Scholar] [CrossRef] [PubMed]
- Sansom, C. Phage therapy for severe infections tested in the first multicentre trial. Lancet Infect. Dis. 2015, 15, 1384–1385. [Google Scholar] [CrossRef]
- NIH. National Institutes of Health Clinical Trials. Available online: https://clinicaltrials.gov/ct2/show/NCT02116010?term=NCT02116010&rank=1 (accessed on 14 June 2017).
- Brown, T.L.; Petrovski, S.; Dyson, Z.A.; Seviour, R.; Tucci, J. The formulation of bacteriophage in a semi solid preparation for control of Propionibacterium acnes growth. PLoS ONE 2016, 11, e0151184. [Google Scholar] [CrossRef] [PubMed]
- Biemer, J.J. Antimicrobial susceptibility testing by the kirby-bauer disc diffusion method. Ann. Clin. Lab. Sci. 1973, 3, 135–140. [Google Scholar] [PubMed]
- Hudzicki, J. Kirby-Bauer Disk Diffusion Suseptibility Test Protocol. Available online: http://www.asmscience.org/content/education/protocol/protocol.3189 (Accessed on 18 December 2017).
- European Committee on Antimicrobial Suseptability Testing. Eucast Disk Diffusion Test Methodology. Available online: http://www.eucast.org/ast_of_bacteria/disk_diffusion_methodology/ (accessed on 18 December 2017).
- U.S. Department of Heath and Human Services Food and Drug Administration. Guide for Industry: Potency Test for Cellular and Gene Therapy Products. Available online: https://www.fda.gov/downloads/biologicsbloodvaccines/guidancecomplianceregulatoryinformation/guidances/cellularandgenetherapy/ucm243392.pdf (accessed on 18 December 2017).
- Brown, T.L.; Petrovski, S.; Hoyle, D.; Chan, H.T.; Lock, P.; Tucci, J. Characterization and formulation into solid dosage forms of a novel bacteriophage lytic against klebsiella oxytoca. PLoS ONE 2017, 12, e0183510. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.L.; Tucci, J.; Dyson, Z.A.; Lock, P.; Adda, C.G.; Petrovski, S. Dynamic interactions between prophages induce lysis in propionibacterium acnes. Res. Microbiol. 2017, 168, 103–112. [Google Scholar] [CrossRef] [PubMed]
- Todd, C. On the electrical behaviour of the bacteriophage. Brit. J. Exp. Path. 1927, 8, 369–376. [Google Scholar]
- Serwer, P.; Pichler, M.E. Electrophoresis of bacteriophage T7 and T7 capsids in agarose gels. J. Virol. 1978, 28, 917–928. [Google Scholar] [PubMed]
- Serwer, P.; Hayes, S.J. Agarose gel electrophoresis of bacteriophages and related particles. I. Avoidance of binding to the gel and recognizing of particles with packaged DNA. Electrophoresis 1982, 3, 76–80. [Google Scholar] [CrossRef]
- Cademartiri, R.; Anany, H.; Gross, I.; Bhayani, R.; Griffiths, M.; Brook, M.A. Immobilization of bacteriophages on modified silica particles. Biomaterials 2010, 31, 1904–1910. [Google Scholar] [CrossRef] [PubMed]
- Anany, H.; Chen, W.; Pelton, R.; Griffiths, M.W. Biocontrol of listeria monocytogenes and Escherichia coli O157:H7 in meat by using phages immobilized on modified cellulose membranes. Appl. Environ. Microbiol. 2011, 77, 6379–6387. [Google Scholar] [CrossRef] [PubMed]
- Piret, J.; Desormeaux, A.; Bergeron, M.G. Sodium lauryl sulfate, a microbicide effective against enveloped and nonenveloped viruses. Curr. Drug Targets 2002, 3, 17–30. [Google Scholar] [CrossRef] [PubMed]
- Sansom, L. Australian Pharmaceutical Formulary and Handbook, 23rd ed.; Pharmaceutical Society of Australia: Canberra, Australia, 2015; pp. 36–48. [Google Scholar]
- Abedon, S.T.; Yin, J. Bacteriophage plaques: Theory and analysis. Methods Mol. Biol. 2009, 501, 161–174. [Google Scholar] [PubMed]
- Thai, C.K.; Dai, H.; Sastry, M.S.; Sarikaya, M.; Schwartz, D.T.; Baneyx, F. Identification and characterization of cu(2)o- and zno-binding polypeptides by escherichia coli cell surface display: Toward an understanding of metal oxide binding. Biotechnol. Bioeng. 2004, 87, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kutateladze, M.; Adamia, R. Phage therapy experience at the eliava institute. Med. Mal. Infect. 2008, 38, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Ryan, E.M.; Gorman, S.P.; Donnelly, R.F.; Gilmore, B.F. Recent advances in bacteriophage therapy: How delivery routes, formulation, concentration and timing influence the success of phage therapy. J. Pharm. Pharmacol. 2011, 63, 1253–1264. [Google Scholar] [CrossRef] [PubMed]
- Fish, R.; Kutter, E.; Wheat, G.; Blasdel, B.; Kutateladze, M.; Kuhl, S. Bacteriophage treatment of intransigent diabetic toe ulcers: A case series. J. Wound Care 2016, 25, S27–S33. [Google Scholar] [CrossRef] [PubMed]
- Lyu, X.; Zhao, C.; Yan, Z.M.; Hua, H. Efficacy of nystatin for the treatment of oral candidiasis: A systematic review and meta-analysis. Drug Des. Devel. Ther. 2016, 10, 1161–1171. [Google Scholar] [CrossRef] [PubMed]
- Miedzybrodzki, R.; Klak, M.; Jonczyk-Matysiak, E.; Bubak, B.; Wojcik, A.; Kaszowska, M.; Weber-Dabrowska, B.; Lobocka, M.; Gorski, A. Means to facilitate the overcoming of gastric juice barrier by a therapeutic staphylococcal bacteriophage a5/80. Front. Microbiol. 2017, 8, 467. [Google Scholar] [CrossRef] [PubMed]
- Netzer, P.; Gaia, C.; Sandoz, M.; Huluk, T.; Gut, A.; Halter, F.; Husler, J.; Inauen, W. Effect of repeated injection and continuous infusion of omeprazole and ranitidine on intragastric pH over 72 h. Am. J. Gastroenterol. 1999, 94, 351–357. [Google Scholar] [PubMed]
- Tennant, S.M.; Hartland, E.L.; Phumoonna, T.; Lyras, D.; Rood, J.I.; Robins-Browne, R.M.; van Driel, I.R. Influence of gastric acid on susceptibility to infection with ingested bacterial pathogens. Infect. Immun. 2008, 76, 639–645. [Google Scholar] [CrossRef] [PubMed]
- Laheij, R.J.; Sturkenboom, M.C.; Hassing, R.J.; Dieleman, J.; Stricker, B.H.; Jansen, J.B. Risk of community-acquired pneumonia and use of gastric acid-suppressive drugs. JAMA 2004, 292, 1955–1960. [Google Scholar] [CrossRef] [PubMed]
- Malik, D.J.; Sokolov, I.J.; Vinner, G.K.; Mancuso, F.; Cinquerrui, S.; Vladisavljevic, G.T.; Clokie, M.R.J.; Garton, N.J.; Stapley, A.G.F.; Kirpichnikova, A. Formulation, stabilisation and encapsulation of bacteriophage for phage therapy. Adv. Colloid Interface Sci. 2017, 249, 100–133. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Pacan, J.C.; Wang, Q.; Xu, Y.; Huang, X.; Korenevsky, A.; Sabour, P.M. Microencapsulation of bacteriophage felix O1 into chitosan-alginate microspheres for oral delivery. Appl. Environ. Microbiol. 2008, 74, 4799–4805. [Google Scholar] [CrossRef] [PubMed]
- Koo, J.; DePaola, A.; Marshall, D.L. Effect of simulated gastric fluid and bile on survival of vibrio vulnificus and vibrio vulnificus phage. J. Food Prot. 2000, 63, 1665–1669. [Google Scholar] [CrossRef] [PubMed]
- Verthe, K.; Possemiers, S.; Boon, N.; Vaneechoutte, M.; Verstraete, W. Stability and activity of an enterobacter aerogenes-specific bacteriophage under simulated gastro-intestinal conditions. Appl. Microbiol. Biotechnol. 2004, 65, 465–472. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Cream (I) | Cream (J) | Mean Difference (I–J) | Standard Error | p-Value |
|---|---|---|---|---|
| Aqueous | Cetomacrogol | −1.329 | 0.080 | <0.001 |
| Aqueous | Cetrimide | −0.868 | 0.080 | <0.001 |
| Cetomacrogol | Cetrimide | 0.461 | 0.078 | <0.001 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Brown, T.L.; Petrovski, S.; Chan, H.T.; Angove, M.J.; Tucci, J. Semi-Solid and Solid Dosage Forms for the Delivery of Phage Therapy to Epithelia. Pharmaceuticals 2018, 11, 26. https://doi.org/10.3390/ph11010026
Brown TL, Petrovski S, Chan HT, Angove MJ, Tucci J. Semi-Solid and Solid Dosage Forms for the Delivery of Phage Therapy to Epithelia. Pharmaceuticals. 2018; 11(1):26. https://doi.org/10.3390/ph11010026
Chicago/Turabian StyleBrown, Teagan L., Steve Petrovski, Hiu Tat Chan, Michael J. Angove, and Joseph Tucci. 2018. "Semi-Solid and Solid Dosage Forms for the Delivery of Phage Therapy to Epithelia" Pharmaceuticals 11, no. 1: 26. https://doi.org/10.3390/ph11010026
APA StyleBrown, T. L., Petrovski, S., Chan, H. T., Angove, M. J., & Tucci, J. (2018). Semi-Solid and Solid Dosage Forms for the Delivery of Phage Therapy to Epithelia. Pharmaceuticals, 11(1), 26. https://doi.org/10.3390/ph11010026