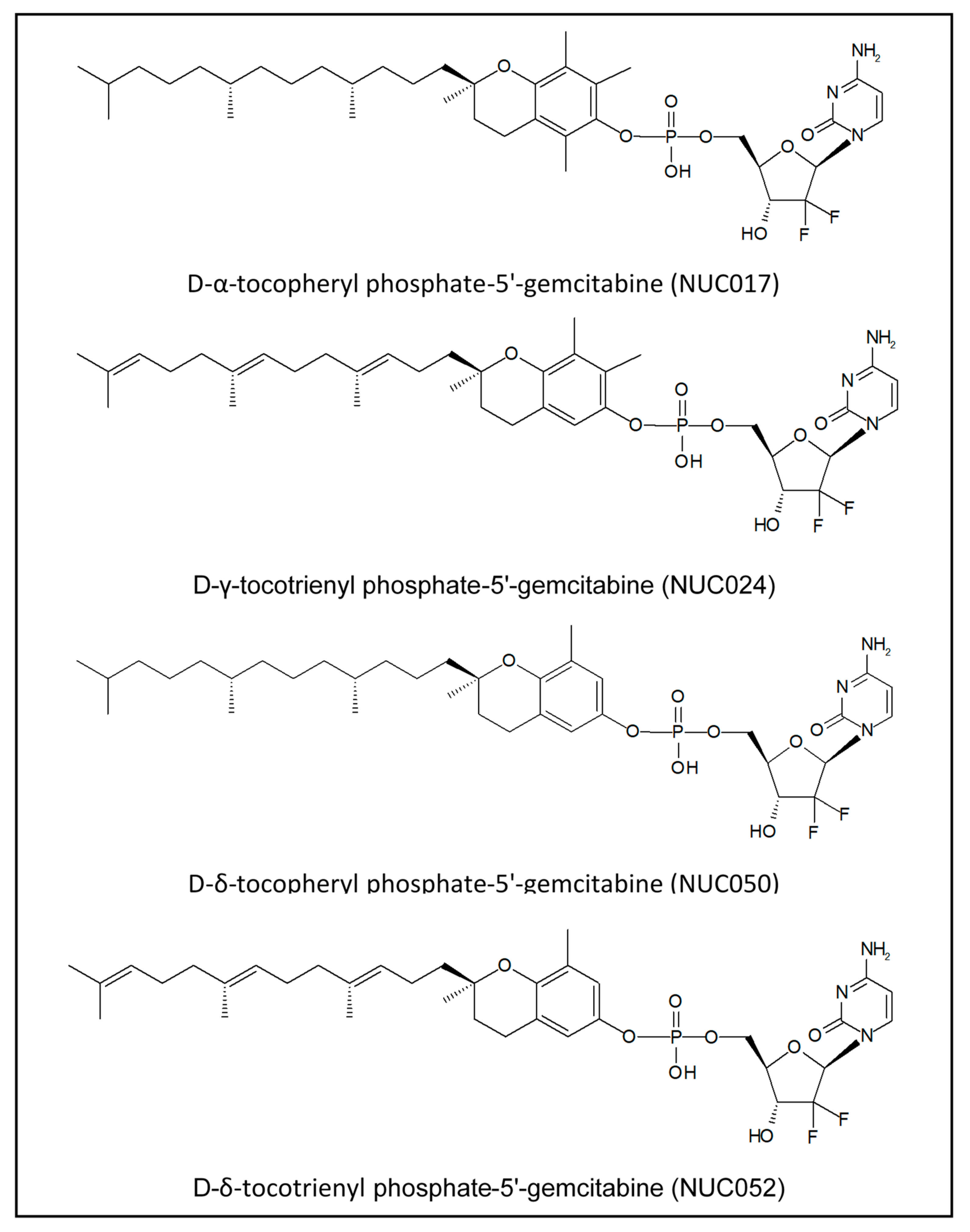
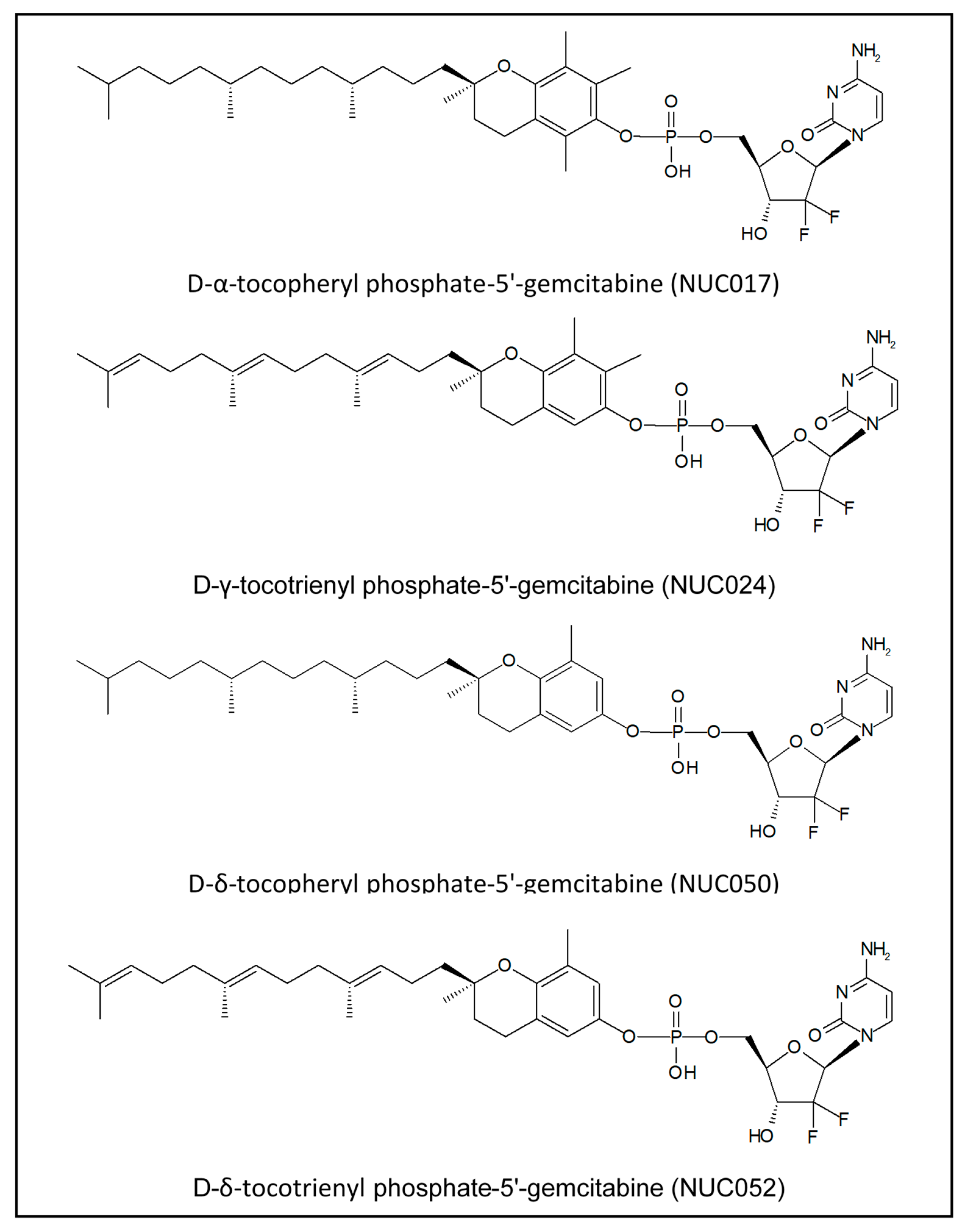
Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides

Abstract
:1. Introduction
2. Results
2.1. In Vitro Studies with VEP-Gemcitabine
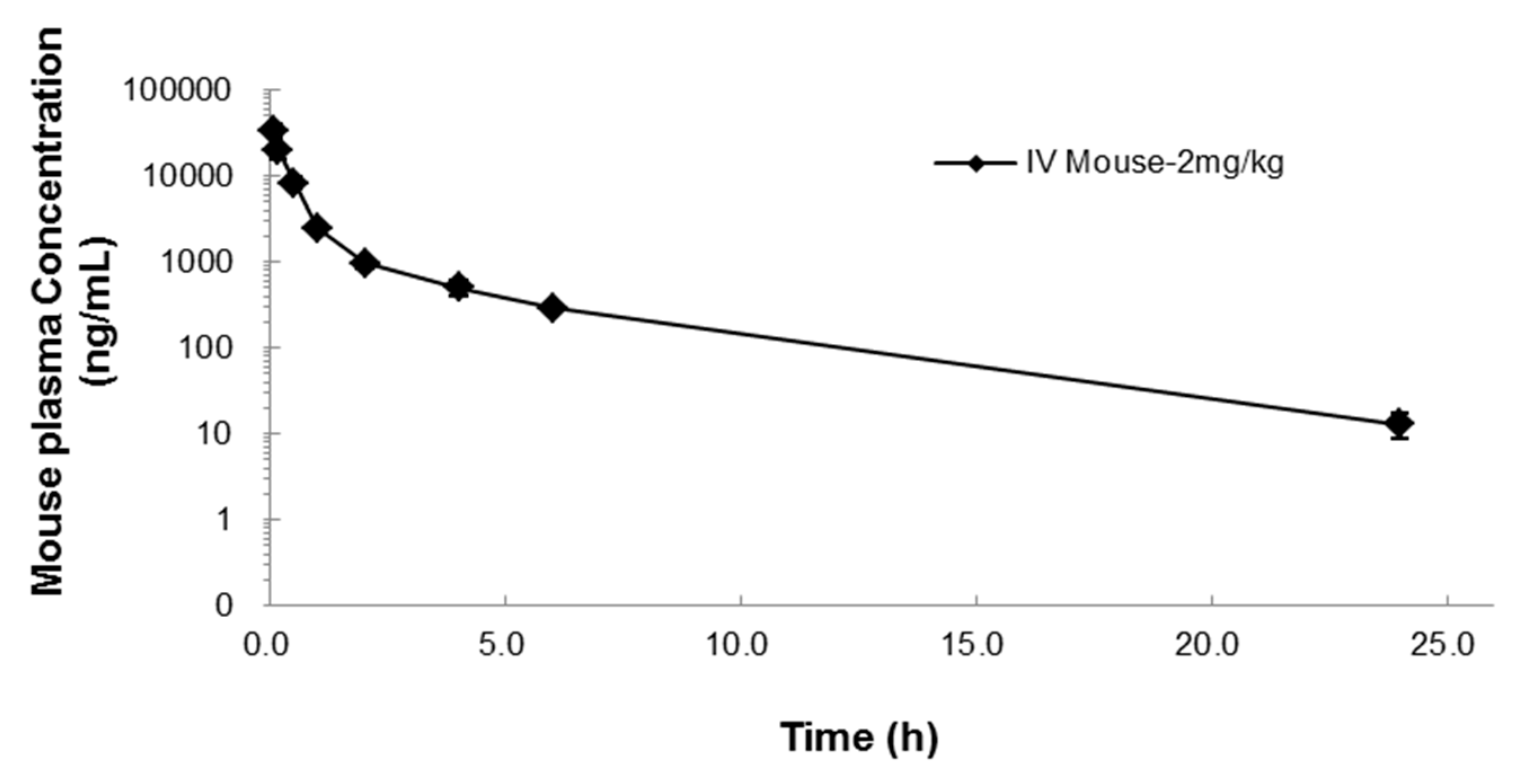
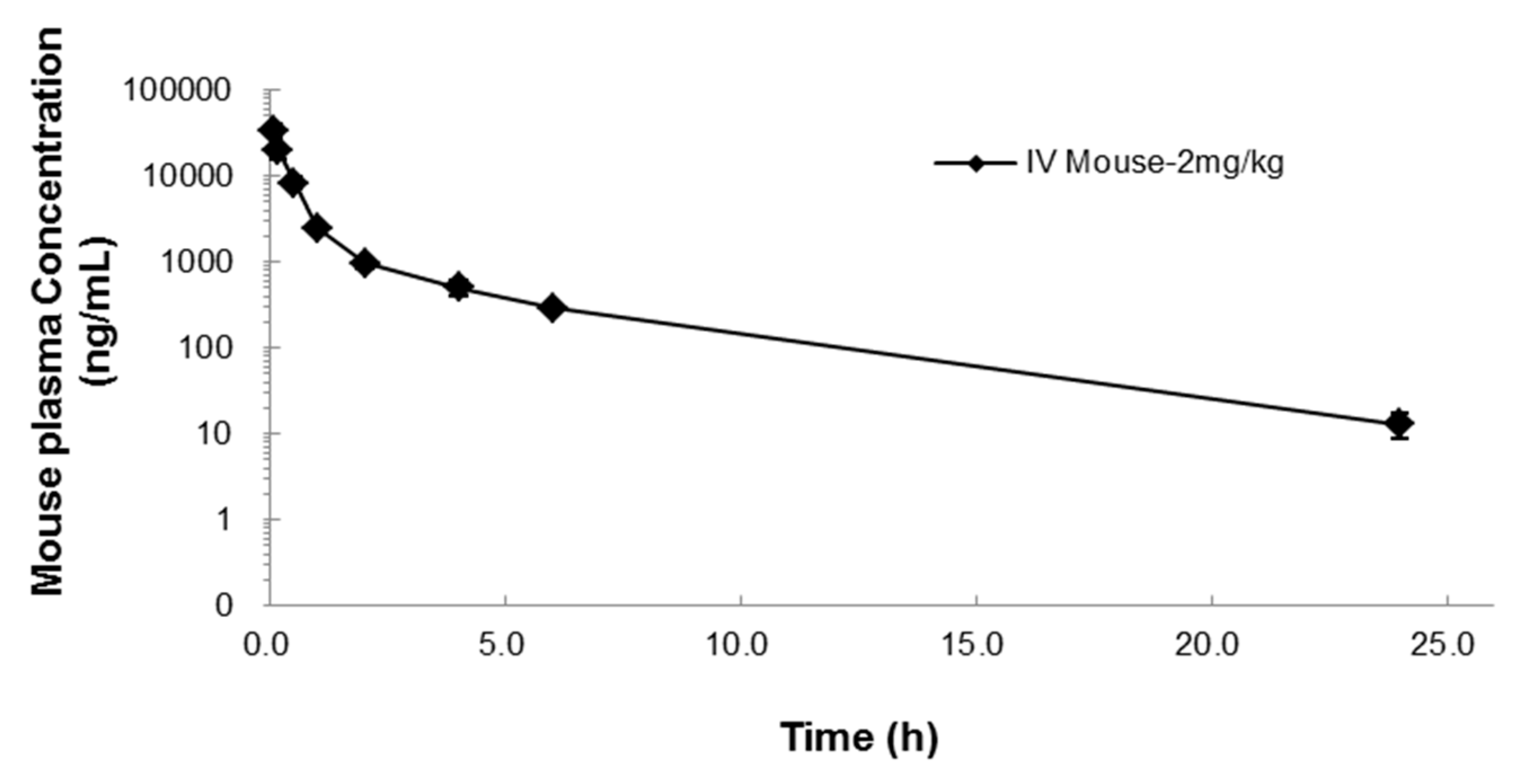
2.2. Pharmacokinetics of NUC050
2.3. Determination of NUC050 Maximum Tolerated Dose (MTD)
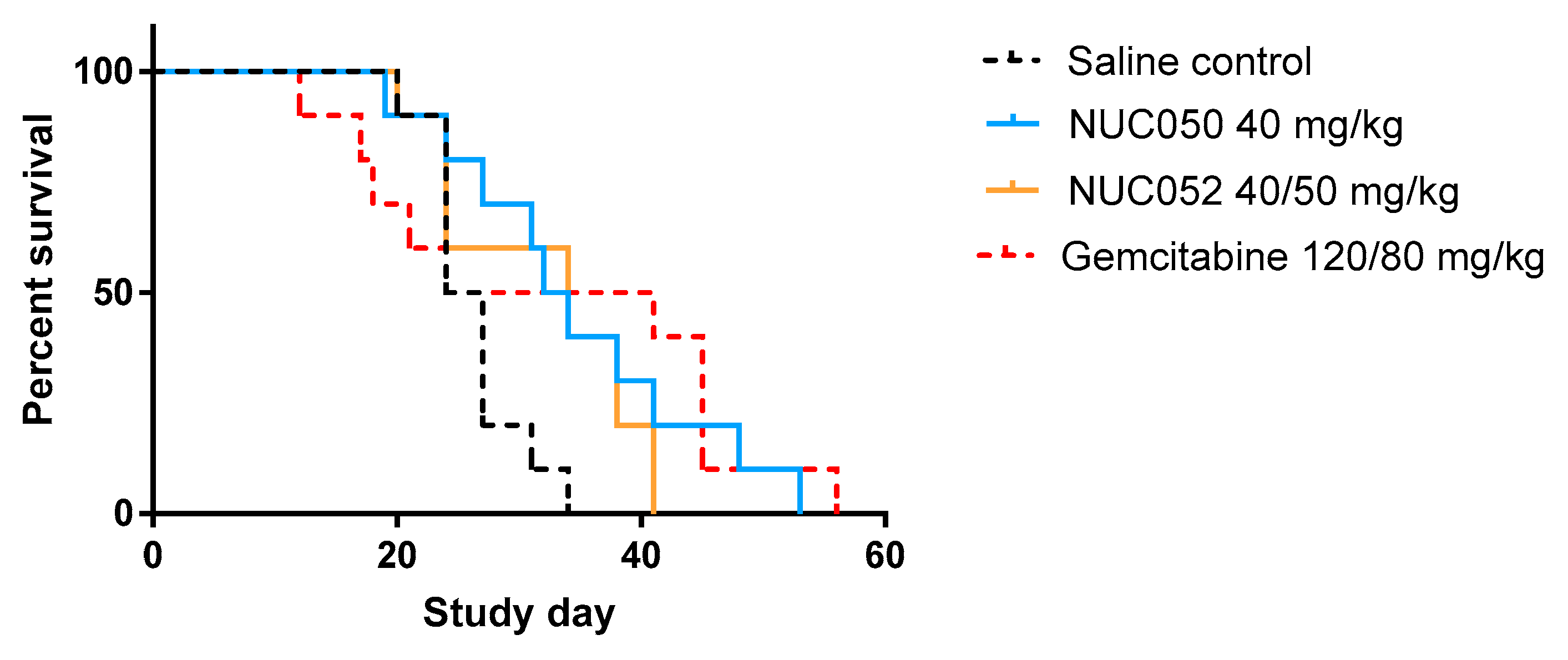
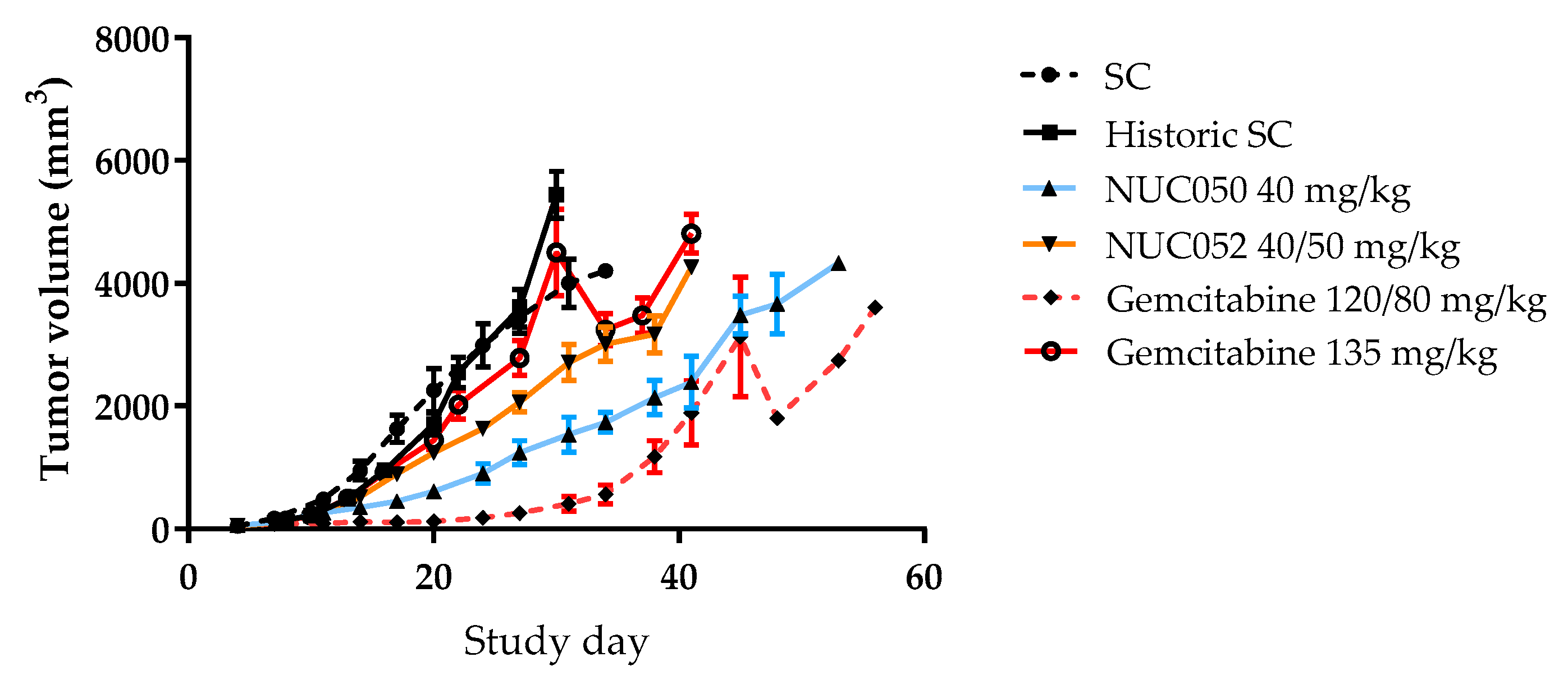
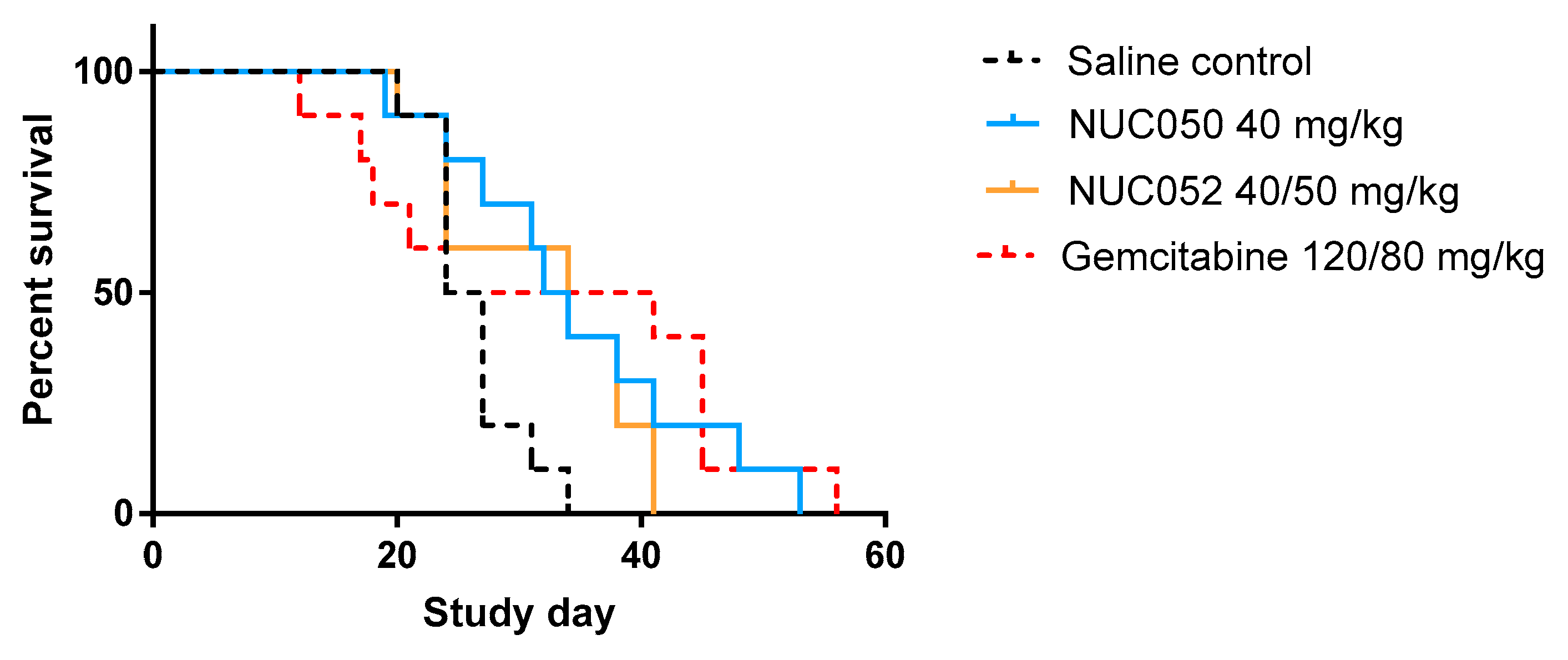
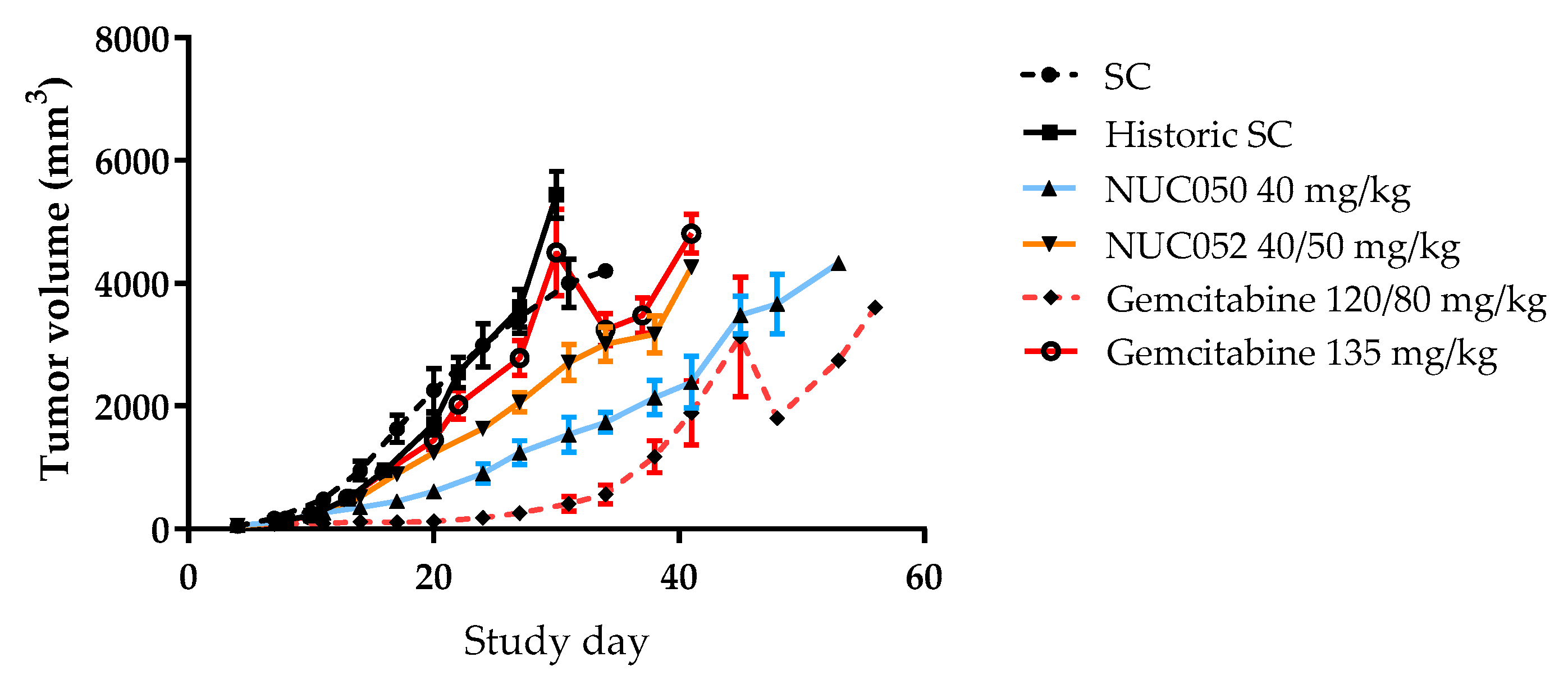
2.4. Testing of NUC050 and NUC052 in a Xenograft of Human NSCLC
3. Discussion
4. Materials and Methods
4.1. VEP-Gemcitabine Synthesis
4.2. Tumor Xenograft Studies
4.3. In Vitro Activity
4.4. Pharmacokinetic Study
4.5. Nano-Emulsion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Ciccolini, J.; Serdjebi, C.; Peters, G.J.; Giovanneti, E. Pharmacokinetics and pharmacogenetics of gemcitabine as a mainstay in adult and pediatric oncology: An EORTC-PAMM perspective. Cancer Chemother. Pharmacol. 2016, 78, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Momparler, R.L. Pharmacology of 5-Aza-2′-deoxycytidine (decitabine). Semin. Hematol. 2005, 42 (Suppl. 2), S9–S16. [Google Scholar] [CrossRef] [PubMed]
- Damaraju, V.L.; Damaraju, S.; Young, J.D.; Baldwin, S.A.; Mackey, J.; Sawyer, M.B.; Cass, C.E. Nucleoside anticancer drugs: The role of nucleoside transporters in resistance to cancer chemotherapy. Oncogene 2003, 22, 7524–7536. [Google Scholar] [CrossRef] [PubMed]
- Jordheim, L.P.; Galmarini, C.M.; Dumontet, C. Gemcitabine resistance due to deoxycytidine kinase deficiency can be reverted by fruitfly deoxynucleoside kinase, DmdNK, in human uterine sarcoma cells. Cancer Chemother. Pharmacol. 2006, 58, 547–554. [Google Scholar] [CrossRef] [PubMed]
- Bergman, A.M.; Adema, A.D.; Balzarini, J.; Bruheim, S.; Fichtner, I.; Noordhuis, P.; Fodstad, O.; Myhren, F.; Sandvold, M.L.; Hendriks, H.R.; et al. Antiproliferative activity, mechanism of action and oral antitumor activity of CP-4126, a fatty acid derivative of gemcitabine, in in vitro and in vivo tumor models. Investig. New Drugs. 2011, 29, 456–466. [Google Scholar] [CrossRef] [PubMed]
- Moysan, E.; Bastiat, G.; Benoit, J.P. Gemcitabine versus modified gemcitabine: A review of several promising chemical modifications. Mol. Pharm. 2013, 10, 430–444. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, W.; Sigmond, J.; Peters, G.J.; Borch, R.F. Synthesis and biological activity of a gemcitabine phosphoramidate prodrug. J. Med. Chem. 2007, 50, 3743–3746. [Google Scholar] [CrossRef] [PubMed]
- Slusarczyk, M.; Lopez, M.H.; Balzarini, J.; Mason, M.; Jiang, W.G.; Blagden, S.; Thompson, E.; Ghazaly, E.; McGuigan, C. Application of ProTide technology to gemcitabine: A successful approach to overcome the key cancer resistance mechanisms leads to a new agent (NUC-1031) in clinical development. J. Med. Chem. 2014, 57, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Sylvester, P.W.; Kaddoumi, A.; Nazzal, S.; El Sayed, K.A. The value of tocotrienols in the prevention and treatment of cancer. J. Am. Coll. Nutr. 2010, 29 (Suppl. 3), 324S–333S. [Google Scholar] [CrossRef] [PubMed]
- Smolarek, A.K.; Suh, N. Chemopreventive activity of vitamin E in breast cancer: A focus on γ- and δ-tocopherol. Nutrients 2011, 3, 962–986. [Google Scholar] [CrossRef] [PubMed]
- Ayoub, N.M.; Bachawal, S.V.; Sylvester, P.W. γ-Tocotrienol inhibits HGF-dependent mitogenesis and Met activation in highly malignant mammary tumour cells. Cell Prolif. 2011, 44, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Loganathan, R.; Selvaduray, K.R.; Nesaretnam, K.; Radhakrishnan, A.K. Tocotrienols promote apoptosis in human breast cancer cells by inducing poly(ADP-ribose) polymerase cleavage and inhibiting nuclear factor kappa-B activity. Cell Prolif. 2013, 46, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Inoue, A.; Takitani, K.; Koh, M.; Kawakami, C.; Kuno, T.; Tamai, H. Induction of apoptosis by γ-tocotrienol in human cancer cell lines and leukemic blasts from patients: Dependency on Bid, cytochrome c, and caspase pathway. Nutr. Cancer 2011, 63, 763–770. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.S.; Radhakrishnan, A.K.; Ibrahim, T.A.; Cheong, S.K. δ- and γ-tocotrienols induce classical ultrastructural apoptotic changes in human T lymphoblastic leukemic cells. Microsc. Microanal. 2012, 18, 462–469. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Li, F.; Manu, K.A.; Shanmugam, M.K.; Loo, S.Y.; Kumar, A.P.; Sethi, G. γ-Tocotrienol is a novel inhibitor of constitutive and inducible STAT3 signalling pathway in human hepatocellular carcinoma: Potential role as an antiproliferative, pro-apoptotic and chemosensitizing agent. Br. J. Pharmacol. 2011, 163, 283–298. [Google Scholar] [CrossRef] [PubMed]
- Kunnumakkara, A.B.; Sung, B.; Ravindran, J.; Diagaradjane, P.; Deorukhkar, A.; Dey, S.; Koca, C.; Yadav, V.R.; Tong, Z.; Gelovani, J.G.; et al. γ-tocotrienol inhibits pancreatic tumors and sensitizes them to gemcitabine treatment by modulating the inflammatory microenvironment. Cancer Res. 2010, 70, 8695–8705. [Google Scholar] [CrossRef] [PubMed]
- Husain, K.; Francois, R.A.; Yamauchi, T.; Perez, M.; Sebti, S.M.; Malafa, M.P. Vitamin E δ-tocotrienol augments the antitumor activity of gemcitabine and suppresses constitutive NF-κB activation in pancreatic cancer. Mol. Cancer Ther. 2011, 10, 2363–2372. [Google Scholar] [CrossRef] [PubMed]
- Yap, W.N.; Chang, P.N.; Han, H.Y.; Lee, D.T.; Ling, M.T.; Wong, Y.C.; Yap, Y.L. Gamma-tocotrienol suppresses prostate cancer cell proliferation and invasion through multiple-signalling pathways. Br. J. Cancer 2008, 99, 1832–1841. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, B.S.; Briski, K.P.; Gapor, A.; Sylvester, P.W. Antiproliferative and apoptotic effects of tocopherols and tocotrienols on preneoplastic and neoplastic mouse mammary epithelial cells. Proc. Soc. Exp. Biol. Med. 2000, 224, 292–301. [Google Scholar] [CrossRef] [PubMed]
- Ling, M.T.; Luk, S.U.; Al-Ejeh, F.; Khanna, K.K. Tocotrienol as a potential anticancer agent. Carcinogenesis 2012, 33, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Galmarini, C.M.; Mackey, J.R.; Dumontet, C. Nucleoside analogues: Mechanisms of drug resistance and reversal strategies. Leukemia 2001, 15, 875–890. [Google Scholar] [CrossRef] [PubMed]
- Shipley, L.A.; Brown, T.J.; Cornpropst, J.D.; Hamilton, M.; Daniels, W.D.; Culp, H.W. Metabolism and disposition of gemcitabine, and oncolytic deoxycytidine analog, in mice, rats, and dogs. Drug Metab. Dispos. 1992, 20, 849–855. [Google Scholar] [PubMed]
- Hamed, S.S.; Straubinger, R.M.; Jusko, W.J. Pharmacodynamic modeling of cell cycle and apoptotic effects of gemcitabine on pancreatic adenocarcinoma cells. Cancer Chemother. Pharmacol. 2013, 72, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Lee, Y.B.; Ahn, C.H.; Kaye, J.; Fine, T.; Kashi, R.; Ohne, O.; Smid, K.; Peters, G.J.; Kim, D.J. A novel cytidine analog, RX-3117, shows potent efficacy in xenograft models, even in tumors that are resistant to gemcitabine. Anticancer Res. 2014, 34, 6951–6959. [Google Scholar] [PubMed]
- Yoon, K.A.; Woo, S.M.; Hong, E.K.; Jung, M.K.; Park, W.S.; Bae, K.; Han, S.S.; Kim, T.H.; Koh, Y.H.; Park, S.J.; et al. Cytidine deaminase as a molecular predictor of gemcitabine response in patients with biliary tract cancer. Oncology 2015, 89, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Mahfouz, R.Z.; Jankowska, A.; Ebrahem, Q.; Gu, X.; Visconte, V.; Tabarroki, A.; Terse, P.; Covey, J.; Chan, K.; Ling, Y.; et al. Increased CDA expression/activity in males contributes to decreased cytidine analog half-life and likely contributes to worse outcomes with 5-azacytidine or decitabine therapy. Clin. Cancer Res. 2013, 19, 938–948. [Google Scholar] [CrossRef] [PubMed]
- Xue, Y.; Zhang, S.; Tang, M.; Zhang, T.; Wang, Y.; Hieda, Y.; Takeshita, H. Comparative study on toxic effects induced by oral or intravascular administration of commonly used disinfectants and surfactants in rats. J. Appl. Toxicol. 2012, 32, 480–487. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, H.; Watanabe, R.; Choyke, P.L. Improving conventional enhanced permeability and retention (EPR) effects; what is the appropriate target? Theranostics 2013, 4, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.S.; Dees, E.C.; Wang, A.Z. Nanoparticle-Delivered Chemotherapy: Old Drugs in New Packages. Oncology 2017, 31, 198–208. [Google Scholar] [PubMed]
- Clavis Pharma Announces Negative Outcome of Pivotal LEAP Trial with CP-4126 in Patients with Metastatic Pancreatic Cancer. Available online: http://www.evaluategroup.com/Universal/View.aspx?type=Story&id=374865 (accessed on 11 December 2017).
- U.S. National Library of Medicine. ClinicalTrials.gov. Available online: https://clinicaltrials.gov/ct2/results?cond=&term=nuc-1031&cntry=&state=&city=&dist=&Search=Search (accessed on 20 December 2017).
- Daifuku, R.; Hu, Z.; Saunthararajah, Y. 5-Aza-2′,2′-difluroro deoxycytidine (NUC013): A novel nucleoside DNA methyl transferase inhibitor and ribonucleotide reductase inhibitor for the treatment of cancer. Pharmaceuticals 2017, 10, 65. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Cell Line | |||
|---|---|---|---|
| Compound | Breast MDA-MB-231 (μM) | Non-Small Cell Lung NCI-H460 (μM) | Colon HCT-116 (μM) |
| Gemcitabine | 0.11 | 0.02 | 0.01 |
| α-tocopheryl phosphate | 23.40 | 52.24 | 48.86 |
| NUC017 | 22.70 | 23.75 | 26.13 |
| δ-tocopheryl phosphate | 29.56 | 69.67 | 70.58 |
| NUC050 | 5.08 | 1.69 | 3.67 |
| γ-tocotrienyl phosphate | 26.42 | 69.14 | 55.71 |
| NUC024 | 4.90 | 4.75 | 4.01 |
| GI50 (μM) | ||||||
|---|---|---|---|---|---|---|
| Breast MDA-MB-231 | Non-Small Cell Lung NCI-H460 | Colon HCT116 | ||||
| Compound | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) | DP (−) | DP (20 μM) |
| Gemcitabine | 3.08 | 56.77 | 0.02 | 0.82 | 0.03 | 2.39 |
| NUC050 | 17.16 | 23.30 | 2.14 | 1.47 | 3.07 | 6.74 |
| NUC024 | 30.34 | 27.77 | 7.16 | 15.98 | 5.55 | 12.61 |
| GI50 (μM) | ||
|---|---|---|
| Cell Line | Gemcitabine | NUC050 |
| CEM WT | 0.002 | 0.59 |
| CEM dCK (−) | 124.5 | 19.2 |
| T1/2 (h) | C0 (ng/mL) | AUClast (h·ng/mL) | AUCinf (h·ng/mL) | AUC Ext (%) | Vss (L/kg) | CL (mL/min/kg) | MRT (h) |
|---|---|---|---|---|---|---|---|
| 3.9 | 42,351 | 19,028 | 19,101 | 0.38 | 0.2 | 0.8 | 1.8 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Daifuku, R.; Koratich, M.; Stackhouse, M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals 2018, 11, 16. https://doi.org/10.3390/ph11010016
Daifuku R, Koratich M, Stackhouse M. Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals. 2018; 11(1):16. https://doi.org/10.3390/ph11010016
Chicago/Turabian StyleDaifuku, Richard, Michael Koratich, and Murray Stackhouse. 2018. "Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides" Pharmaceuticals 11, no. 1: 16. https://doi.org/10.3390/ph11010016
APA StyleDaifuku, R., Koratich, M., & Stackhouse, M. (2018). Vitamin E Phosphate Nucleoside Prodrugs: A Platform for Intracellular Delivery of Monophosphorylated Nucleosides. Pharmaceuticals, 11(1), 16. https://doi.org/10.3390/ph11010016