Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment

 ,
,  and
and 
{kind=link}
{kind=link}
Abstract
1. Introduction
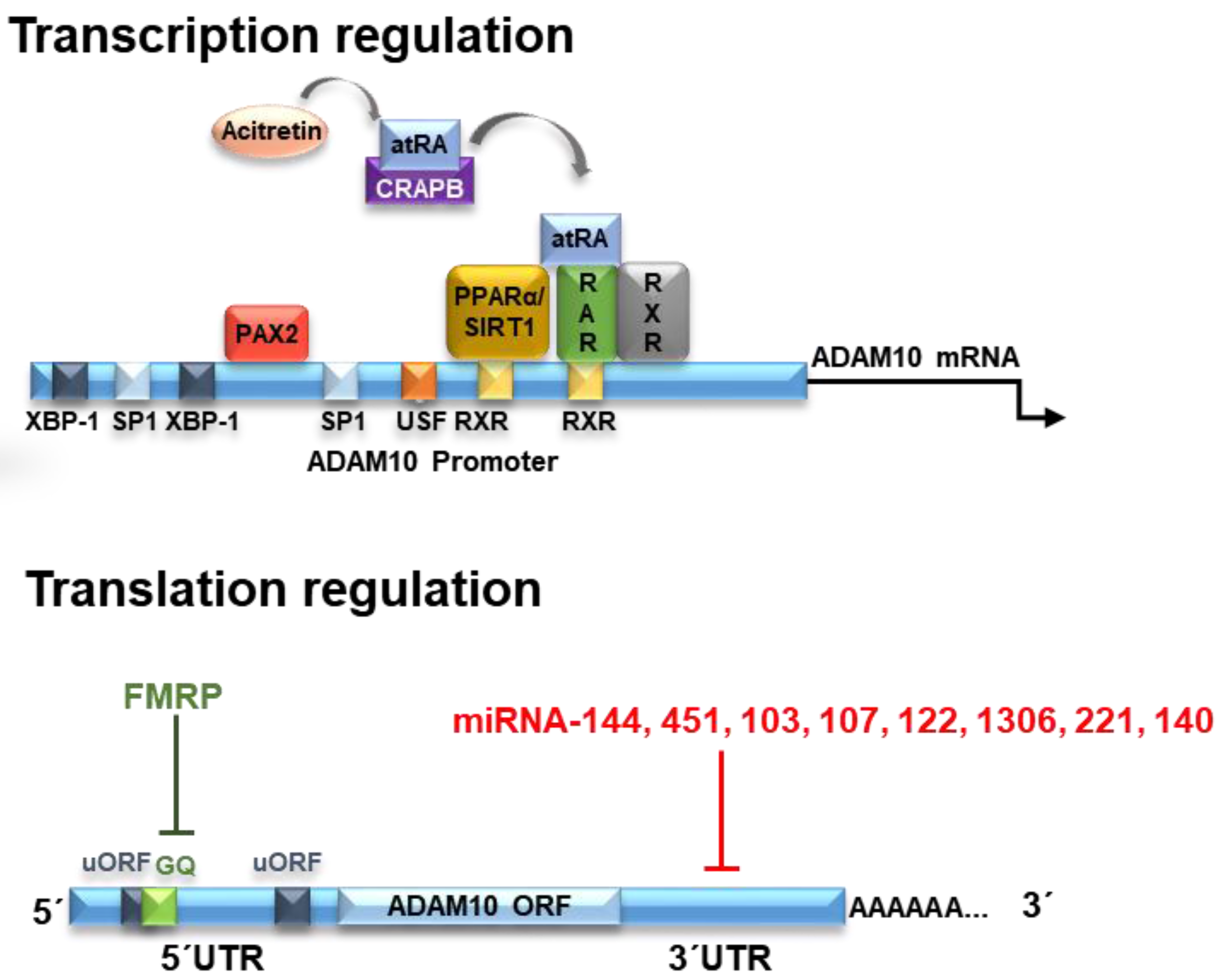
2. Transcriptional Regulators
2.1. Retinoic Acid
2.2. Sirtuins
2.3. XBP-1
2.4. Melatonin
2.5. SOX-2
2.6. PAX2
3. Translational Regulators
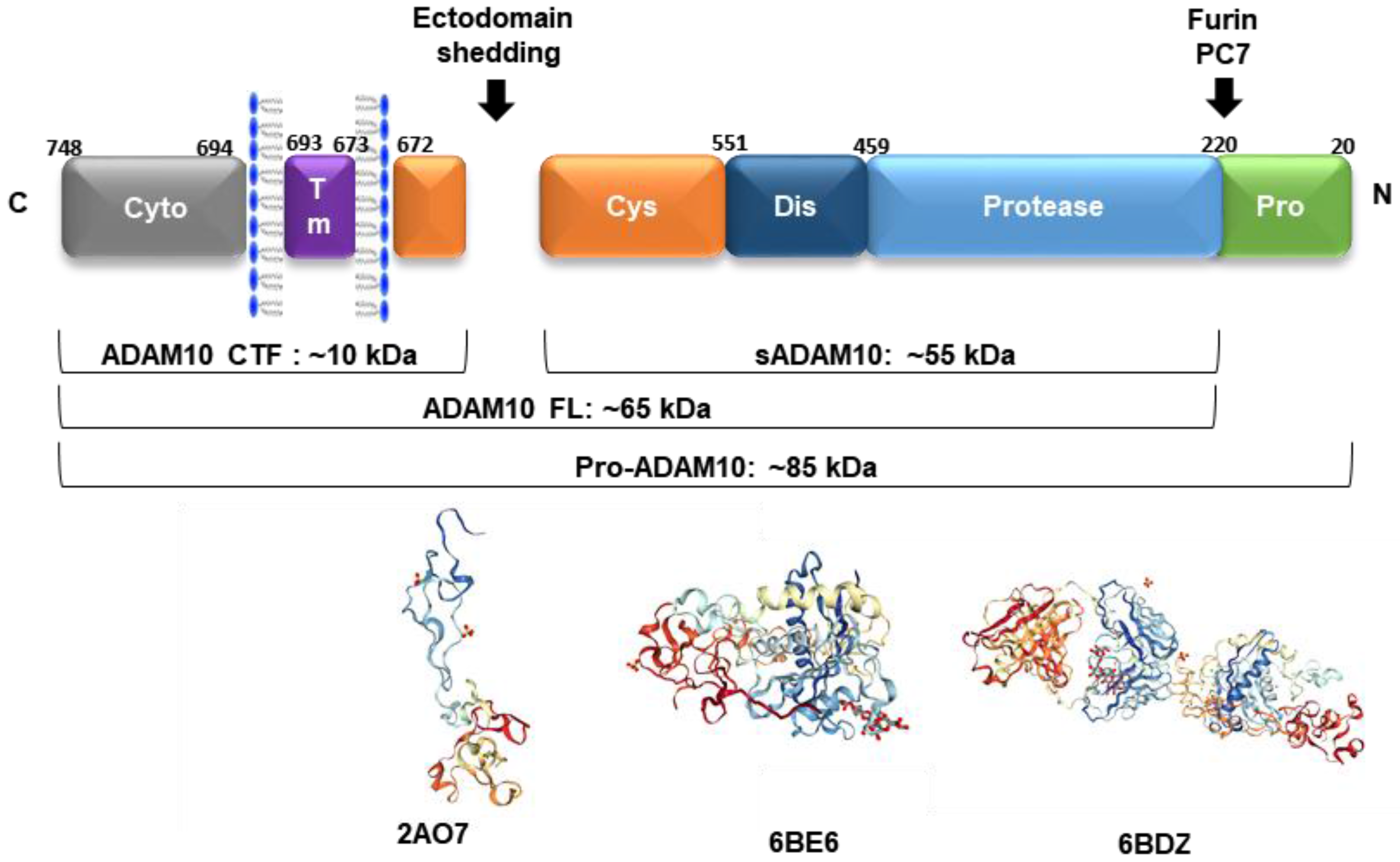
4. Post-Translational Regulators
4.1. Metallothioneins
4.2. Cellular Trafficking Regulators: Tetraspanins, SAP97 and AP2
4.3. Acetylcholinesterase Inhibitors (AChEIs)
4.4. Natural Products
4.5. Statins
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Huovila, A.P.J.; Turner, A.J.; Pelto-Huikko, M.; Karkkainen, L.; Ortiz, R.M. Shedding light on ADAM metalloproteinases. Trends Biochem. Sci. 2005, 30, 413–422. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Borroni, B.; Pelucchi, S.; Gardoni, F.; Di Luca, M. ADAM10 as a therapeutic target for brain diseases: From developmental disorders to Alzheimer’s disease. Expert Opin. Ther. Targets 2017, 21, 1017–1026. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B.; Checler, F. alpha-Secretase in Alzheimer’s disease and beyond: Mechanistic, regulation and function in the shedding of membrane proteins. Curr. Alzheimer Res. 2012, 9, 140–156. [Google Scholar] [CrossRef] [PubMed]
- Prinzen, C.; Muller, U.; Endres, K.; Fahrenholz, F.; Postina, R. Genomic structure and functional characterization of the human ADAM10 promoter. FASEB J. 2005, 19, 1522–1524. [Google Scholar] [CrossRef] [PubMed]
- Sandell, L.L.; Sanderson, B.W.; Moiseyev, G.; Johnson, T.; Mushegian, A.; Young, K.; Rey, J.P.; Ma, J.X.; Staehling-Hampton, K.; Trainor, P.A. RDH10 is essential for synthesis of embryonic retinoic acid and is required for limb, craniofacial, and organ development. Gene Dev. 2007, 21, 1113–1124. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.P.; Casadesus, G.; Zhu, X.W.; Lee, H.G.; Perry, G.; Smith, M.A.; Gustaw-Rothenberg, K.; Lerner, A. All-trans retinoic acid as a novel therapeutic strategy for Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 1615–1621. [Google Scholar] [CrossRef] [PubMed]
- Mangelsdorf, D.J.; Evans, R.M. The RXR heterodimers and orphan receptors. Cell 1995, 83, 841–850. [Google Scholar] [CrossRef]
- Saftig, P.; Lichtenthaler, S.F. The alpha secretase ADAM10: A metalloprotease with multiple functions in the brain. Prog. Neurobiol. 2015, 135, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Vincent, B. Regulation of the alpha-secretase ADAM10 at transcriptional, translational and post-translational levels. Brain Res. Bull. 2016, 126, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Sun, Q.; Chen, S. Oxidative stress: A major pathogenesis and potential therapeutic target of antioxidative agents in Parkinson’s disease and Alzheimer’s disease. Prog. Neurobiol. 2016, 147, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Singh, A. A review on mitochondrial restorative mechanism of antioxidants in Alzheimer’s disease and other neurological conditions. Front. Pharmacol. 2015, 6, 206. [Google Scholar] [CrossRef] [PubMed]
- Di Domenico, F.; Barone, E.; Perluigi, M.; Butterfield, D.A. Strategy to reduce free radical species in Alzheimer’s disease: An update of selected antioxidants. Expert Rev. Neurother. 2015, 15, 19–40. [Google Scholar] [CrossRef] [PubMed]
- Koryakina, A.; Aeberhard, J.; Kiefer, S.; Hamburger, M.; Kuenzi, P. Regulation of secretases by all-trans-retinoic acid. FEBS J. 2009, 276, 2645–2655. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Endres, K.; Fahrenholz, F.; Lotz, J.; Hiemke, C.; Teipel, S.; Lieb, K.; Tuscher, O.; Fellgiebel, A. Increased CSF APPs-alpha levels in patients with Alzheimer disease treated with acitretin. Neurology 2014, 83, 1930–1935. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Grimm, M.O.; Stahlmann, C.; Hartmann, T.; Shudo, K.; Tomita, T.; Endres, K. Rescue of Hypovitaminosis A Induces Non-Amyloidogenic Amyloid Precursor Protein (APP) Processing. Curr. Alzheimer Res. 2016, 13, 1277–1289. [Google Scholar] [CrossRef] [PubMed]
- Roy, A.; Jana, M.; Corbett, G.T.; Ramaswamy, S.; Kordower, J.H.; Gonzalez, F.J.; Pahan, K. Regulation of cyclic AMP response element binding and hippocampal plasticity-related genes by peroxisome proliferator-activated receptor alpha. Cell Rep. 2013, 4, 724–737. [Google Scholar] [CrossRef] [PubMed]
- Corbett, G.T.; Gonzalez, F.J.; Pahan, K. Activation of peroxisome proliferator-activated receptor alpha stimulates ADAM10-mediated proteolysis of APP. Proc. Natl. Acad. Sci. USA 2015, 112, 8445–8450. [Google Scholar] [CrossRef] [PubMed]
- Remington, R.; Bechtel, C.; Larsen, D.; Samar, A.; Page, R.; Morrell, C.; Shea, T.B. Maintenance of Cognitive Performance and Mood for Individuals with Alzheimer’s Disease Following Consumption of a Nutraceutical Formulation: A One-Year, Open-Label Study. J. Alzheimers Dis. 2016, 51, 991–995. [Google Scholar] [CrossRef] [PubMed]
- Carafa, V.; Rotili, D.; Forgione, M.; Cuomo, F.; Serretiello, E.; Hailu, G.S.; Jarho, E.; Lahtela-Kakkonen, M.; Mai, A.; Altucci, L. Sirtuin functions and modulation: From chemistry to the clinic. Clin. Epigenet. 2016, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Donmez, G.; Wang, D.; Cohen, D.E.; Guarente, L. SIRT1 suppresses beta-amyloid production by activating the alpha-secretase gene ADAM10. Cell 2010, 142, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Theendakara, V.; Patent, A.; Peters Libeu, C.A.; Philpot, B.; Flores, S.; Descamps, O.; Poksay, K.S.; Zhang, Q.; Cailing, G.; Hart, M.; et al. Neuroprotective Sirtuin ratio reversed by ApoE4. Proc. Natl. Acad. Sci. USA 2013, 110, 18303–18308. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.B.; Pardee, A.B. Evidence for defective retinoid transport and function in late onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 2901–2905. [Google Scholar] [CrossRef] [PubMed]
- Goodman, A.B. Retinoid receptors, transporters, and metabolizers as therapeutic targets in late onset Alzheimer disease. J. Cell. Physiol. 2006, 209, 598–603. [Google Scholar] [CrossRef] [PubMed]
- Corcoran, J.P.; So, P.L.; Maden, M. Disruption of the retinoid signalling pathway causes a deposition of amyloid beta in the adult rat brain. Eur. J. Neurosci. 2004, 20, 896–902. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.S. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: Part 2. Altern. Med. Rev. 2010, 15, 313–328. [Google Scholar] [PubMed]
- Kelly, G. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: Part 1. Altern. Med. Rev. 2010, 15, 245–263. [Google Scholar] [PubMed]
- Morselli, E.; Maiuri, M.C.; Markaki, M.; Megalou, E.; Pasparaki, A.; Palikaras, K.; Criollo, A.; Galluzzi, L.; Malik, S.A.; Vitale, I.; et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Dis. 2010, 1, e10. [Google Scholar] [CrossRef] [PubMed]
- Nikolaev, A.; McLaughlin, T.; O’Leary, D.D.; Tessier-Lavigne, M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature 2009, 457, 981–989. [Google Scholar] [CrossRef] [PubMed]
- Reinhardt, S.; Schuck, F.; Grosgen, S.; Riemenschneider, M.; Hartmann, T.; Postina, R.; Grimm, M.; Endres, K. Unfolded protein response signaling by transcription factor XBP-1 regulates ADAM10 and is affected in Alzheimer’s disease. FASEB J. 2014, 28, 978–997. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.H.; Scapa, E.F.; Cohen, D.E.; Glimcher, L.H. Regulation of hepatic lipogenesis by the transcription factor XBP1. Science 2008, 320, 1492–1496. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.; Liu, H.; Li, L.; Liu, H.; Yang, J.; Shi, W.; Feng, Y.; Huang, H.; Wu, L. The Roles of Endoplasmic Reticulum Stress in the Pathophysiological Development of Cartilage and Chondrocytes. Curr. Pharm. Des. 2017, 23, 1693–1704. [Google Scholar] [CrossRef] [PubMed]
- Kaneko, M.; Koike, H.; Saito, R.; Kitamura, Y.; Okuma, Y.; Nomura, Y. Loss of HRD1-mediated protein degradation causes amyloid precursor protein accumulation and amyloid-beta generation. J. Neurosci. 2010, 30, 3924–3932. [Google Scholar] [CrossRef] [PubMed]
- Dunys, J.; Duplan, E.; Checler, F. The transcription factor X-box binding protein-1 in neurodegenerative diseases. Mol. Neurodegener. 2014, 9, 35. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Htoo, H.H.; Wintachai, P.; Hernandez, J.F.; Dubois, C.; Postina, R.; Xu, H.; Checler, F.; Smith, D.R.; Govitrapong, P.; et al. Melatonin stimulates the nonamyloidogenic processing of betaAPP through the positive transcriptional regulation of ADAM10 and ADAM17. J. Pineal Res. 2015, 58, 151–165. [Google Scholar] [CrossRef] [PubMed]
- Shukla, M.; Govitrapong, P.; Boontem, P.; Reiter, R.J.; Satayavivad, J. Mechanisms of Melatonin in Alleviating Alzheimer’s Disease. Curr. Neuropharmacol. 2017, 15, 1010–1031. [Google Scholar] [CrossRef] [PubMed]
- Waldhauser, F.; Steger, H. Changes in melatonin secretion with age and pubescence. J. Neural Transm. Suppl. 1986, 21, 183–197. [Google Scholar] [PubMed]
- Waldhauser, F.; Weiszenbacher, G.; Tatzer, E.; Gisinger, B.; Waldhauser, M.; Schemper, M.; Frisch, H. Alterations in nocturnal serum melatonin levels in humans with growth and aging. J. Clin. Endocrinol. Metab. 1988, 66, 648–652. [Google Scholar] [CrossRef] [PubMed]
- Skene, D.J.; Vivien-Roels, B.; Sparks, D.L.; Hunsaker, J.C.; Pevet, P.; Ravid, D.; Swaab, D.F. Daily variation in the concentration of melatonin and 5-methoxytryptophol in the human pineal gland: Effect of age and Alzheimer’s disease. Brain Res. 1990, 528, 170–174. [Google Scholar] [CrossRef]
- Mishima, K.; Tozawa, T.; Satoh, K.; Matsumoto, Y.; Hishikawa, Y.; Okawa, M. Melatonin secretion rhythm disorders in patients with senile dementia of Alzheimer’s type with disturbed sleep-waking. Biol. Psychiatry 1999, 45, 417–421. [Google Scholar] [CrossRef]
- Zhou, J.N.; Liu, R.Y.; Kamphorst, W.; Hofman, M.A.; Swaab, D.F. Early neuropathological Alzheimer’s changes in aged individuals are accompanied by decreased cerebrospinal fluid melatonin levels. J. Pineal Res. 2003, 35, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. Melatonin as a natural ally against oxidative stress: A physicochemical examination. J. Pineal Res. 2011, 51, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Tan, D.X.; Reiter, R.J. On the free radical scavenging activities of melatonin’s metabolites, AFMK and AMK. J. Pineal Res. 2013, 54, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.A.; Acuna-Castroviejo, D.; Coto-Montes, A.; Boga, J.A.; Manchester, L.C.; Fuentes-Broto, L.; Korkmaz, A.; Ma, S.; Tan, D.X.; Reiter, R.J. Alzheimer’s disease: Pathological mechanisms and the beneficial role of melatonin. J. Pineal Res. 2012, 52, 167–202. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.; Bozner, P.; Soto, C.; Shao, H.; Robakis, N.K.; Zagorski, M.; Frangione, B.; Ghiso, J. Inhibition of Alzheimer beta-fibrillogenesis by melatonin. J. Biol. Chem. 1998, 273, 7185–7188. [Google Scholar] [CrossRef] [PubMed]
- Pappolla, M.A.; Sos, M.; Omar, R.A.; Bick, R.J.; Hickson-Bick, D.L.; Reiter, R.J.; Efthimiopoulos, S.; Robakis, N.K. Melatonin prevents death of neuroblastoma cells exposed to the Alzheimer amyloid peptide. J. Neurosci. 1997, 17, 1683–1690. [Google Scholar] [PubMed]
- Feng, Z.; Chang, Y.; Cheng, Y.; Zhang, B.L.; Qu, Z.W.; Qin, C.; Zhang, J.T. Melatonin alleviates behavioral deficits associated with apoptosis and cholinergic system dysfunction in the APP 695 transgenic mouse model of Alzheimer’s disease. J. Pineal Res. 2004, 37, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Feng, Z.; Zhang, J.T. Protective effect of melatonin on beta-amyloid-induced apoptosis in rat astroglioma C6 cells and its mechanism. Free Radic. Biol. Med. 2004, 37, 1790–1801. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.H.; Jung, S.B.; Lee, M.H.; Kim, C.J.; Oh, Y.T.; Kang, I.; Kim, J.; Kim, E.H. Melatonin attenuates amyloid beta25-35-induced apoptosis in mouse microglial BV2 cells. Neurosci. Lett. 2005, 380, 26–31. [Google Scholar] [CrossRef] [PubMed]
- Matsubara, E.; Bryant-Thomas, T.; Pacheco Quinto, J.; Henry, T.L.; Poeggeler, B.; Herbert, D.; Cruz-Sanchez, F.; Chyan, Y.J.; Smith, M.A.; Perry, G.; et al. Melatonin increases survival and inhibits oxidative and amyloid pathology in a transgenic model of Alzheimer’s disease. J. Neurochem. 2003, 85, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Olcese, J.M.; Cao, C.; Mori, T.; Mamcarz, M.B.; Maxwell, A.; Runfeldt, M.J.; Wang, L.; Zhang, C.; Lin, X.; Zhang, G.; et al. Protection against cognitive deficits and markers of neurodegeneration by long-term oral administration of melatonin in a transgenic model of Alzheimer disease. J. Pineal Res. 2009, 47, 82–96. [Google Scholar] [CrossRef] [PubMed]
- Quinn, J.; Kulhanek, D.; Nowlin, J.; Jones, R.; Pratico, D.; Rokach, J.; Stackman, R. Chronic melatonin therapy fails to alter amyloid burden or oxidative damage in old Tg2576 mice: Implications for clinical trials. Brain Res. 2005, 1037, 209–213. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.Y.; Zheng, W.; Ng, C.H.; Ungvari, G.S.; Wei, W.; Xiang, Y.T. Meta-analysis of randomized, double-blind, placebo-controlled trials of melatonin in Alzheimer’s disease. Int. J. Geriatr. Psychiatry 2017, 32, 50–57. [Google Scholar] [CrossRef] [PubMed]
- Sarlak, G.; Vincent, B. The Roles of the Stem Cell-Controlling SOX2 Transcription Factor: From Neuroectoderm Development to Alzheimer’s Disease? Mol. Neurobiol. 2016, 53, 1679–1698. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.L.; Cavallaro, M.; Braida, D.; Di Cristofano, A.; Canta, A.; Vezzani, A.; Ottolenghi, S.; Pandolfi, P.P.; Sala, M.; DeBiasi, S.; et al. SOX2 deficiency causes neurodegeneration and impaired neurogenesis in the adult mouse brain. Development 2004, 131, 3805–3819. [Google Scholar] [CrossRef] [PubMed]
- Crews, L.; Adame, A.; Patrick, C.; Delaney, A.; Pham, E.; Rockenstein, E.; Hansen, L.; Masliah, E. Increased BMP6 levels in the brains of Alzheimer’s disease patients and APP transgenic mice are accompanied by impaired neurogenesis. J. Neurosci. 2010, 30, 12252–12262. [Google Scholar] [CrossRef] [PubMed]
- Arnold, K.; Sarkar, A.; Yram, M.A.; Polo, J.M.; Bronson, R.; Sengupta, S.; Seandel, M.; Geijsen, N.; Hochedlinger, K. SOX2(+) adult stem and progenitor cells are important for tissue regeneration and survival of mice. Cell Stem Cell 2011, 9, 317–329. [Google Scholar] [CrossRef] [PubMed]
- Suh, H.; Consiglio, A.; Ray, J.; Sawai, T.; D’Amour, K.A.; Gage, F.H. In vivo fate analysis reveals the multipotent and self-renewal capacities of SOX2+ neural stem cells in the adult hippocampus. Cell Stem Cell 2007, 1, 515–528. [Google Scholar] [CrossRef] [PubMed]
- Sarlak, G.; Htoo, H.H.; Hernandez, J.F.; Iizasa, H.; Checler, F.; Konietzko, U.; Song, W.; Vincent, B. SOX2 functionally interacts with βAPP, the βAPP intracellular domain and ADAM10 at a transcriptional level in human cells. Neuroscience 2016, 312, 153–164. [Google Scholar] [CrossRef] [PubMed]
- Demars, M.P.; Bartholomew, A.; Strakova, Z.; Lazarov, O. Soluble amyloid precursor protein: A novel proliferation factor of adult progenitor cells of ectodermal and mesodermal origin. Stem Cell Res. Ther. 2011, 2, 36. [Google Scholar] [CrossRef] [PubMed]
- Passer, B.; Pellegrini, L.; Russo, C.; Siegel, R.M.; Lenardo, M.J.; Schettini, G.; Bachmann, M.; Tabaton, M.; D’Adamio, L. Generation of an apoptotic intracellular peptide by gamma-secretase cleavage of Alzheimer’s amyloid beta protein precursor. J. Alzheimer Dis. 2000, 2, 289–301. [Google Scholar] [CrossRef]
- Alves da Costa, C.; Sunyach, C.; Pardossi-Piquard, R.; Sevalle, J.; Vincent, B.; Boyer, N.; Kawarai, T.; Girardot, N.; St George-Hyslop, P.; Checler, F. Presenilin-dependent gamma-secretase-mediated control of p53-associated cell death in Alzheimer’s disease. J. Neurosci. 2006, 26, 6377–6385. [Google Scholar] [CrossRef] [PubMed]
- Flammang, B.; Pardossi-Piquard, R.; Sevalle, J.; Debayle, D.; Dabert-Gay, A.S.; Thevenet, A.; Lauritzen, I.; Checler, F. Evidence that the amyloid-beta protein precursor intracellular domain, AICD, derives from beta-secretase-generated C-terminal fragment. J. Alzheimer Dis. 2012, 30, 145–153. [Google Scholar]
- Wan, W.; Xia, S.; Kalionis, B.; Liu, L.; Li, Y. The Role of Wnt Signaling in the Development of Alzheimer’s Disease: A Potential Therapeutic Target? BioMed Res. Int. 2014, 2014, 301575. [Google Scholar] [CrossRef] [PubMed]
- Dahl, E.; Koseki, H.; Balling, R. Pax genes and organogenesis. BioEssays 1997, 19, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Doberstein, K.; Pfeilschifter, J.; Gutwein, P. The transcription factor PAX2 regulates ADAM10 expression in renal cell carcinoma. Carcinogenesis 2011, 32, 1713–1723. [Google Scholar] [CrossRef] [PubMed]
- Epstein, J.; Cai, J.; Glaser, T.; Jepeal, L.; Maas, R. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J. Biol. Chem. 1994, 269, 8355–8361. [Google Scholar] [PubMed]
- Robson, E.J.; He, S.J.; Eccles, M.R. A PANorama of PAX genes in cancer and development. Nat. Rev. Cancer 2006, 6, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Li, C.G.; Eccles, M.R. PAX Genes in Cancer; Friends or Foes? Front. Genet. 2012, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.B.; Doberstein, K.; Baumgarten, P.; Wieland, A.; Ungerer, C.; Burger, C.; Hardt, K.; Boehncke, W.H.; Pfeilschifter, J.; Mihic-Probst, D.; et al. PAX2 regulates ADAM10 expression and mediates anchorage-independent cell growth of melanoma cells. PLoS ONE 2011, 6, e22312. [Google Scholar] [CrossRef] [PubMed]
- Endres, K.; Deller, T. Regulation of Alpha-Secretase ADAM10 In vitro and In vivo: Genetic, Epigenetic, and Protein-Based Mechanisms. Front. Mol. Neurosci. 2017, 10, 56. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Buell, D.; Zilow, S.; Ludwig, A.K.; Nuscher, B.; Lichtenthaler, S.F.; Prinzen, C.; Fahrenholz, F.; Haass, C. Expression of the anti-amyloidogenic secretase ADAM10 is suppressed by its 5’-untranslated region. J. Biol. Chem. 2010, 285, 15753–15760. [Google Scholar] [CrossRef] [PubMed]
- Lammich, S.; Kamp, F.; Wagner, J.; Nuscher, B.; Zilow, S.; Ludwig, A.K.; Willem, M.; Haass, C. Translational repression of the disintegrin and metalloprotease ADAM10 by a stable G-quadruplex secondary structure in its 5’-untranslated region. J. Biol. Chem. 2011, 286, 45063–45072. [Google Scholar] [CrossRef] [PubMed]
- Huppert, J.L.; Bugaut, A.; Kumari, S.; Balasubramanian, S. G-quadruplexes: The beginning and end of UTRs. Nucleic Acids Res. 2008, 36, 6260–6268. [Google Scholar] [CrossRef] [PubMed]
- Dai, J.; Liu, Z.Q.; Wang, X.Q.; Lin, J.; Yao, P.F.; Huang, S.L.; Ou, T.M.; Tan, J.H.; Li, D.; Gu, L.Q.; et al. Discovery of Small Molecules for Up-Regulating the Translation of Antiamyloidogenic Secretase, a Disintegrin and Metalloproteinase 10 (ADAM10), by Binding to the G-Quadruplex-Forming Sequence in the 5’ Untranslated Region (UTR) of Its mRNA. J. Med. Chem. 2015, 58, 3875–3891. [Google Scholar] [CrossRef] [PubMed]
- Pasciuto, E.; Ahmed, T.; Wahle, T.; Gardoni, F.; D’Andrea, L.; Pacini, L.; Jacquemont, S.; Tassone, F.; Balschun, D.; Dotti, C.G.; et al. Dysregulated ADAM10-Mediated Processing of APP during a Critical Time Window Leads to Synaptic Deficits in Fragile X Syndrome. Neuron 2015, 87, 382–398. [Google Scholar] [CrossRef] [PubMed]
- Ambros, V. The functions of animal microRNAs. Nature 2004, 431, 350–355. [Google Scholar] [CrossRef] [PubMed]
- Krol, J.; Loedige, I.; Filipowicz, W. The widespread regulation of microRNA biogenesis, function and decay. Nat. Rev. Genet. 2010, 11, 597–610. [Google Scholar] [CrossRef] [PubMed]
- Berezikov, E.; Thuemmler, F.; van Laake, L.W.; Kondova, I.; Bontrop, R.; Cuppen, E.; Plasterk, R.H. Diversity of microRNAs in human and chimpanzee brain. Nat. Genet. 2006, 38, 1375–1377. [Google Scholar] [CrossRef] [PubMed]
- Delay, C.; Mandemakers, W.; Hébert, S.S. MicroRNAs in Alzheimer’s disease. Neurobiol. Dis. 2012, 46, 285–290. [Google Scholar] [CrossRef] [PubMed]
- Schonrock, N.; Matamales, M.; Ittner, L.M.; Götz, J. MicroRNA networks surrounding APP and amyloid-β metabolism—Implications for Alzheimer’s disease. Exp. Neurol. 2012, 235, 447–454. [Google Scholar] [CrossRef] [PubMed]
- Bai, S.; Nasser, M.W.; Wang, B.; Hsu, S.H.; Datta, J.; Kutay, H.; Yadav, A.; Nuovo, G.; Kumar, P.; Ghoshal, K. MicroRNA-122 inhibits tumorigenic properties of hepatocellular carcinoma cells and sensitizes these cells to sorafenib. J. Biol. Chem. 2009, 284, 32015–32027. [Google Scholar] [CrossRef] [PubMed]
- Barao, S.; Zhou, L.; Adamczuk, K.; Vanhoutvin, T.; van Leuven, F.; Demedts, D.; Vijverman, A.C.; Bossuyt, X.; Vandenberghe, R.; De Strooper, B. BACE1 levels correlate with phospho-tau levels in human cerebrospinal fluid. Curr. Alzheimer Res. 2013, 10, 671–678. [Google Scholar] [CrossRef] [PubMed]
- Manzine, P.R.; Pelucchi, S.; Horst, M.A.; Vale, F.A.C.; Pavarini, S.C.I.; Audano, M.; Mitro, N.; Di Luca, M.; Marcello, E.; Cominetti, M.R. microRNA 221 Targets ADAM10 mRNA and is Downregulated in Alzheimer’s Disease. J. Alzheimer Dis. 2017. [Google Scholar] [CrossRef] [PubMed]
- Augustin, R.; Endres, K.; Reinhardt, S.; Kuhn, P.H.; Lichtenthaler, S.F.; Hansen, J.; Wurst, W.; Trumbach, D. Computational identification and experimental validation of microRNAs binding to the Alzheimer-related gene ADAM10. BMC Med. Genet. 2012, 13, 35. [Google Scholar] [CrossRef] [PubMed]
- Akhter, R.; Shao, Y.; Shaw, M.; Formica, S.; Khrestian, M.; Leverenz, J.B.; Bekris, L.M. Regulation of ADAM10 by miR-140-5p and potential relevance for Alzheimer’s disease. Neurobiol. Aging 2017, 63, 110–119. [Google Scholar] [CrossRef] [PubMed]
- Tousseyn, T.; Thathiah, A.; Jorissen, E.; Raemaekers, T.; Konietzko, U.; Reiss, K.; Maes, E.; Snellinx, A.; Serneels, L.; Nyabi, O.; et al. ADAM10, the rate-limiting protease of regulated intramembrane proteolysis of Notch and other proteins, is processed by ADAMS-9, ADAMS-15, and the gamma-secretase. J. Biol. Chem. 2009, 284, 11738–11747. [Google Scholar] [CrossRef] [PubMed]
- Mancia, F.; Shapiro, L. ADAM and Eph: How Ephrin-signaling cells become detached. Cell 2005, 123, 185–187. [Google Scholar] [CrossRef] [PubMed]
- Seegar, T.C.M.; Killingsworth, L.B.; Saha, N.; Meyer, P.A.; Patra, D.; Zimmerman, B.; Janes, P.W.; Rubinstein, E.; Nikolov, D.B.; Skiniotis, G.; et al. Structural Basis for Regulated Proteolysis by the alpha-Secretase ADAM10. Cell 2017, 171, 1638–1648.e1637. [Google Scholar] [CrossRef] [PubMed]
- Anders, A.; Gilbert, S.; Garten, W.; Postina, R.; Fahrenholz, F. Regulation of the alpha-secretase ADAM10 by its prodomain and proprotein convertases. FASEB J. 2001, 15, 1837–1839. [Google Scholar] [CrossRef] [PubMed]
- Cisse, M.A.; Sunyach, C.; Lefranc-Jullien, S.; Postina, R.; Vincent, B.; Checler, F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J. Biol. Chem. 2005, 280, 40624–40631. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.; Maretzky, T.; Peleg, Y.; Blobel, C.P.; Sagi, I. The Functional Maturation of A Disintegrin and Metalloproteinase (ADAM) 9, 10, and 17 Requires Processing at a Newly Identified Proprotein Convertase (PC) Cleavage Site. J. Biol. Chem. 2015, 290, 12135–12146. [Google Scholar] [CrossRef] [PubMed]
- Park, B.H.; Kim, H.G.; Jin, S.W.; Song, S.G.; Jeong, H.G. Metallothionein-III increases ADAM10 activity in association with furin, PC7, and PKCalpha during non-amyloidogenic processing. FEBS Lett. 2014, 588, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Meloni, G.; Polanski, T.; Braun, O.; Vasak, M. Effects of Zn(2+), Ca(2+), and Mg(2+) on the structure of Zn(7)metallothionein-3: Evidence for an additional zinc binding site. Biochemistry 2009, 48, 5700–5707. [Google Scholar] [CrossRef] [PubMed]
- Yu, W.H.; Lukiw, W.J.; Bergeron, C.; Niznik, H.B.; Fraser, P.E. Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 2001, 894, 37–45. [Google Scholar] [CrossRef]
- Erickson, J.C.; Sewell, A.K.; Jensen, L.T.; Winge, D.R.; Palmiter, R.D. Enhanced neurotrophic activity in Alzheimer’s disease cortex is not associated with down-regulation of metallothionein-III (GIF). Brain Res. 1994, 649, 297–304. [Google Scholar] [CrossRef]
- Amoureux, M.C.; Van Gool, D.; Herrero, M.T.; Dom, R.; Colpaert, F.C.; Pauwels, P.J. Regulation of metallothionein-III (GIF) mRNA in the brain of patients with Alzheimer disease is not impaired. Mol. Chem. Neuropathol. 1997, 32, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Carrasco, J.; Adlard, P.; Cotman, C.; Quintana, A.; Penkowa, M.; Xu, F.; Van Nostrand, W.E.; Hidalgo, J. Metallothionein-I and -III expression in animal models of Alzheimer disease. Neuroscience 2006, 143, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Irie, Y.; Keung, W.M. Anti-amyloid beta activity of metallothionein-III is different from its neuronal growth inhibitory activity: Structure-activity studies. Brain Res. 2003, 960, 228–234. [Google Scholar] [CrossRef]
- Irie, Y.; Keung, W.M. Metallothionein-III antagonizes the neurotoxic and neurotrophic effects of amyloid beta peptides. Biochem. Biophs. Res. Commun. 2001, 282, 416–420. [Google Scholar] [CrossRef] [PubMed]
- Saint-Pol, J.; Eschenbrenner, E.; Dornier, E.; Boucheix, C.; Charrin, S.; Rubinstein, E. Regulation of the trafficking and the function of the metalloprotease ADAM10 by tetraspanins. Biochem. Soc. Trans. 2017, 45, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Hemler, M.E. Tetraspanin proteins promote multiple cancer stages. Nat. Rev. Cancer 2014, 14, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Charrin, S.; Jouannet, S.; Boucheix, C.; Rubinstein, E. Tetraspanins at a glance. J. Cell Sci. 2014, 127, 3641–3648. [Google Scholar] [CrossRef] [PubMed]
- Lichtenthaler, S.F. Alpha-Secretase Cleavage of the Amyloid Precursor Protein: Proteolysis Regulated by Signaling Pathways and Protein Trafficking. Curr. Alzheimer Res. 2012, 9, 165–177. [Google Scholar] [CrossRef] [PubMed]
- Arduise, C.; Abache, T.; Li, L.; Billard, M.; Chabanon, A.; Ludwig, A.; Mauduit, P.; Boucheix, C.; Rubinstein, E.; Le Naour, F. Tetraspanins regulate ADAM10-mediated cleavage of TNF-alpha and epidermal growth factor. J. Immunol. 2008, 181, 7002–7013. [Google Scholar] [CrossRef] [PubMed]
- Haining, E.J.; Yang, J.; Bailey, R.L.; Khan, K.; Collier, R.; Tsai, S.; Watson, S.P.; Frampton, J.; Garcia, P.; Tomlinson, M.G. The TspanC8 Subgroup of Tetraspanins Interacts with A Disintegrin and Metalloprotease 10 (ADAM10) and Regulates Its Maturation and Cell Surface Expression. J. Biol. Chem. 2012, 287, 39753–39765. [Google Scholar] [CrossRef] [PubMed]
- Dornier, E.; Coumailleau, F.; Ottavi, J.F.; Moretti, J.; Boucheix, C.; Mauduit, P.; Schweisguth, F.; Rubinstein, E. TspanC8 tetraspanins regulate ADAM10/Kuzbanian trafficking and promote Notch activation in flies and mammals. J. Cell Biol. 2012, 199, 481–496. [Google Scholar] [CrossRef] [PubMed]
- Matthews, A.L.; Szyroka, J.; Collier, R.; Noy, P.J.; Tomlinson, M.G. Scissor sisters: Regulation of ADAM10 by the TspanC8 tetraspanins. Biochem. Soc. Trans. 2017, 45, 719–730. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Sheng, M. PDZ domain proteins of synapses. Nat. Rev. Neurosci. 2004, 5, 771–781. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Gardoni, F.; Mauceri, D.; Romorini, S.; Jeromin, A.; Epis, R.; Borroni, B.; Cattabeni, F.; Sala, C.; Padovani, A.; et al. Synapse-associated protein-97 mediates alpha-secretase ADAM10 trafficking and promotes its activity. J. Neurosci. 2007, 27, 1682–1691. [Google Scholar] [CrossRef] [PubMed]
- Saraceno, C.; Marcello, E.; Di Marino, D.; Borroni, B.; Claeysen, S.; Perroy, J.; Padovani, A.; Tramontano, A.; Gardoni, F.; Di Luca, M. SAP97-mediated ADAM10 trafficking from Golgi outposts depends on PKC phosphorylation. Cell Death Dis. 2014, 5, e1547. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Epis, R.; Saraceno, C.; Gardoni, F.; Borroni, B.; Cattabeni, F.; Padovani, A.; Di Luca, M. SAP97-mediated local trafficking is altered in Alzheimer disease patients’ hippocampus. Neurobiol. Aging 2012, 33, 422.e1–422.e10. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Z.; Ewers, M.; Teipel, S.; Burger, K.; Wallin, A.; Blennow, K.; He, P.; McAllister, C.; Hampel, H.; Shen, Y. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch. Gen. Psychiatry 2007, 64, 718–726. [Google Scholar] [CrossRef] [PubMed]
- Marcello, E.; Saraceno, C.; Musardo, S.; Vara, H.; de la Fuente, A.G.; Pelucchi, S.; Di Marino, D.; Borroni, B.; Tramontano, A.; Pérez-Otaño, I.; et al. Endocytosis of synaptic ADAM10 in neuronal plasticity and Alzheimer’s disease. J. Clin. Investig. 2013, 123, 2523–2538. [Google Scholar] [CrossRef] [PubMed]
- Canevelli, M.; Quarata, F.; Remiddi, F.; Lucchini, F.; Lacorte, E.; Vanacore, N.; Bruno, G.; Cesari, M. Sex and gender differences in the treatment of Alzheimer’s disease: A systematic review of randomized controlled trials. Pharmacol. Res. 2017, 115, 218–223. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Gardoni, F.; Marcello, E.; Colciaghi, F.; Borroni, B.; Padovani, A.; Cattabeni, F.; Di Luca, M. Acetylcholinesterase inhibitors increase ADAM10 activity by promoting its trafficking in neuroblastoma cell lines. J. Neurochem. 2004, 90, 1489–1499. [Google Scholar] [CrossRef] [PubMed]
- Sarno, T.A.; Talib, L.L.; Joaquim, H.P.; Bram, J.M.; Gattaz, W.F.; Forlenza, O.V. Protein Expression of BACE1 is Downregulated by Donepezil in Alzheimer’s Disease Platelets. J. Alzheimer Dis. 2017, 55, 1445–1451. [Google Scholar] [CrossRef] [PubMed]
- Bianco, O.A.F.M.; Manzine, P.R.; Nascimento, C.M.C.; Vale, F.A.C.; Pavarini, S.C.I.; Cominetti, M.R. Serotoninergic antidepressants positively affect platelet ADAM10 expression in patients with Alzheimer’s disease. Int. Psychogeriatr. 2016, 28, 939–944. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.L.; Guo, Z.; Halagappa, V.M.; Pearson, M.; Gray, A.J.; Matsuoka, Y.; Brown, M.; Martin, B.; Iyun, T.; Maudsley, S.; et al. Prophylactic treatment with paroxetine ameliorates behavioral deficits and retards the development of amyloid and tau pathologies in 3xTgAD mice. Exp. Neurol. 2007, 205, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-beta levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [PubMed]
- Ullah, F.; Liang, A.; Rangel, A.; Gyengesi, E.; Niedermayer, G.; Munch, G. High bioavailability curcumin: An anti-inflammatory and neurosupportive bioactive nutrient for neurodegenerative diseases characterized by chronic neuroinflammation. Arch. Toxicol. 2017, 91, 1623–1634. [Google Scholar] [CrossRef] [PubMed]
- Narasingappa, R.B.; Javagal, M.R.; Pullabhatla, S.; Htoo, H.H.; Rao, J.K.; Hernandez, J.F.; Govitrapong, P.; Vincent, B. Activation of alpha-secretase by curcumin-aminoacid conjugates. Biochem. Biophys. Res. Commun. 2012, 424, 691–696. [Google Scholar] [CrossRef] [PubMed]
- Sun, Q.; Jia, N.; Wang, W.; Jin, H.; Xu, J.; Hu, H. Activation of SIRT1 by curcumin blocks the neurotoxicity of amyloid-beta25-35 in rat cortical neurons. Biochem. Biophys. Res. Commun. 2014, 448, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Poltronieri, J.; Becceneri, A.B.; Fuzer, A.M.; Cesar, J.C.; Martin, A.C.B.M.; Vieira, P.C.; Pouliot, N.; Cominetti, M.R. [6]-gingerol as a Cancer Chemopreventive Agent: A Review of Its Activity on Different Steps of the Metastatic Process. Mini-Rev. Med. Chem. 2014, 14, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Wiart, C. A note on the relevance of [6]-Gingerol for the prevention and/or treatment of Alzheimer’s disease. Food Chem. Toxicol. 2013, 51, 456. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.; Park, G.H.; Kim, C.Y.; Jang, J.H. [6]-Gingerol attenuates beta-amyloid-induced oxidative cell death via fortifying cellular antioxidant defense system. Food Chem. Toxicol. 2011, 49, 1261–1269. [Google Scholar] [CrossRef] [PubMed]
- Karthick, C.; Periyasamy, S.; Jayachandran, K.S.; Anusuyadevi, M. Intrahippocampal Administration of Ibotenic Acid Induced Cholinergic Dysfunction via NR2A/NR2B Expression: Implications of Resveratrol against Alzheimer Disease Pathophysiology. Front. Mol. Neurosci. 2016, 9, 28. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Sathya, M.; Moorthi, P.; Premkumar, P.; Kandasamy, M.; Jayachandran, K.S.; Anusuyadevi, M. Resveratrol Intervenes Cholesterol- and Isoprenoid-Mediated Amyloidogenic Processing of AbetaPP in Familial Alzheimer’s Disease. J. Alzheimers Dis. 2016. [Google Scholar] [CrossRef]
- Abdul, H.M.; Calabrese, V.; Calvani, M.; Butterfield, D.A. Acetyl-l-carnitine-induced up-regulation of heat shock proteins protects cortical neurons against amyloid-beta peptide 1-42-mediated oxidative stress and neurotoxicity: Implications for Alzheimer’s disease. J. Neurosci. Res. 2006, 84, 398–408. [Google Scholar] [CrossRef] [PubMed]
- Pettegrew, J.W.; Levine, J.; McClure, R.J. Acetyl-l-carnitine physical-chemical, metabolic, and therapeutic properties: Relevance for its mode of action in Alzheimer’s disease and geriatric depression. Mol. Psychiatry 2000, 5, 616–632. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, M.; Borroni, B.; Cattabeni, F.; Padovani, A.; Di Luca, M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol. Dis. 2005, 19, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Adlard, P.A.; Perreau, V.M.; Pop, V.; Cotman, C.W. Voluntary exercise decreases amyloid load in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 4217–4221. [Google Scholar] [CrossRef] [PubMed]
- Kamenetz, F.; Tomita, T.; Hsieh, H.; Seabrook, G.; Borchelt, D.; Iwatsubo, T.; Sisodia, S.; Malinow, R. APP processing and synaptic function. Neuron 2003, 37, 925–937. [Google Scholar] [CrossRef]
- Epis, R.; Marcello, E.; Gardoni, F.; Longhi, A.; Calvani, M.; Iannuccelli, M.; Cattabeni, F.; Canonico, P.L.; Di Luca, M. Modulatory effect of acetyl-l-carnitine on amyloid precursor protein metabolism in hippocampal neurons. Eur. J. Pharmacol. 2008, 597, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.C.; Liu, C.C.; Kanekiyo, T.; Xu, H.; Bu, G. Apolipoprotein E and Alzheimer disease: Risk, mechanisms and therapy. Nat. Rev. Neurol. 2013, 9, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Kok, E.; Haikonen, S.; Luoto, T.; Huhtala, H.; Goebeler, S.; Haapasalo, H.; Karhunen, P.J. Apolipoprotein E-dependent accumulation of Alzheimer disease-related lesions begins in middle age. Ann. Neurol. 2009, 65, 650–657. [Google Scholar] [CrossRef] [PubMed]
- Shackleton, B.; Crawford, F.; Bachmeier, C. Apolipoprotein E-mediated Modulation of ADAM10 in Alzheimer’s Disease. Curr. Alzheimer Res. 2017, 14, 578–585. [Google Scholar] [CrossRef] [PubMed]
- Yanagisawa, K. Cholesterol and pathological processes in Alzheimer’s disease. J. Neurosci. Res. 2002, 70, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Silva, T.; Teixeira, J.; Remiao, F.; Borges, F. Alzheimer’s disease, cholesterol, and statins: The junctions of important metabolic pathways. Angew. Chem. 2013, 52, 1110–1121. [Google Scholar] [CrossRef] [PubMed]
- Anstey, K.J.; Lipnicki, D.M.; Low, L.F. Cholesterol as a risk factor for dementia and cognitive decline: A systematic review of prospective studies with meta-analysis. Am. J. Geriatr. Psychiatry 2008, 16, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Kivipelto, M.; Helkala, E.L.; Laakso, M.P.; Hanninen, T.; Hallikainen, M.; Alhainen, K.; Soininen, H.; Tuomilehto, J.; Nissien, A. Midlife vascular risk factors and Alzheimer’s disease in later life: Longitudinal, population based study. Br. Med. J. 2001, 322, 1447–1451. [Google Scholar] [CrossRef]
- Kojro, E.; Fuger, P.; Prinzen, C.; Kanarek, A.M.; Rat, D.; Endres, K.; Fahrenholz, F.; Postina, R. Statins and the Squalene Synthase Inhibitor Zaragozic Acid Stimulate the Non-Amyloidogenic Pathway of Amyloid-beta Protein Precursor Processing by Suppression of Cholesterol Synthesis. J. Alzheimers Dis. 2010, 20, 1215–1231. [Google Scholar] [CrossRef] [PubMed]
- Mason, J.C. Statins and their role in vascular protection. Clin. Sci. 2003, 105, 251–266. [Google Scholar] [CrossRef] [PubMed]
- Cole, S.L.; Grudzien, A.; Manhart, I.O.; Kelly, B.L.; Oakley, H.; Vassar, R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J. Biol. Chem. 2005, 280, 18755–18770. [Google Scholar] [CrossRef] [PubMed]
- Ostrowski, S.M.; Wilkinson, B.L.; Golde, T.E.; Landreth, G. Statins reduce amyloid-beta production through inhibition of protein Isoprenylation. J. Biol. Chem. 2007, 282, 26832–26844. [Google Scholar] [CrossRef] [PubMed]
- Friedhoff, L.T.; Cullen, E.I.; Geoghagen, N.S.; Buxbaum, J.D. Treatment with controlled-release lovastatin decreases serum concentrations of human beta-amyloid (A beta) peptide. Int. J. Neuropsychopharmacol. 2001, 4, 127–130. [Google Scholar] [CrossRef] [PubMed]
- Wolazin, B.; Manger, J.; Bryant, R.; Cordy, J.; Green, R.C.; McKee, A. Re-assessing the relationship between cholesterol, statins and Alzheimer’s disease. Acta Neurol. Scand. 2006, 114, 63–70. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. https://doi.org/10.3390/ph11010012
Peron R, Vatanabe IP, Manzine PR, Camins A, Cominetti MR. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals. 2018; 11(1):12. https://doi.org/10.3390/ph11010012
Chicago/Turabian StylePeron, Rafaela, Izabela Pereira Vatanabe, Patricia Regina Manzine, Antoni Camins, and Márcia Regina Cominetti. 2018. "Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment" Pharmaceuticals 11, no. 1: 12. https://doi.org/10.3390/ph11010012
APA StylePeron, R., Vatanabe, I. P., Manzine, P. R., Camins, A., & Cominetti, M. R. (2018). Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals, 11(1), 12. https://doi.org/10.3390/ph11010012