Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions


{kind=link}
Abstract
1. Introduction
2. Persistence as an Evolutionary Adaptation
3. Biofilms Can Promote Antibiotic Persistence in Clinical Settings
4. Growth, Metabolism, and ATP Production
5. Carbon Catabolite Repression Systems Coordinate Antibiotic Persistence and Tolerance
6. Sugar Metabolism and the Eradication of Persisters
7. Cellular Permeability, Proton Motive Force, and Persistence
8. Stress Responses and Persistence: The Stringent Response
9. Stress Responses and Persistence: The SOS Response
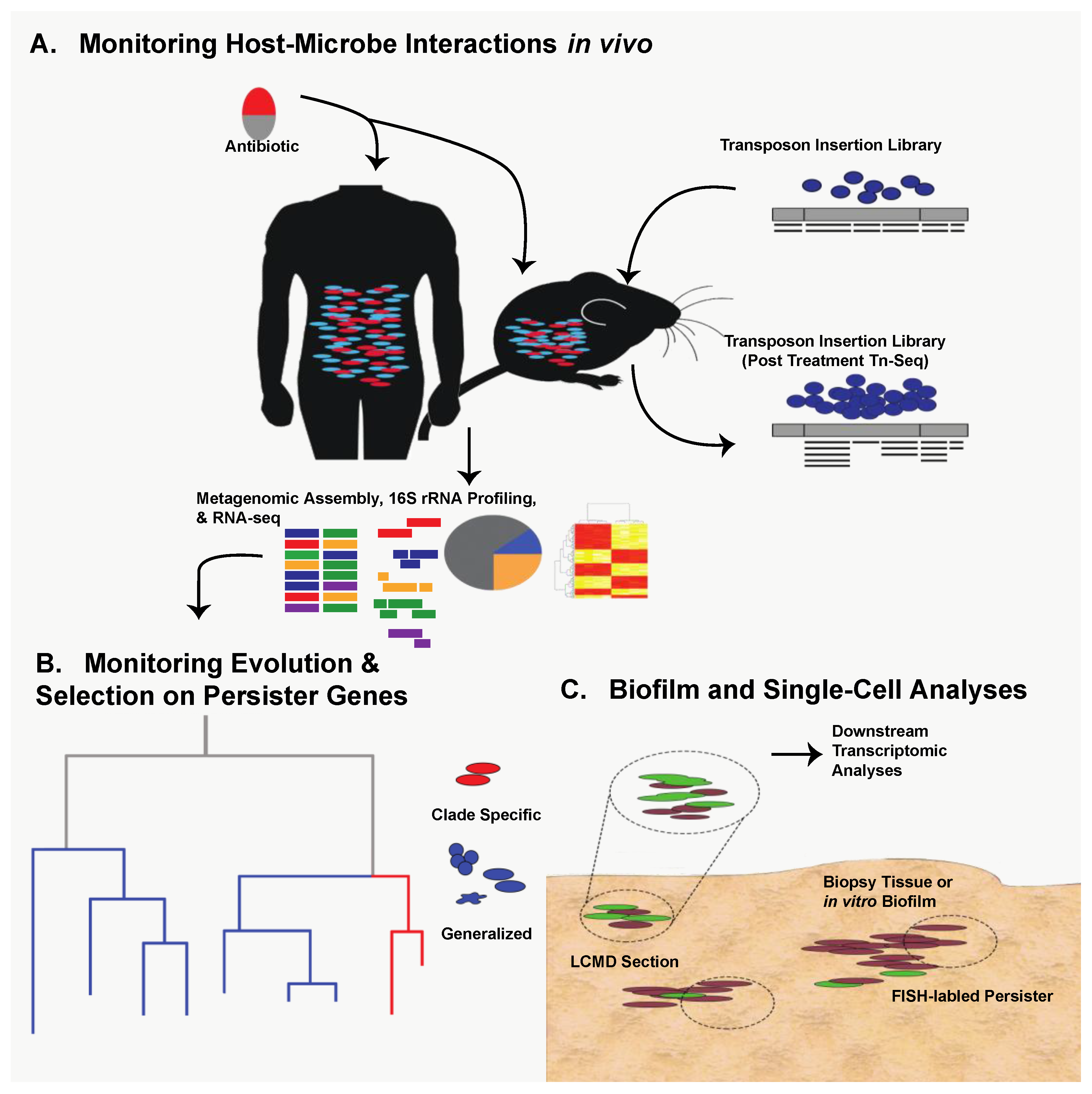
10. Future Directions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Michiels, J.E.; Van den Bergh, B.; Verstraeten, N.; Michiels, J. Molecular mechanisms and clinical implications of bacterial persistence. Drug Resist. Updat. 2016, 29, 76–89. [Google Scholar] [CrossRef] [PubMed]
- Ventola, C.L. The Antibiotic Resistance Crisis. Pharm. Ther. 2015, 40, 277–283. [Google Scholar]
- Bigger, J.W. Treatment of Staphylococcal Infections with Penicillin by Intermittent Sterilisation. Lancet 1944, 244, 497–500. [Google Scholar] [CrossRef]
- Brauner, A.; Fridman, O.; Gefen, O.; Balaban, N.Q. Distinguishing between resistance, tolerance and persistence to antibiotic treatment. Nat. Rev. Microbiol. 2016, 14, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Olive, A.J.; Sassetti, C.M. Metabolic crosstalk between host and pathogen: Sensing, adapting and competing. Nat. Rev. Microbiol. 2016, 14, 221–234. [Google Scholar] [CrossRef] [PubMed]
- Harms, A.; Maisonneuve, E.; Gerdes, K. Mechanisms of bacterial persistence during stress and antibiotic exposure. Science 2016, 354, aaf4268. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells. Annu. Rev. Microbiol. 2010, 64, 357–372. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Persister cells, dormancy and infectious disease. Nat. Rev. Microbiol. 2007, 5, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [PubMed]
- Balaban, N.Q.; Merrin, J.; Chait, R.; Kowalik, L.; Leibler, S. Bacterial persistence as a phenotypic switch. Science 2004, 305, 1622–1625. [Google Scholar] [CrossRef] [PubMed]
- Nandakumar, M.; Nathan, C.; Rhee, K.Y. Isocitrate lyase mediates broad antibiotic tolerance in Mycobacterium tuberculosis. Nat. Commun. 2014, 5, 4306. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.S.; Hung, D.T. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 2014, 4, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Rotem, E.; Loinger, A.; Ronin, I.; Levin-Reisman, I.; Gabay, C.; Shoresh, N.; Biham, O.; Balaban, N.Q. Regulation of phenotypic variability by a threshold-based mechanism underlies bacterial persistence. Proc. Natl. Acad. Sci. USA 2010, 107, 12541–12546. [Google Scholar] [CrossRef] [PubMed]
- Keren, I.; Kaldalu, N.; Spoering, A.; Wang, Y.P.; Lewis, K. Persister cells and tolerance to antimicrobials. FEMS Microbiol. Lett. 2004, 230, 13–18. [Google Scholar] [CrossRef]
- Levin, B.R.; Rozen, D.E. Non-inherited antibiotic resistance. Nat. Rev. Microbiol. 2006, 4, 556–562. [Google Scholar] [CrossRef] [PubMed]
- MacLean, R.C.; Vogwill, T. Limits to compensatory adaptation and the persistence of antibiotic resistance in pathogenic bacteria. Evol. Med. Public Health 2014, 2015, 4–12. [Google Scholar] [CrossRef] [PubMed]
- Vogwill, T.; Comfort, A.C.; Furió, V.; MacLean, R.C. Persistence and resistance as complementary bacterial adaptations to antibiotics. J. Evol. Biol. 2016, 29, 1223–1233. [Google Scholar] [CrossRef] [PubMed]
- Valentino, M.D.; Foulston, L.; Sadaka, A.; Kos, V.N.; Villet, R.A.; Santa Maria, J.; Lazinski, D.W.; Camilli, A.; Walker, S.; Hooper, D.C.; et al. Genes contributing to Staphylococcus aureus fitness in abscess- and infection-related ecologies. mBio 2014, 5, e01729-14. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Leonard, S.P.; Kwong, W.K.; Engel, P.; Moran, N.A. Genome-wide screen identifies host colonization determinants in a bacterial gut symbiont. Proc. Natl. Acad. Sci. USA 2016, 113, 13887–13892. [Google Scholar] [CrossRef] [PubMed]
- Santiago, M.; Matano, L.M.; Moussa, S.H.; Gilmore, M.S.; Walker, S.; Meredith, T.C. A new platform for ultra-high density Staphylococcus aureus transposon libraries. BMC Genomics 2015, 16, 252. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K. Riddle of biofilm resistance. Antimicrob. Agents Chemother. 2001, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Cohen, N.R.; Lobritz, M.A.; Collins, J.J. Microbial persistence and the road to drug resistance. Cell Host Microbe 2013, 13, 632–642. [Google Scholar] [CrossRef] [PubMed]
- Goswami, M.; Bhattacharyya, P.; Tribedi, P. Allee effect: The story behind the stabilization or extinction of microbial ecosystem. Arch. Microbiol. 2017, 199, 185–190. [Google Scholar] [CrossRef] [PubMed]
- Davey, M.E.; O’toole, G.A. Microbial biofilms: From ecology to molecular genetics. Microbiol. Mol. Biol. Rev. 2000, 64, 847–867. [Google Scholar] [CrossRef] [PubMed]
- Des Roches, S.; Post, D.M.; Turley, N.E.; Bailey, J.K.; Hendry, A.P.; Kinnison, M.T.; Schweitzer, J.A.; Palkovacs, E.P. The ecological importance of intraspecific variation. Nat. Ecol. Evol. 2018, 2, 57–64. [Google Scholar] [CrossRef] [PubMed]
- Goneau, L.W.; Yeoh, N.S.; MacDonald, K.W.; Cadieux, P.A.; Burton, J.P.; Razvi, H.; Reid, G. Selective Target Inactivation Rather than Global Metabolic Dormancy Causes Antibiotic Tolerance in Uropathogens. Antimicrob. Agents Chemother. 2014, 58, 2089–2097. [Google Scholar] [CrossRef] [PubMed]
- Perez, A.C.; Pang, B.; King, L.B.; Tan, L.; Murrah, K.A.; Reimche, J.L.; Wren, J.T.; Richardson, S.H.; Ghandi, U.; Swords, W.E. Residence of Streptococcus pneumoniae and Moraxella catarrhaliswithin polymicrobial biofilm promotes antibiotic resistance and bacterial persistence in vivo. Pathog. Dis. 2014, 70, 280–288. [Google Scholar] [CrossRef] [PubMed]
- Lebeaux, D.; Ghigo, J.M.; Beloin, C. Biofilm-Related Infections: Bridging the Gap between Clinical Management and Fundamental Aspects of Recalcitrance toward Antibiotics. Microbiol. Mol. Biol. Rev. 2014, 78, 510–543. [Google Scholar] [CrossRef] [PubMed]
- Dörr, T.; Vulic, M.; Lewis, K. Ciprofloxacin causes persister formation by inducing the TisB toxin in Escherichia coli. PLoS Biol. 2010, 8, e1000317. [Google Scholar] [CrossRef] [PubMed]
- Kudrin, P.; Varik, V.; Oliveira, S.R.A.; Beljantseva, J.; Del Peso Santos, T.; Dzhygyr, I.; Rejman, D.; Cava, F.; Tenson, T.; Hauryliuk, V. Subinhibitory Concentrations of Bacteriostatic Antibiotics Induce relA-Dependent and relA-Independent Tolerance to β-Lactams. Antimicrob. Agents Chemother. 2017, 61, e02173-16. [Google Scholar] [CrossRef] [PubMed]
- Shah, D.; Zhang, Z.; Khodursky, A.; Kaldalu, N.; Kurg, K.; Lewis, K. Persisters: A distinct physiological state of E. coli. BMC Microbiol. 2006, 6, 53. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.H.; Molla, M.N.; Cantor, C.R.; Collins, J.J. Bacterial charity work leads to population-wide resistance. Nature 2010, 467, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Jefferson, K.K. What drives bacteria to produce a biofilm? FEMS Microbiol. Lett. 2004, 236, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Blango, M.G.; Mulvey, M.A. Persistence of Uropathogenic Escherichia coli in the Face of Multiple Antibiotics. Antimicrob. Agents Chemother. 2010, 54, 1855–1863. [Google Scholar] [CrossRef] [PubMed]
- Parsek, M.R.; Singh, P.K. Bacterial Biofilms: An Emerging Link to Disease Pathogenesis. Annu. Rev. Microbiol. 2003, 57, 677–701. [Google Scholar] [CrossRef] [PubMed]
- Urish, K.L.; DeMuth, P.W.; Kwan, B.W.; Craft, D.W.; Ma, D.; Haider, H.; Tuan, R.S.; Wood, T.K.; Davis, C.M. Antibiotic-tolerant Staphylococcus aureus Biofilm Persists on Arthroplasty Materials. Clin. Orthop. Relat. Res. 2016, 474, 1649–1656. [Google Scholar] [CrossRef] [PubMed]
- Conlon, B.P.; Rowe, S.E.; Gandt, A.B.; Nuxoll, A.S.; Donegan, N.P.; Zalis, E.A.; Clair, G.; Adkins, J.N.; Cheung, A.L.; Lewis, K. Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 2016, 1, 16051. [Google Scholar] [CrossRef] [PubMed]
- Nistico, L.; Gieseke, A.; Stoodley, P.; Hall-Stoodley, L.; Kerschner, J.E.; Ehrlich, G.D. Fluorescence “in situ” hybridization for the detection of biofilm in the middle ear and upper respiratory tract mucosa. Methods Mol. Biol. 2009, 493, 191–213. [Google Scholar] [CrossRef] [PubMed]
- Brileya, K.A.; Camilleri, L.B.; Fields, M.W. 3D-fluorescence in situ hybridization of intact, anaerobic biofilm. Methods Mol. Biol. 2014, 1151, 189–197. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Brynildsen, M.P. Establishment of a method to rapidly assay bacterial persister metabolism. Antimicrob. Agents Chemother. 2013, 57, 4398–4409. [Google Scholar] [CrossRef] [PubMed]
- Henry, T.C.; Brynildsen, M.P. Development of Persister-FACSeq: A method to massively parallelize quantification of persister physiology and its heterogeneity. Sci. Rep. 2016, 6, 25100. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.-E.C.; Santos, S.; Vogel, J. New RNA-seq approaches for the study of bacterial pathogens. Curr. Opin. Microbiol. 2017, 35, 78–87. [Google Scholar] [CrossRef] [PubMed]
- Saliba, A.-E.; Li, L.; Westermann, A.J.; Appenzeller, S.; Stapels, D.A.C.; Schulte, L.N.; Helaine, S.; Vogel, J. Single-cell RNA-seq ties macrophage polarization to growth rate of intracellular Salmonella. Nat. Microbiol. 2016, 2, 16206. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Haseley, N.; Brown, D.; Penaranda, C.; Jijon, H.B.; Trombetta, J.J.; Satija, R.; Shalek, A.K.; Xavier, R.J.; Regev, A.; et al. Pathogen Cell-to-Cell Variability Drives Heterogeneity in Host Immune Responses. Cell 2015, 162, 1309–1321. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.; McMillan, I.; Norris, M.H.; Hoang, T.T. Single prokaryotic cell isolation and total transcript amplification protocol for transcriptomic analysis. Nat. Protoc. 2015, 10, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Heacock-Kang, Y.; Sun, Z.; Zarzycki-Siek, J.; McMillan, I.A.; Norris, M.H.; Bluhm, A.P.; Cabanas, D.; Fogen, D.; Vo, H.; Donachie, S.P.; et al. Spatial transcriptomes within the Pseudomonas aeruginosa biofilm architecture. Mol. Microbiol. 2017, 106, 976–985. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Chen, L.; Chen, Z.; Zhang, W. RNA-seq based transcriptomic analysis of single bacterial cells. Integr. Biol. (Camb.) 2015, 7, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Hör, J.; Gorski, S.A.; Vogel, J. Bacterial RNA Biology on a Genome Scale. Mol. Cell 2018. [Google Scholar] [CrossRef] [PubMed]
- Walters, M.C.; Roe, F.; Bugnicourt, A.; Franklin, M.J.; Stewart, P.S. Contributions of Antibiotic Penetration, Oxygen Limitation, and Low Metabolic Activity to Tolerance of Pseudomonas aeruginosa Biofilms to Ciprofloxacin and Tobramycin. Antimicrob. Agents Chemother. 2003, 47, 317–323. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, G.; Shigeta, M.; Komatsuzawa, H.; Sugai, M.; Suginaka, H.; Usui, T. Effect of the Growth Rate of Pseudomonas aeruginosa Biofilms on the Susceptibility to Antimicrobial Agents: β-Lactams and Fluoroquinolones. Chemotherapy 1999, 45, 28–36. [Google Scholar] [CrossRef] [PubMed]
- Jõers, A.; Kaldalu, N.; Tenson, T. The frequency of persisters in Escherichia coli reflects the kinetics of awakening from dormancy. J. Bacteriol. 2010, 192, 3379–3384. [Google Scholar] [CrossRef] [PubMed]
- Lobritz, M.A.; Belenky, P.; Porter, C.B.M.; Gutierrez, A.; Yang, J.H.; Schwarz, E.G.; Dwyer, D.J.; Khalil, A.S.; Collins, J.J. Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 2015, 112, 8173–8180. [Google Scholar] [CrossRef] [PubMed]
- Betts, J.C.; Lukey, P.T.; Robb, L.C.; McAdam, R.A.; Duncan, K. Evaluation of a nutrient starvation model of Mycobacterium tuberculosis persistence by gene and protein expression profiling. Mol. Microbiol. 2002, 43, 717–731. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.; Lemire, J.; Mailloux, R.J.; Chenier, D.; Hamel, R.; Appanna, V.D. An ATP and Oxalate Generating Variant Tricarboxylic Acid Cycle Counters Aluminum Toxicity in Pseudomonas fluorescens. PLoS ONE 2009, 4. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez, A.; Jain, S.; Bhargava, P.; Hamblin, M.; Lobritz, M.A.; Collins, J.J. Understanding and Sensitizing Density-Dependent Persistence to Quinolone Antibiotics. Mol. Cell 2017, 68, 1147–1154.e3. [Google Scholar] [CrossRef] [PubMed]
- Fung, D.K.C.; Chan, E.W.C.; Chin, M.L.; Chan, R.C.Y. Delineation of a bacterial starvation stress response network which can mediate antibiotic tolerance development. Antimicrob. Agents Chemother. 2010, 54, 1082–1093. [Google Scholar] [CrossRef] [PubMed]
- Fridman, O.; Goldberg, A.; Ronin, I.; Shoresh, N.; Balaban, N.Q. Optimization of lag time underlies antibiotic tolerance in evolved bacterial populations. Nature 2014, 513, 418–421. [Google Scholar] [CrossRef] [PubMed]
- Amato, S.M.; Orman, M.A.; Brynildsen, M.P. Metabolic control of persister formation in Escherichia coli. Mol. Cell 2013, 50, 475–487. [Google Scholar] [CrossRef] [PubMed]
- Bernier, S.P.; Lebeaux, D.; DeFrancesco, A.S.; Valomon, A.; Soubigou, G.; Coppée, J.-Y.; Ghigo, J.-M.; Beloin, C. Starvation, together with the SOS response, mediates high biofilm-specific tolerance to the fluoroquinolone ofloxacin. PLoS Genet. 2013, 9, e1003144. [Google Scholar] [CrossRef] [PubMed]
- Bailey, A.M.; Webber, M.A.; Piddock, L.J.V. Medium plays a role in determining expression of acrB, marA, and soxS in Escherichia coli. Antimicrob. Agents Chemother. 2006, 50, 1071–1074. [Google Scholar] [CrossRef] [PubMed]
- Kornberg, H.L.; Madsen, N.B. Synthesis of C4-dicarboxylic acids from acetate by a “glyoxylate bypass” of the tricarboxylic acid cycle. Biochim. Biophys. Acta 1957, 24, 651–653. [Google Scholar] [CrossRef]
- Kornberg, H.L.; KREBS, H.A. Synthesis of Cell Constituents from C2-Units by a Modified Tricarboxylic Acid Cycle. Nature 1957, 179, 988–991. [Google Scholar] [CrossRef] [PubMed]
- Dunn, M.F.; Ramírez-Trujillo, J.A.; Hernández-Lucas, I. Major roles of isocitrate lyase and malate synthase in bacterial and fungal pathogenesis. Microbiology 2009, 155, 3166–3175. [Google Scholar] [CrossRef] [PubMed]
- Ahn, S.; Jung, J.; Jang, I.-A.; Madsen, E.L.; Park, W. Role of Glyoxylate Shunt in Oxidative Stress Response. J. Biol. Chem. 2016, 291, 11928–11938. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Zhang, Y. PhoU is a persistence switch involved in persister formation and tolerance to multiple antibiotics and stresses in Escherichia coli. Antimicrob. Agents Chemother. 2007, 51, 2092–2099. [Google Scholar] [CrossRef] [PubMed]
- Orman, M.A.; Brynildsen, M.P. Dormancy is not necessary or sufficient for bacterial persistence. Antimicrob. Agents Chemother. 2013, 57, 3230–3239. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Bhargava, P.; McCloskey, D.; Mao, N.; Palsson, B.O.; Collins, J.J. Antibiotic-Induced Changes to the Host Metabolic Environment Inhibit Drug Efficacy and Alter Immune Function. Cell Host Microbe 2017, 22, 757–765.e3. [Google Scholar] [CrossRef] [PubMed]
- Carvalho, S.M.; Kloosterman, T.G.; Kuipers, O.P.; Neves, A.R. CcpA Ensures Optimal Metabolic Fitness of Streptococcus pneumoniae. PLoS ONE 2011, 6, e26707. [Google Scholar] [CrossRef] [PubMed]
- Görke, B.; Stülke, J. Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nat. Rev. Microbiol. 2008, 6, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Bizzini, A.; Entenza, J.M.; Moreillon, P. Loss of penicillin tolerance by inactivating the carbon catabolite repression determinant CcpA in Streptococcus gordonii. J. Antimicrob. Chemother. 2007, 59, 607–615. [Google Scholar] [CrossRef] [PubMed]
- Willenborg, J.R.; Willms, D.; Bertram, R.; Goethe, R.; Valentin-Weigand, P. Characterization of multi-drug tolerant persister cells in Streptococcus suis. BMC Microbiol. 2014, 14, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Sadykov, M.R.; Hartmann, T.; Mattes, T.A.; Hiatt, M.; Jann, N.J.; Zhu, Y.; Ledala, N.; Landmann, R.; Herrmann, M.; Rohde, H.; et al. CcpA coordinates central metabolism and biofilm formation in Staphylococcus epidermidis. Microbiology 2011, 157, 3458–3468. [Google Scholar] [CrossRef] [PubMed]
- Thomas, V.C.; Chittezham Thomas, V.; Kinkead, L.C.; Janssen, A.; Schaeffer, C.R.; Woods, K.M.; Lindgren, J.K.; Peaster, J.M.; Chaudhari, S.S.; Sadykov, M.; et al. A dysfunctional tricarboxylic acid cycle enhances fitness of Staphylococcus epidermidis during β-lactam stress. mBio 2013, 4, e00437-13. [Google Scholar] [CrossRef] [PubMed]
- Mansour, S.C.; Pletzer, D.; de la Fuente-Núñez, C.; Kim, P.; Cheung, G.Y.C.; Joo, H.-S.; Otto, M.; Hancock, R.E.W. Bacterial Abscess Formation Is Controlled by the Stringent Stress Response and Can Be Targeted Therapeutically. EBioMedicine 2016, 12, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Meylan, S.; Porter, C.B.M.; Yang, J.H.; Belenky, P.; Gutierrez, A.; Lobritz, M.A.; Park, J.; Kim, S.H.; Moskowitz, S.M.; Collins, J.J. Carbon Sources Tune Antibiotic Susceptibility in Pseudomonas aeruginosa via Tricarboxylic Acid Cycle Control. Cell Chem. Biol. 2017, 24, 195–206. [Google Scholar] [CrossRef] [PubMed]
- Barraud, N.; Buson, A.; Jarolimek, W.; Rice, S.A. Mannitol enhances antibiotic sensitivity of persister bacteria in Pseudomonas aeruginosa biofilms. PLoS ONE 2013, 8, e84220. [Google Scholar] [CrossRef] [PubMed]
- Allison, K.R.; Brynildsen, M.P.; Collins, J.J. Metabolite-enabled eradication of bacterial persisters by aminoglycosides. Nature 2011, 473, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Taber, H.W.; Mueller, J.P.; Miller, P.F.; Arrow, A.S. Bacterial uptake of aminoglycoside antibiotics. Microbiol. Rev. 1987, 51, 439–457. [Google Scholar] [PubMed]
- Eswaran, J.; Koronakis, E.; Higgins, M.K.; Hughes, C.; Koronakis, V. Three’s company: Component structures bring a closer view of tripartite drug efflux pumps. Curr. Opin. Struct. Biol. 2004, 14, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Fraimow, H.S.; Greenman, J.B.; Leviton, I.M.; Dougherty, T.J.; Miller, M.H. Tobramycin uptake in Escherichia coli is driven by either electrical potential or ATP. J. Bacteriol. 1991, 173, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Webber, M.A. The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 2002, 51, 9–11. [Google Scholar] [CrossRef]
- Paulsen, I.T.; Brown, M.H.; Skurray, R.A. Proton-dependent multidrug efflux systems. Microbiol. Rev. 1996, 60, 575–608. [Google Scholar] [PubMed]
- Jiafeng, L.; Fu, X.; Chang, Z. Hypoionic shock treatment enables aminoglycosides antibiotics to eradicate bacterial persisters. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Hocquet, D.; Vogne, C.; El Garch, F.; Vejux, A.; Gotoh, N.; Lee, A.; Lomovskaya, O.; Plesiat, P. MexXY-OprM Efflux Pump Is Necessary for Adaptive Resistance of Pseudomonas aeruginosa to Aminoglycosides. Antimicrob. Agents Chemother. 2003, 47, 1371–1375. [Google Scholar] [CrossRef] [PubMed]
- De Kievit, T.R.; Parkins, M.D.; Gillis, R.J.; Srikumar, R.; Ceri, H.; Poole, K.; Iglewski, B.H.; Storey, D.G. Multidrug efflux pumps: Expression patterns and contribution to antibiotic resistance in Pseudomonas aeruginosa biofilms. Antimicrob. Agents Chemother. 2001, 45, 1761–1770. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Mah, T.-F. Involvement of a novel efflux system in biofilm-specific resistance to antibiotics. J. Bacteriol. 2008, 190, 4447–4452. [Google Scholar] [CrossRef] [PubMed]
- Soto, S.M. Role of efflux pumps in the antibiotic resistance of bacteria embedded in a biofilm. Virulence 2013, 4, 223–229. [Google Scholar] [CrossRef] [PubMed]
- Colangeli, R.; Helb, D.; Sridharan, S.; Sun, J.; Varma-Basil, M.; Hazbón, M.H.; Harbacheuski, R.; Megjugorac, N.J.; Jacobs, W.R.; Holzenburg, A.; et al. The Mycobacterium tuberculosis iniA gene is essential for activity of an efflux pump that confers drug tolerance to both isoniazid and ethambutol. Mol. Microbiol. 2005, 55, 1829–1840. [Google Scholar] [CrossRef] [PubMed]
- Lomovskaya, O.; Warren, M.S.; Lee, A.; Galazzo, J.; Fronko, R.; Lee, M.; Blais, J.; Cho, D.; Chamberland, S.; Renau, T.; et al. Identification and characterization of inhibitors of multidrug resistance efflux pumps in Pseudomonas aeruginosa: Novel agents for combination therapy. Antimicrob. Agents Chemother. 2001, 45, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Pu, Y.; Zhao, Z.; Li, Y.; Zou, J.; Ma, Q.; Zhao, Y.; Ke, Y.; Zhu, Y.; Chen, H.; Baker, M.A.B.; et al. Enhanced Efflux Activity Facilitates Drug Tolerance in Dormant Bacterial Cells. Mol. Cell 2016, 62, 284–294. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, K.; Semsey, S. Microbiology: Pumping persisters. Nature 2016, 534, 41–42. [Google Scholar] [CrossRef] [PubMed]
- Du Toit, A. Bacterial physiology: Persisters are under the pump. Nat. Rev. Microbiol. 2016, 14, 332–333. [Google Scholar] [CrossRef] [PubMed]
- Mascio, C.T.M.; Alder, J.D.; Silverman, J.A. Bactericidal action of daptomycin against stationary-phase and nondividing Staphylococcus aureus cells. Antimicrob. Agents Chemother. 2007, 51, 4255–4260. [Google Scholar] [CrossRef] [PubMed]
- Herisse, M.; Duverger, Y.; Martin-Verstraete, I.; Barras, F.; Ezraty, B. Silver potentiates aminoglycoside toxicity by enhancing their uptake. Mol. Microbiol. 2017, 105, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Morones-Ramirez, J.R.; Winkler, J.A.; Spina, C.S.; Collins, J.J. Silver enhances antibiotic activity against gram-negative bacteria. Sci. Transl. Med. 2013, 5, 190ra81. [Google Scholar] [CrossRef] [PubMed]
- Gaca, A.O.; Kajfasz, J.K.; Miller, J.H.; Liu, K.; Wang, J.D.; Abranches, J.; Lemos, J.A. Basal Levels of (p)ppGpp in Enterococcus faecalis: The Magic beyond the Stringent Response. mBio 2013, 4, e00646-13. [Google Scholar] [CrossRef] [PubMed]
- Strugeon, E.; Tilloy, V.; Ploy, M.-C.; Da Re, S. The Stringent Response Promotes Antibiotic Resistance Dissemination by Regulating Integron Integrase Expression in Biofilms. mBio 2016, 7, e00868-16. [Google Scholar] [CrossRef] [PubMed]
- Geiger, T.; Kastle, B.; Gratani, F.L.; Goerke, C.; Wolz, C. Two Small (p)ppGpp Synthases in Staphylococcus aureus Mediate Tolerance against Cell Envelope Stress Conditions. J. Bacteriol. 2014, 196, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Atkinson, G.C.; Tenson, T.; Hauryliuk, V. The RelA/SpoT homolog (RSH) superfamily: Distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS ONE 2011, 6, e23479. [Google Scholar] [CrossRef] [PubMed]
- Hauryliuk, V.; Atkinson, G.C.; Murakami, K.S.; Tenson, T.; Gerdes, K. Recent functional insights into the role of (p)ppGpp in bacterial physiology. Nat. Rev. Microbiol. 2015, 13, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Sureka, K.; Ghosh, B.; Bose, I.; Basu, J.; Kundu, M. Phenotypic heterogeneity in mycobacterial stringent response. BMC Syst. Biol. 2011, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Maisonneuve, E.; Castro-Camargo, M.; Gerdes, K. (p)ppGpp controls bacterial persistence by stochastic induction of toxin-antitoxin activity. Cell 2013, 154, 1140–1150. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, D.; Joshi-Datar, A.; Lepine, F.; Bauerle, E.; Olakanmi, O.; Beer, K.; McKay, G.; Siehnel, R.; Schafhauser, J.; Wang, Y.; et al. Active starvation responses mediate antibiotic tolerance in biofilms and nutrient-limited bacteria. Science 2011, 334, 982–986. [Google Scholar] [CrossRef] [PubMed]
- Vega, N.M.; Allison, K.R.; Khalil, A.S.; Collins, J.J. Signaling-mediated bacterial persister formation. Nat. Chem. Biol. 2012, 8, 431–433. [Google Scholar] [CrossRef] [PubMed]
- Fisher, R.A.; Gollan, B.; Helaine, S. Persistent bacterial infections and persister cells. Nat. Rev. Microbiol. 2017, 15, 453–464. [Google Scholar] [CrossRef] [PubMed]
- Mechler, L.; Herbig, A.; Paprotka, K.; Fraunholz, M.; Nieselt, K.; Bertram, R. A novel point mutation promotes growth phase-dependent daptomycin tolerance in Staphylococcus aureus. Antimicrob. Agents Chemother. 2015, 59, 5366–5376. [Google Scholar] [CrossRef] [PubMed]
- Odenholt, I. Pharmacodynamic effects of subinhibitory antibiotic concentrations. Int. J. Antimicrob. Agents 2001, 17, 1–8. [Google Scholar] [CrossRef]
- Schultz, D.; Palmer, A.C.; Kishony, R. Regulatory Dynamics Determine Cell Fate following Abrupt Antibiotic Exposure. Cell Syst. 2017, 5, 509–517.e3. [Google Scholar] [CrossRef] [PubMed]
- Devaraj, S.; Hemarajata, P.; Versalovic, J. The Human Gut Microbiome and Body Metabolism: Implications for Obesity and Diabetes. Clin. Chem. 2013, 59, 617–628. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Tremaroli, V.; Bäckhed, F. Linking Microbiota to Human Diseases: A Systems Biology Perspective. Trends Endocrinol. MeTable 2015, 26, 758–770. [Google Scholar] [CrossRef] [PubMed]
- Lam, Y.Y.; Ravussin, E. Analysis of energy metabolism in humans: A review of methodologies. Mol Metab 2016, 5, 1057–1071. [Google Scholar] [CrossRef] [PubMed]
- Kastenmüller, G.; Raffler, J.; Gieger, C.; Suhre, K. Genetics of human metabolism: An update. Hum. Mol. Genet. 2015, 24, R93–R101. [Google Scholar] [CrossRef] [PubMed]
- Cabral, D.J.; Wurster, J.I.; Flokas, M.E.; Alevizakos, M.; Zabat, M.; Korry, B.J.; Rowan, A.D.; Sano, W.H.; Andreatos, N.; Ducharme, R.B.; et al. The salivary microbiome is consistent between subjects and resistant to impacts of short-term hospitalization. Sci. Rep. 2017, 7, 11040. [Google Scholar] [CrossRef] [PubMed]
- Rodrigues, R.R.; Greer, R.L.; Dong, X.; DSouza, K.N.; Gurung, M.; Wu, J.Y.; Morgun, A.; Shulzhenko, N. Antibiotic-Induced Alterations in Gut Microbiota Are Associated with Changes in Glucose Metabolism in Healthy Mice. Front. Microbiol. 2017, 8, 2306. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Bowman, A.A.; Young, V.B. Antibiotic-Induced Alterations of the Gut Microbiota Alter Secondary Bile Acid Production and Allow for Clostridium difficile Spore Germination and Outgrowth in the Large Intestine. mSphere 2016, 1, e00045-15. [Google Scholar] [CrossRef] [PubMed]
- Ge, X.; Ding, C.; Zhao, W.; Xu, L.; Tian, H.; Gong, J.; Zhu, M.; Li, J.; Li, N. Antibiotics-induced depletion of mice microbiota induces changes in host serotonin biosynthesis and intestinal motility. J. Transl. Med. 2017, 15, 13. [Google Scholar] [CrossRef] [PubMed]
- Sommer, M.O.; Dantas, G. Antibiotics and the resistant microbiome. Curr. Opin. Microbiol. 2011, 14, 556–563. [Google Scholar] [CrossRef] [PubMed]
- Zaura, E.; Brandt, B.W.; Teixeira de Mattos, M.J.; Buijs, M.J.; Caspers, M.P.M.; Rashid, M.U.; Weintraub, A.; Nord, C.E.; Savell, A.; Hu, Y.; et al. Same Exposure but Two Radically Different Responses to Antibiotics: Resilience of the Salivary Microbiome versus Long-Term Microbial Shifts in Feces. mBio 2015, 6, e01693-15. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabral, D.J.; Wurster, J.I.; Belenky, P. Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions. Pharmaceuticals 2018, 11, 14. https://doi.org/10.3390/ph11010014
Cabral DJ, Wurster JI, Belenky P. Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions. Pharmaceuticals. 2018; 11(1):14. https://doi.org/10.3390/ph11010014
Chicago/Turabian StyleCabral, Damien J., Jenna I. Wurster, and Peter Belenky. 2018. "Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions" Pharmaceuticals 11, no. 1: 14. https://doi.org/10.3390/ph11010014
APA StyleCabral, D. J., Wurster, J. I., & Belenky, P. (2018). Antibiotic Persistence as a Metabolic Adaptation: Stress, Metabolism, the Host, and New Directions. Pharmaceuticals, 11(1), 14. https://doi.org/10.3390/ph11010014