Comparison of In Vitro Assays in Selecting Radiotracers for In Vivo P-Glycoprotein PET Imaging

 , , ,
, , ,
Abstract
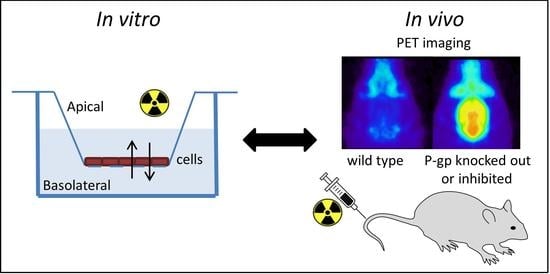
1. Introduction
2. Results
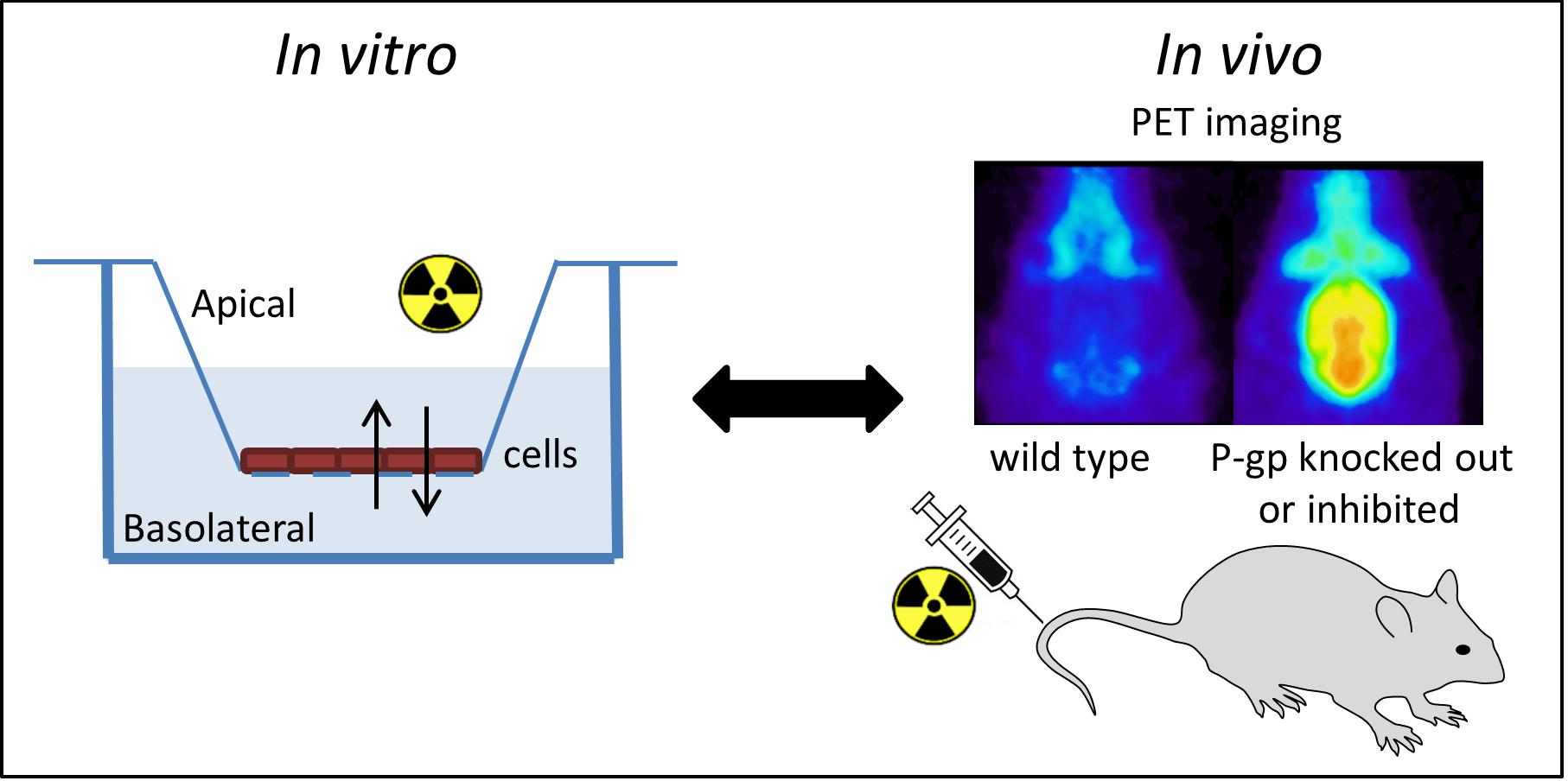
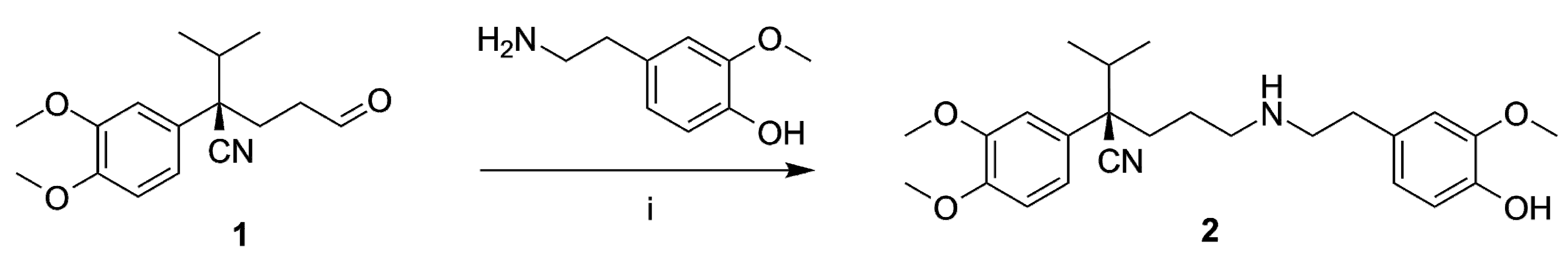
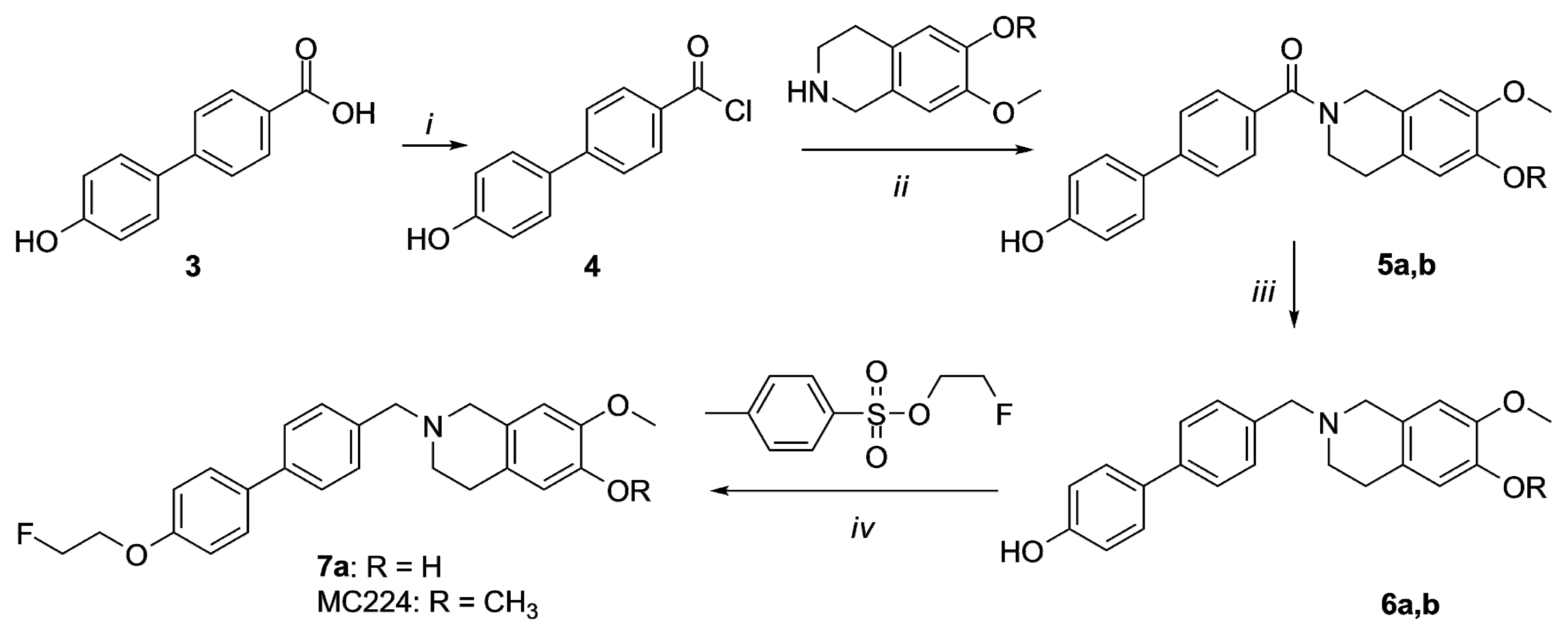
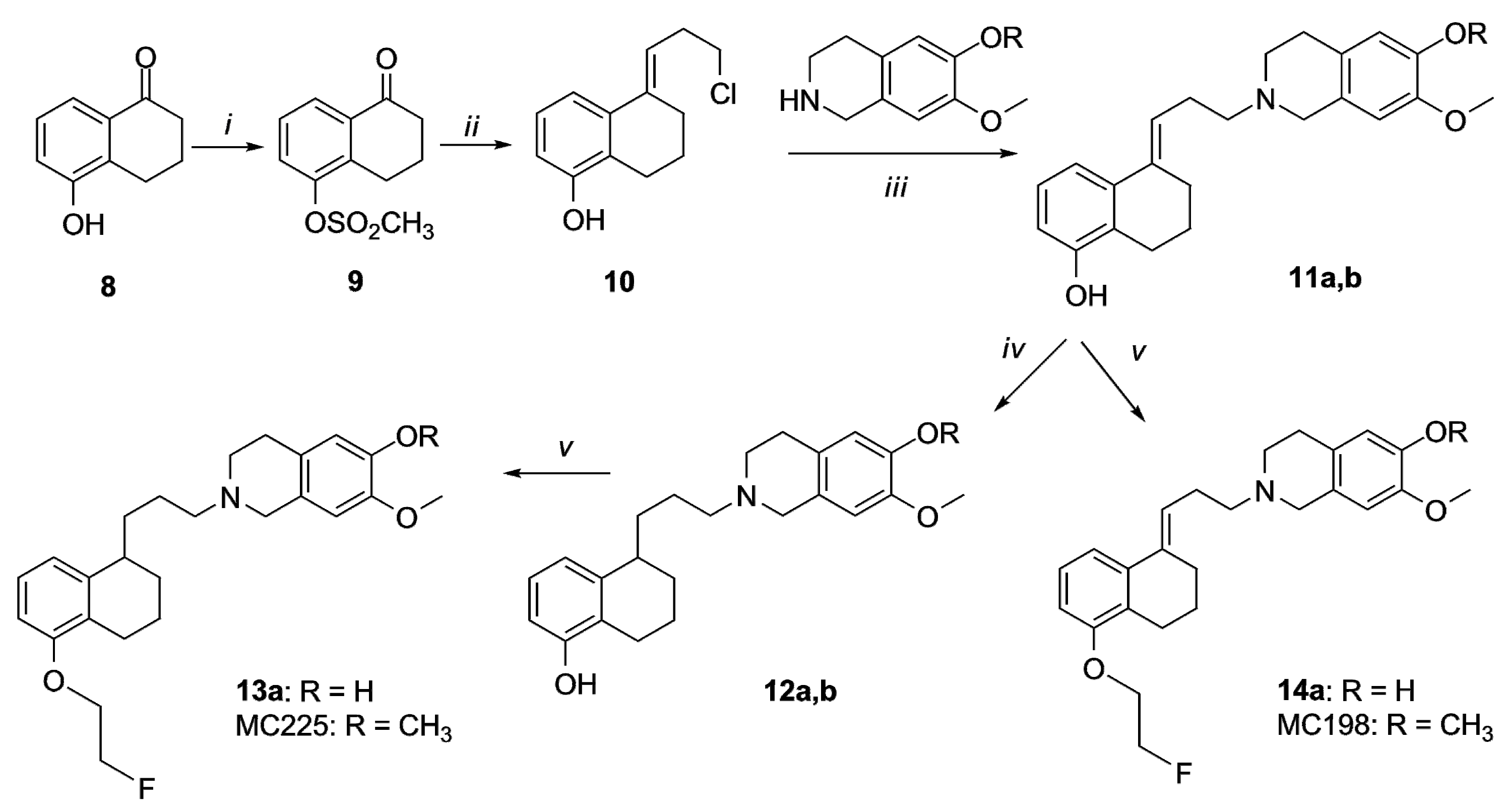
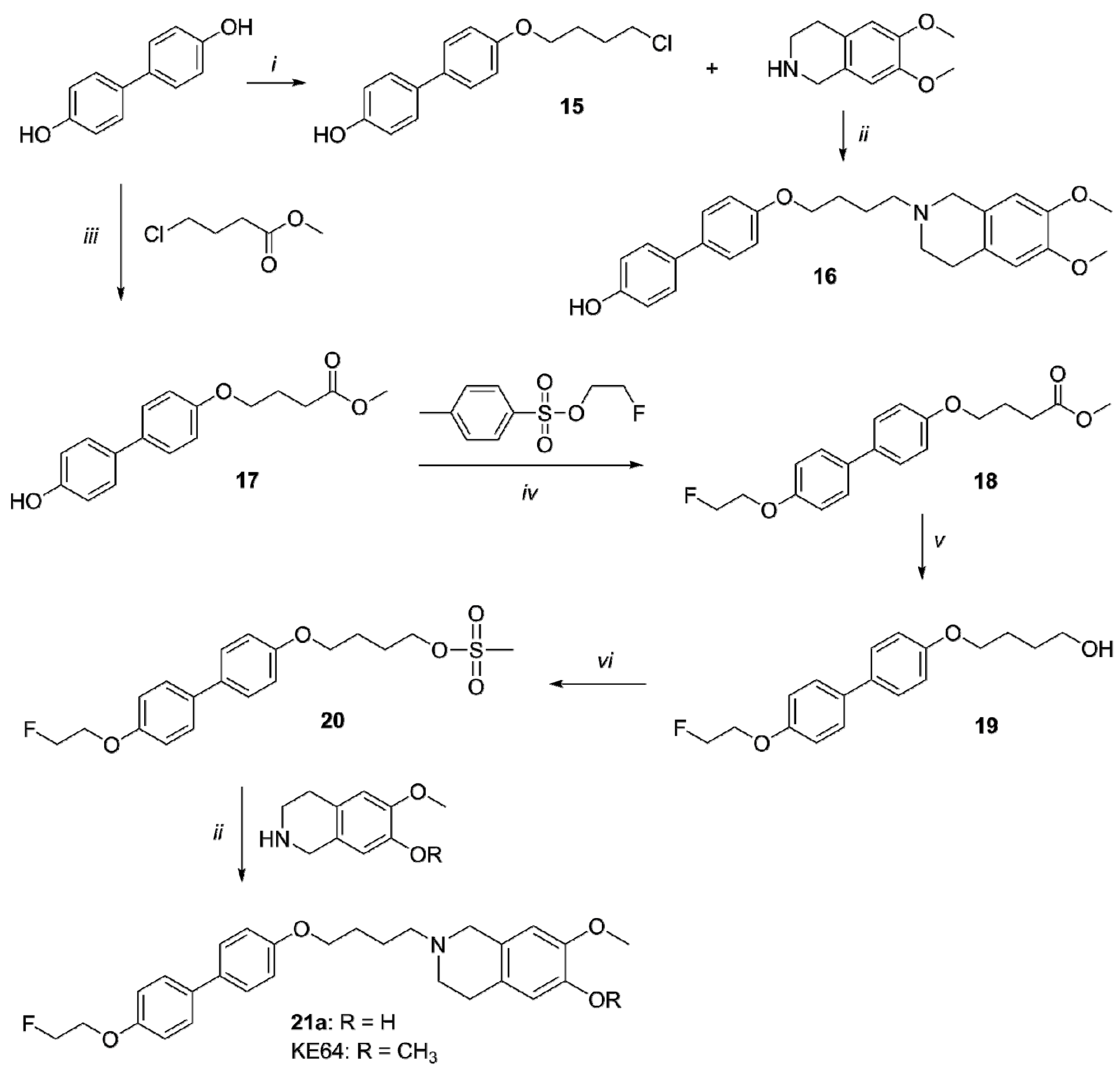
2.1. Organic Synthesis
2.2. Radiosynthesis
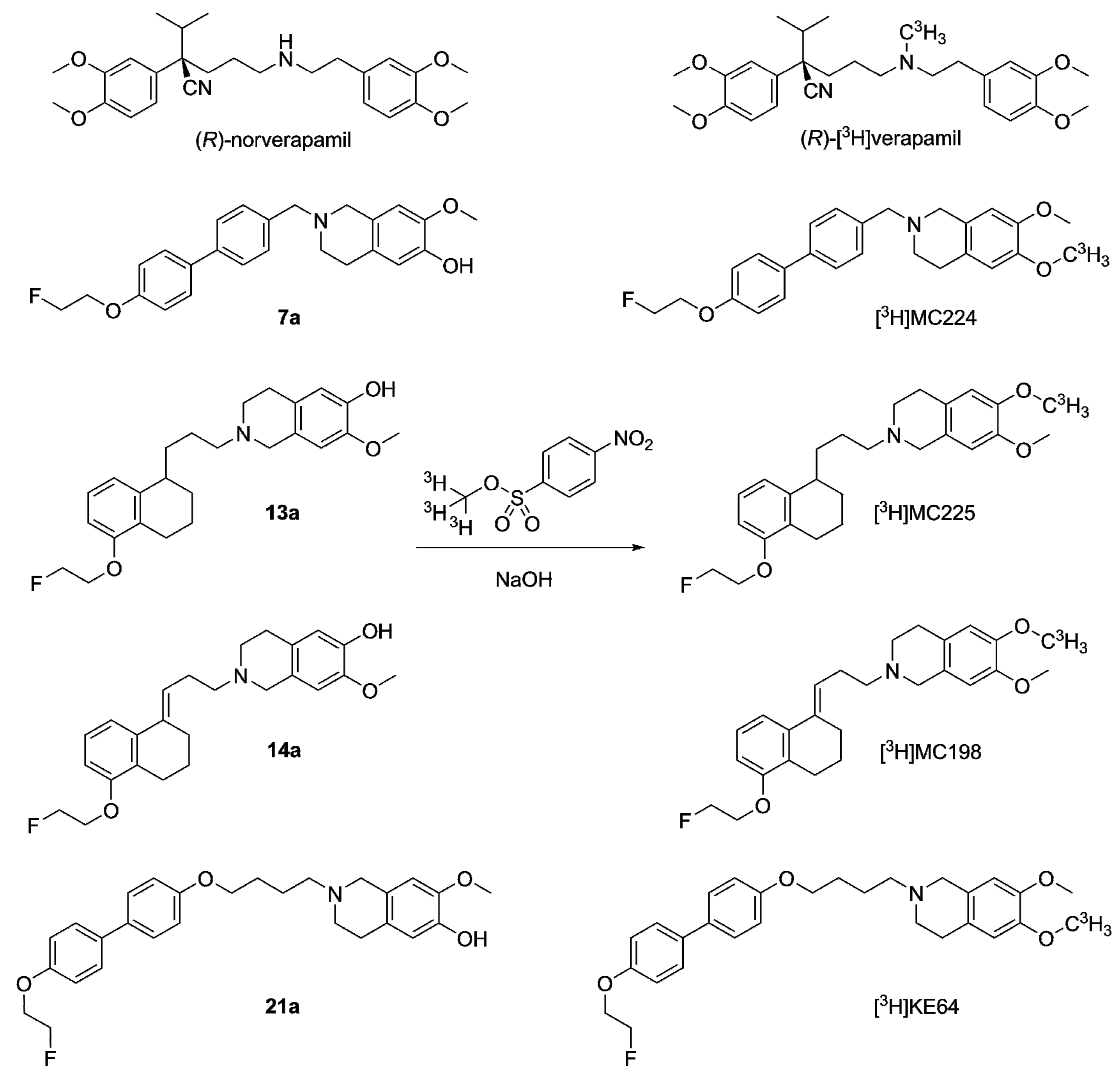
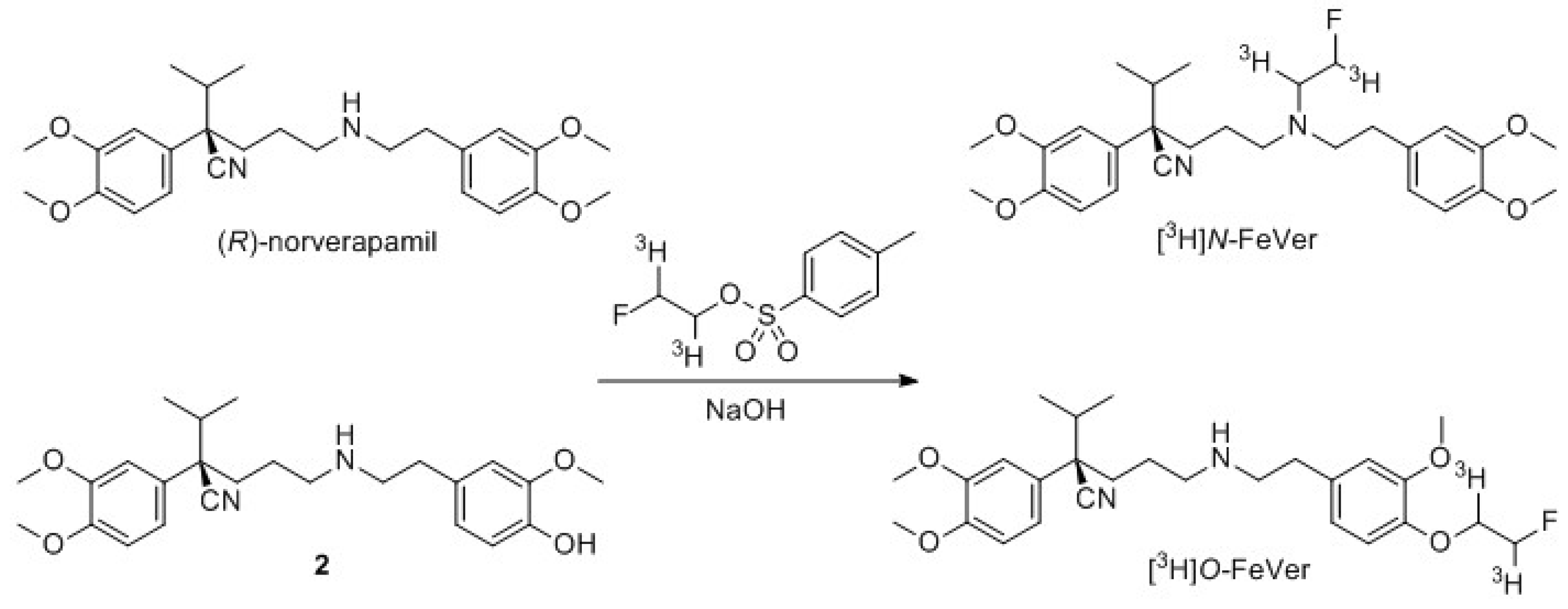
2.2.1. Tritium Labeling
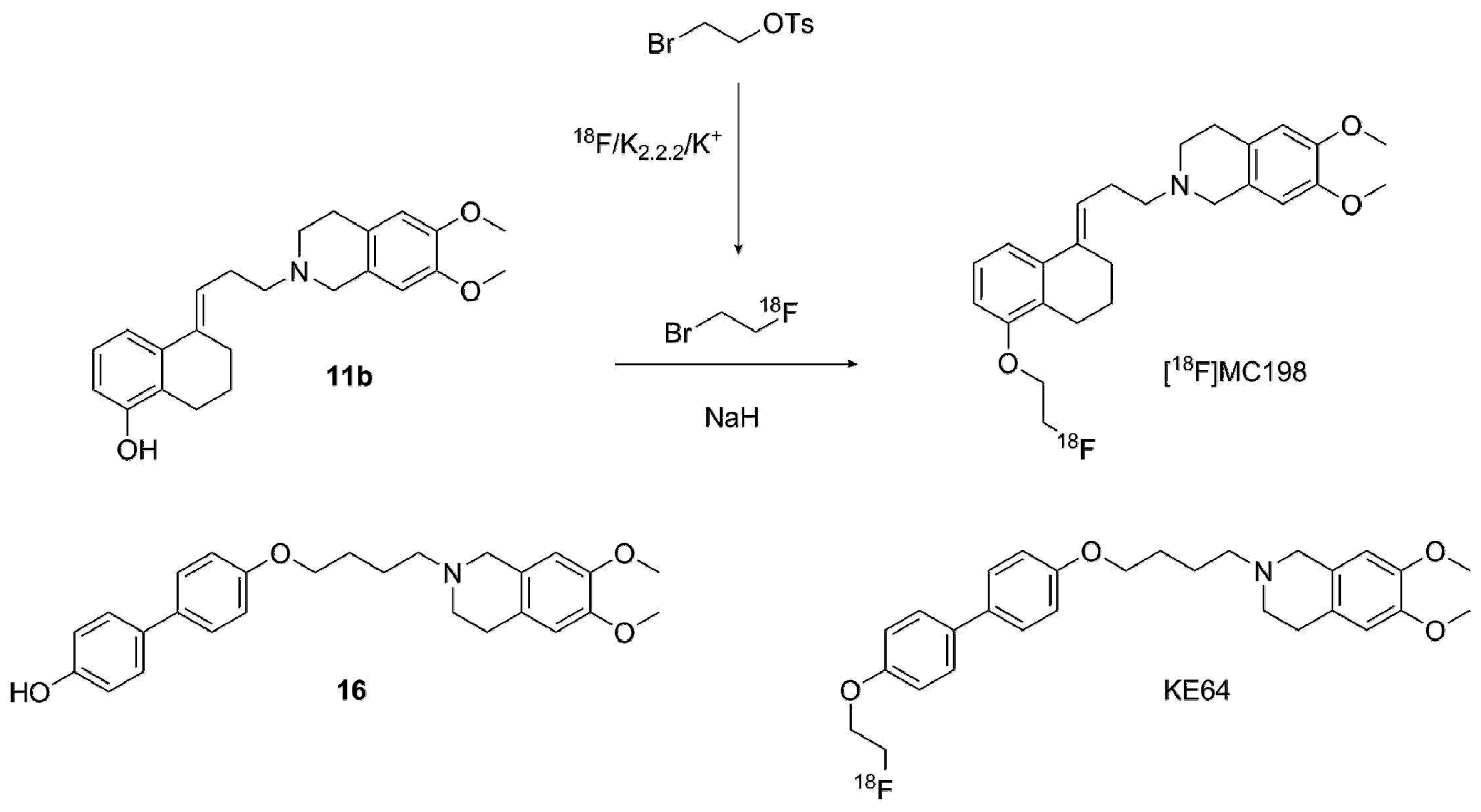
2.2.2. Radiolabeling of [18F]MC198 and [18F]KE64
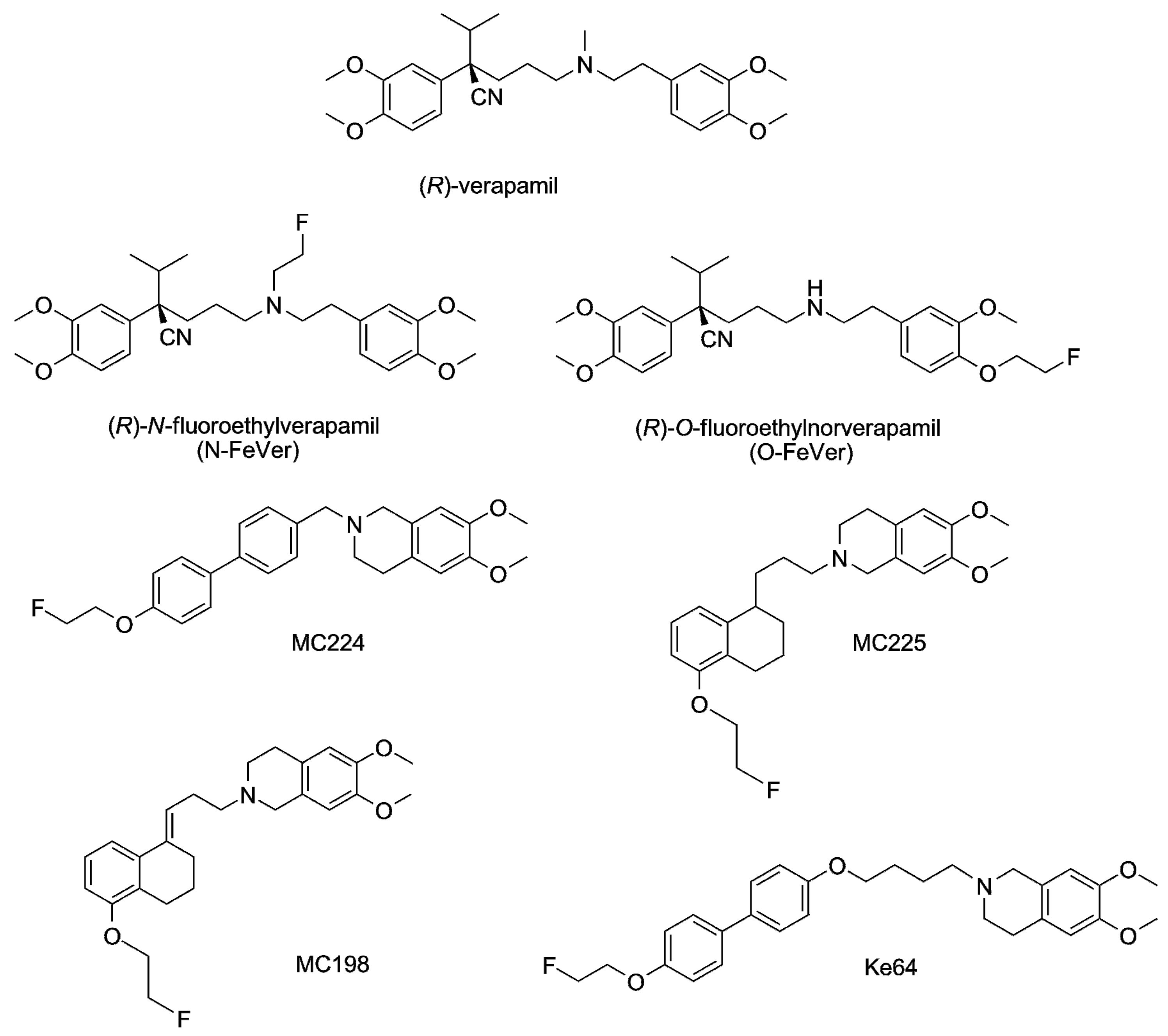
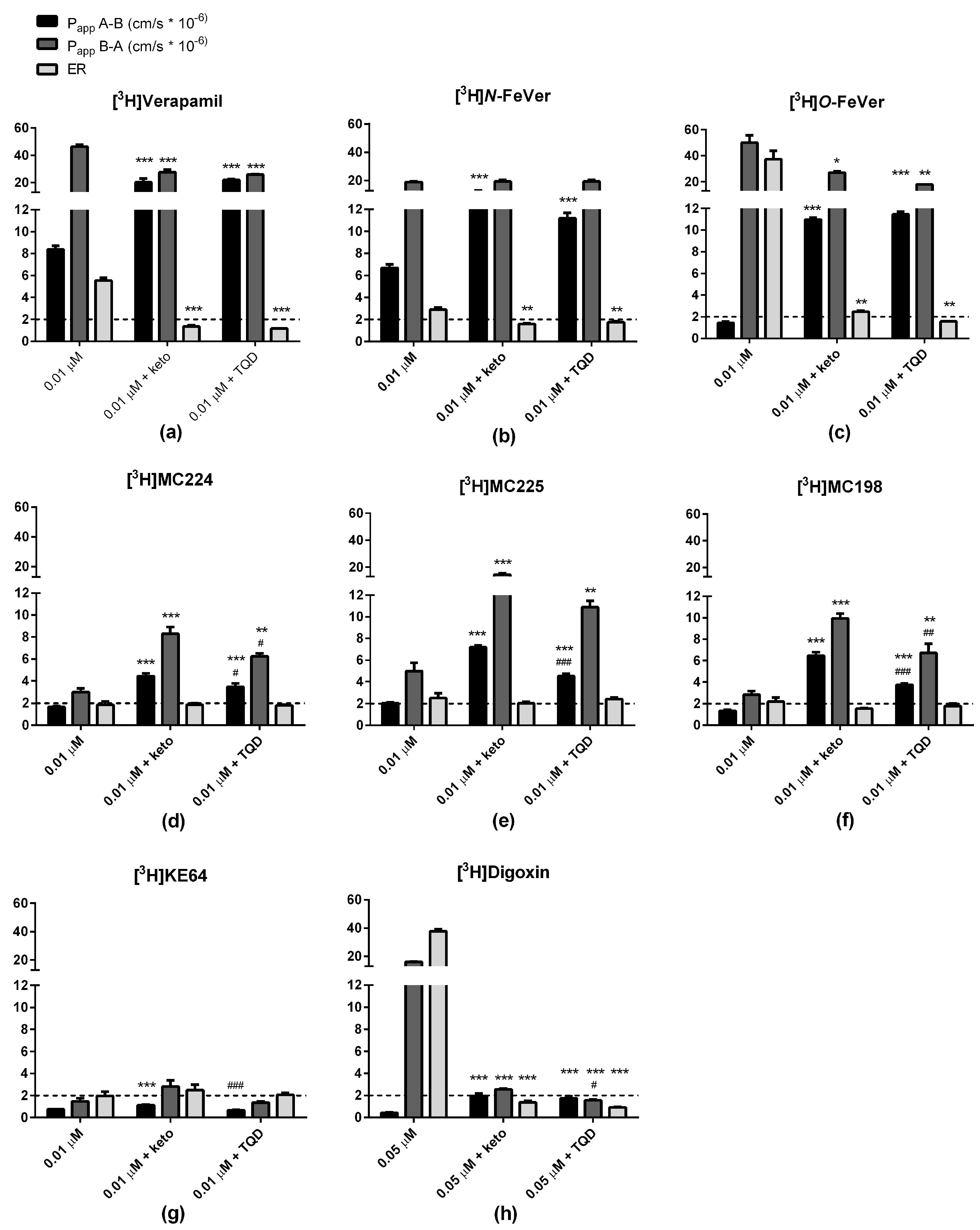
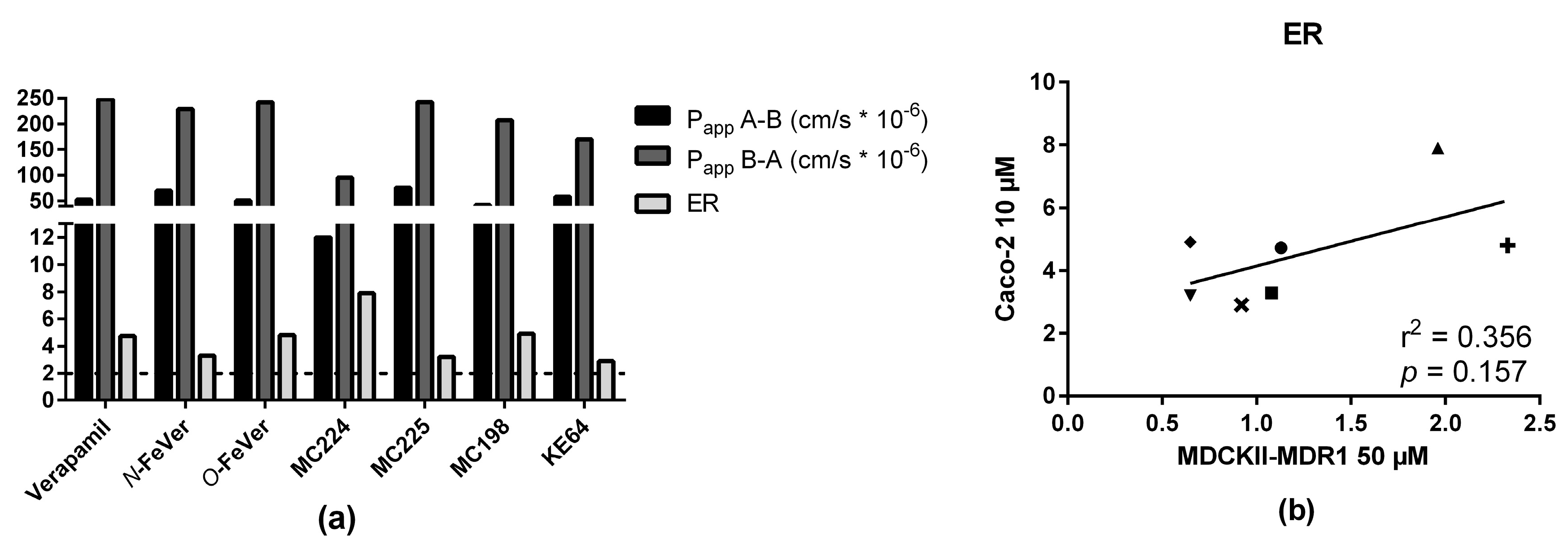
2.3. In Vitro Experiments
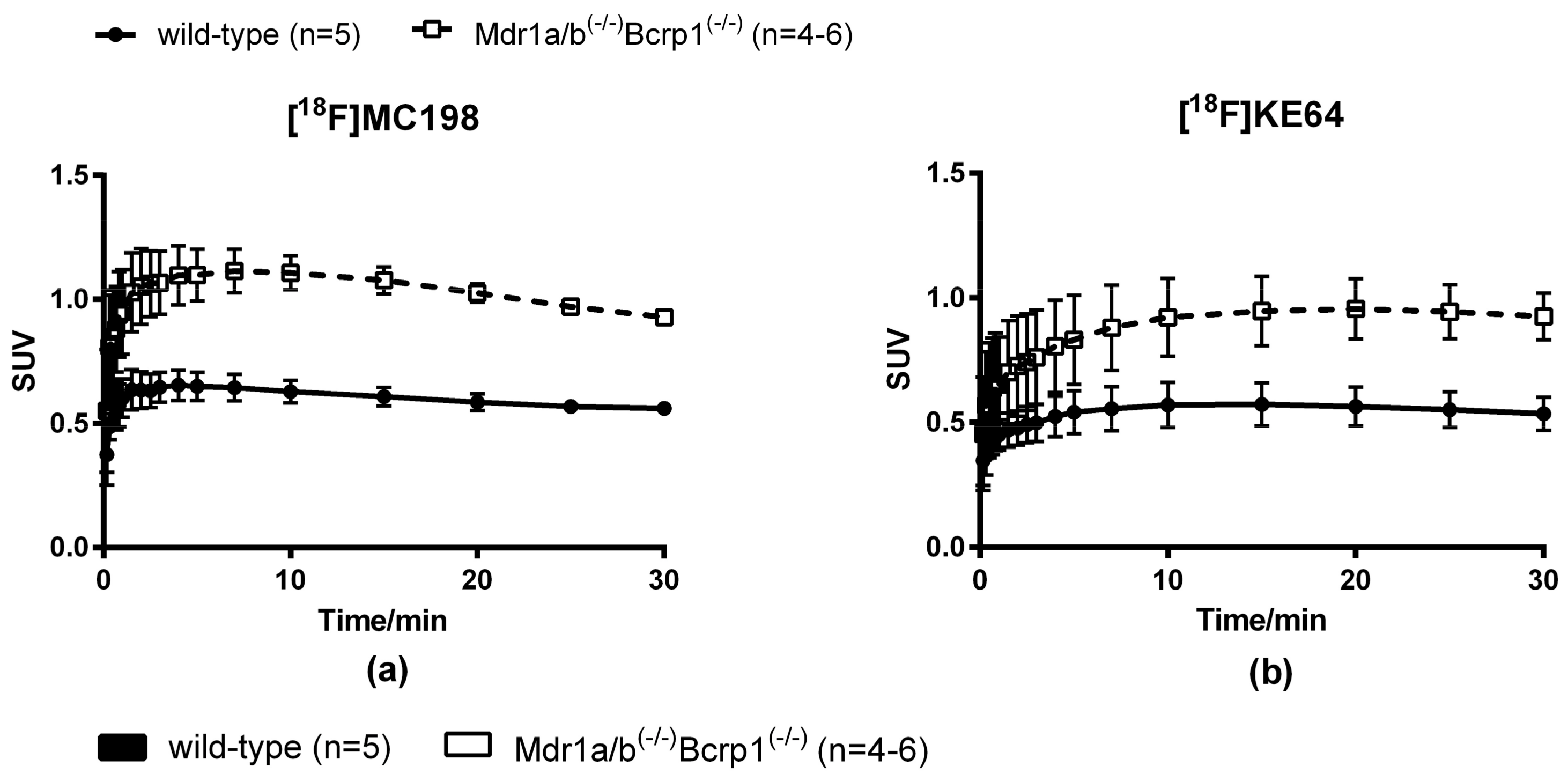
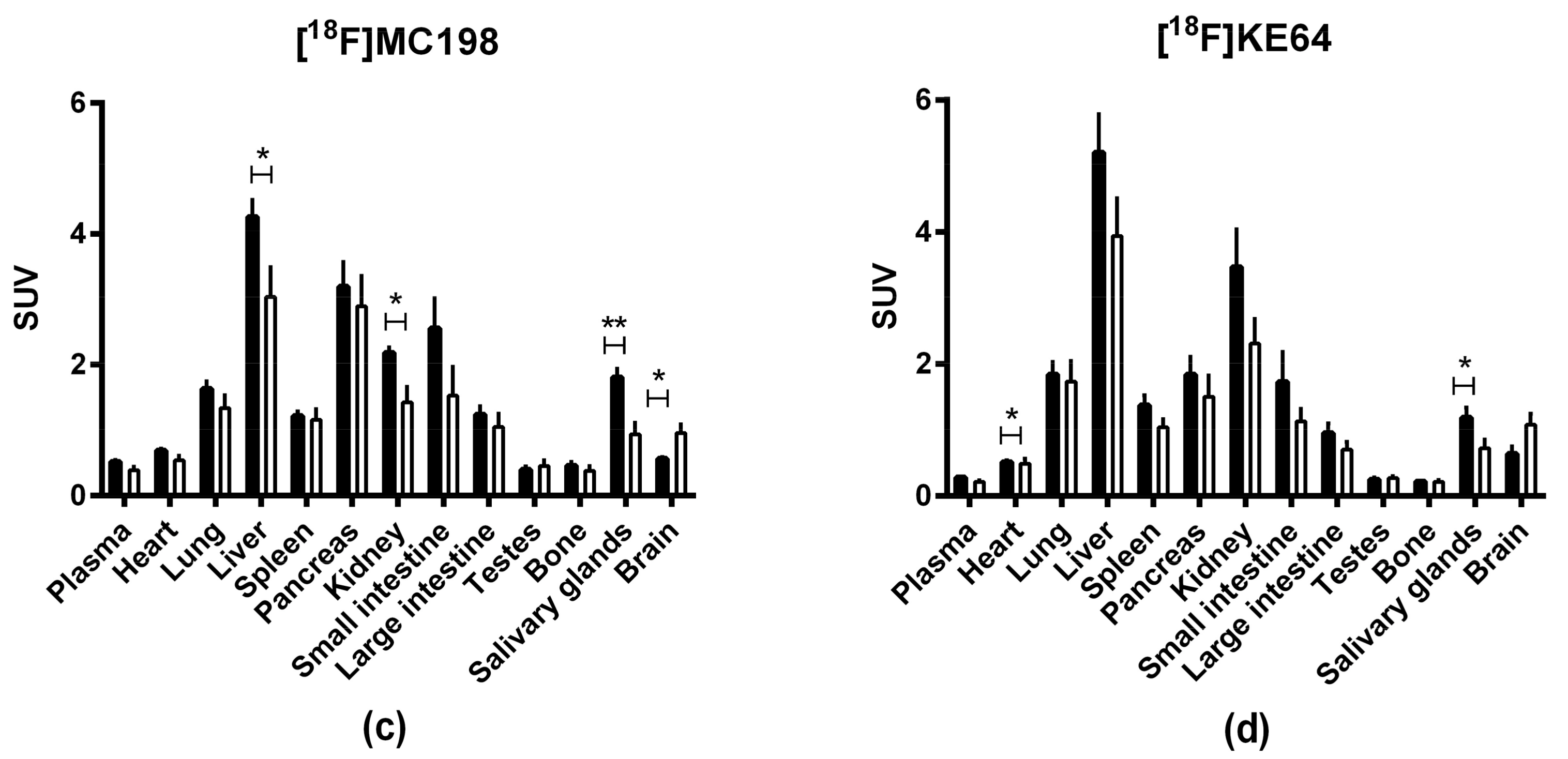
2.4. In Vivo Evaluation of [18F]MC198 and [18F]KE64
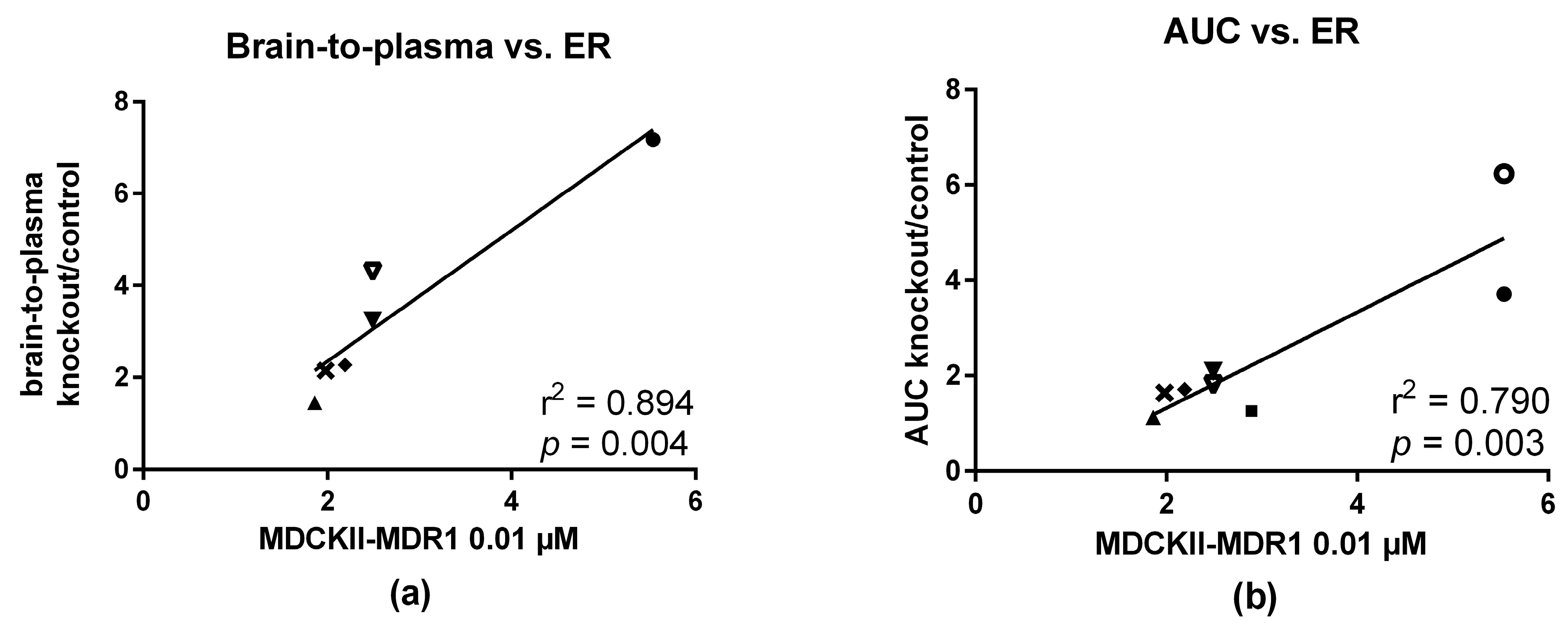
2.5. In Vitro—In Vivo Correlation
3. Discussion
3.1. Verapamil, N-FeVer and O-FeVer Behavior
3.2. MC224, MC225, MC198 and KE64 Behavior
3.3. Calcein-AM Assay
3.4. In Vitro and In Vivo Correlation
4. Materials and Methods
4.1. Chemicals
4.2. Organic Synthesis
General Methods
4.3. Radiochemistry
4.3.1. General Methods in Tritium Labeling
4.3.2. [3H]Verapamil (Scheme 5)
4.3.3. [3H]N-FeVer (Scheme 6)
4.3.4. [3H]O-FeVer (Scheme 6)
4.3.5. [3H]MC224 (Scheme 5)
4.3.6. [3H]MC225 (Scheme 5)
4.3.7. [3H]MC198 (Scheme 5)
4.3.8. [3H]KE64 (Scheme 5)
4.3.9. Radiosynthesis of [18F]MC198 and [18F]KE64 (Scheme 7)
4.4. In Vitro Experiments with Tritiated Molecules
4.4.1. Cell Culture
4.4.2. Assessment of Monolayer Integrity
4.4.3. Bidirectional Transport of Tritiated Compounds in MDCKII-MDR1 Cells
4.4.4. Calculations
4.5. In Vitro Experiments with Non-Radioactive Molecules
4.6. Animal Experiments with [18F]MC198 and [18F]KE64
PET Procedure and Data Analysis
4.7. Statistical Analysis
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhang, L.; Strong, J.M.; Qiu, W.; Lesko, L.J.; Huang, S. Scientific Perspectives on Drug Transporters and Their Role in Drug Interactions. Mol. Pharm. 2006, 3, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Bikadi, Z.; Rosenberg, M.F.; Mao, Q. Structure and function of the human breast cancer resistance protein (BCRP/ABCG2). Curr. Drug Metab. 2010, 11, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Van Waterschoot, R.A.B.; Schinkel, A.H. A critical analysis of the interplay between cytochrome P450 3A and P-glycoprotein: Recent insights from knockout and transgenic mice. Pharmacol. Rev. 2011, 63, 390–410. [Google Scholar] [CrossRef] [PubMed]
- Prueksaritanont, T.; Chu, X.; Gibson, C.; Cui, D.; Yee, K.L.; Ballard, J.; Cabalu, T.; Hochman, J. Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J. 2013, 15, 629–645. [Google Scholar] [CrossRef] [PubMed]
- Bailey, C.A.; Bryla, P.; Malick, A.W. The use of the intestinal epithelial cell culture model, Caco-2, in pharmaceutical development. Adv. Drug Deliv. Rev. 1996, 22, 85–103. [Google Scholar] [CrossRef]
- Tang, F.; Horie, K.; Borchardt, R.T. Are MDCK Cells Transfected with the Human MDR1 Gene a Good Model of the Human Intestinal Mucosa? Pharm. Res. 2002, 19, 765–772. [Google Scholar] [CrossRef] [PubMed]
- Dukes, J.D.; Whitley, P.; Chalmers, A.D. The MDCK variety pack: Choosing the right strain. BMC Cell Biol. 2011, 12, 43. [Google Scholar] [CrossRef] [PubMed]
- Xia, C.C.Q.; Liu, N.; Yang, D.; Miwa, G.; Gan, L.-S. Expression, localization, and functional characteristics of breast cancer resistance protein in Caco-2 cells. Drug Metab. Dispos. 2005, 33, 637–643. [Google Scholar] [CrossRef] [PubMed]
- Walle, U.K.; Galijatovic, A.; Walle, T. Transport of the flavonoid chrysin and its conjugated metabolites by the human intestinal cell line Caco-2. Biochem. Pharmacol. 1999, 58, 431–438. [Google Scholar] [CrossRef]
- Feng, B.; Mills, J.B.; Davidson, R.E.; Mireles, R.J.; Janiszewski, J.S.; Troutman, M.D.; De Morais, S.M. In vitro P-glycoprotein assays to predict the in vivo interactions of P-glycoprotein with drugs in the central nervous system. Drug Metab. Dispos. 2008, 36, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Polli, J.W.; Wring, S.A.; Humphreys, J.E.; Huang, L.; Morgan, J.B.; Webster, L.O.; Serabjit-Singh, C.S. Rational use of in vitro P-glycoprotein assays in drug discovery. J. Pharmacol. Exp. Ther. 2001, 299, 620–628. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, R.; De Morais, S.M.; Kalvass, J.C. In vitro P-glycoprotein efflux ratio can predict the in vivo brain penetration regardless of biopharmaceutics drug disposition classification system class. Drug Metab. Dispos. 2013, 41, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, R.; Windhorst, A.; Elsinga, P.; Colabufo, N.; Lammertsma, A.; Luurtsema, G. Radiopharmaceuticals for assessing ABC transporters at the blood-brain barrier. Clin. Pharmacol. Ther. 2015, 97, 362–371. [Google Scholar] [CrossRef] [PubMed]
- Van Assema, D.M.E.; van Berckel, B.N.M. Blood-Brain Barrier ABC-transporter P-glycoprotein in Alzheimer’s Disease: Still a Suspect? Curr. Pharm. Des. 2016, 22, 1–8. [Google Scholar] [CrossRef]
- Zhang, C.; Kwan, P.; Zuo, Z.; Baum, L. The transport of antiepileptic drugs by P-glycoprotein. Adv. Drug Deliv. Rev. 2012, 64, 930–942. [Google Scholar] [CrossRef] [PubMed]
- Bankstahl, J.P.; Bankstahl, M.; Römermann, K.; Wanek, T.; Stanek, J.; Windhorst, A.D.; Fedrowitz, M.; Erker, T.; Müller, M.; Löscher, W.; et al. Tariquidar and elacridar are dose-dependently transported by P-glycoprotein and Bcrp at the blood-brain barrier: A small-animal positron emission tomography and in vitro study. Drug Metab. Dispos. 2013, 41, 754–762. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, P.; Bui, T.; Ho, R.J.Y.; Unadkat, J.D. In Vitro-to-in Vivo Prediction of P-glycoprotein-Based Drug Interactions at the Human and Rodent Blood-Brain Barrier. Drug Metab. Dispos. 2008, 36, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Römermann, K.; Wanek, T.; Bankstahl, M.; Bankstahl, J.P.; Fedrowitz, M.; Müller, M.; Löscher, W.; Kuntner, C.; Langer, O. (R)-[11C]verapamil is selectively transported by murine and human P-glycoprotein at the blood-brain barrier, and not by MRP1 and BCRP. Nucl. Med. Biol. 2013, 40, 873–878. [Google Scholar] [CrossRef] [PubMed]
- Raaphorst, R.M.; Luurtsema, G.; Schuit, R.C.; Kooijman, E.J.M.; Elsinga, P.H.; Lammertsma Adriaan, A.; Windhorst, A.D. Synthesis and evaluation of new fluorine-18 labeled verapamil analogs to investigate the function of P-glycoprotein in the blod-brain barrier. ACS Chem. Neurosci. 2017, in press. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, H.; Cantore, M.; Colabufo, N.A.; Elsinga, P.H.; Windhorst, A.D.; Luurtsema, G. Synthesis and preclinical evaluation of three novel fluorine-18 labeled radiopharmaceuticals for P-Glycoprotein PET imaging at the blood–brain barrier. Mol. Pharm. 2015, 12, 2265–2275. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, H.; Windhorst, A.D.; Elsinga, P.H.; Cantore, M.; Colabufo, N.A.; Willemsen, A.T.M.; Luurtsema, G. Evaluation of [18F]MC225 as a PET radiotracer for measuring P-glycoprotein function at the blood-brain barrier in rats: Kinetics, metabolism, and selectivity. J. Cereb. Blood Flow Metab. 2017, 37, 1286–1298. [Google Scholar] [CrossRef] [PubMed]
- Savolainen, H.; Meerlo, P.; Elsinga, P.H.; Windhorst, A.D.; Dierckx, R.A.J.O.; Colabufo, N.A.; van Waarde, A.; Luurtsema, G. P-glycoprotein function in the rodent brain displays a daily rhythm, a quantitative in vivo PET study. AAPS J. 2016, 18, 1524–1531. [Google Scholar] [CrossRef] [PubMed]
- Weidner, L.D.; Fung, K.L.; Kannan, P.; Moen, J.K.; Kumar, J.S.; Mulder, J.; Innis, R.B.; Gottesman, M.M.; Hall, M. Tariquidar is an Inhibitor and Not a Substrate of Human and Mouse P-glycoprotein. Drug Metab. Dispos. 2015, 44, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Wang, E.J.; Lew, K.; Casciano, C.N.; Clement, R.P.; Johnson, W.W. Interaction of common azole antifungals with P glycoprotein. Antimicrob. Agents Chemother. 2002, 46, 160–165. [Google Scholar] [CrossRef] [PubMed]
- Keogh, J.P.; Kunta, J.R. Development, validation and utility of an in vitro technique for assessment of potential clinical drug-drug interactions involving P-glycoprotein. Eur. J. Pharm. Sci. 2006, 27, 543–554. [Google Scholar] [CrossRef] [PubMed]
- Van Waarde, A.; Ramakrishnan, N.K.; Rybczynska, A.A.; Elsinga, P.H.; Berardi, F.; de Jong, J.R.; Kwizera, C.; Perrone, R.; Cantore, M.; Sijbesma, J.W.A.; et al. Synthesis and preclinical evaluation of novel PET probes for P-glycoprotein function and expression. J. Med. Chem. 2009, 52, 4524–4532. [Google Scholar] [CrossRef] [PubMed]
- Capparelli, E.; Zinzi, L.; Cantore, M.; Contino, M.; Perrone, M.G.; Luurtsema, G.; Berardi, F.; Perrone, R.; Colabufo, N.A. SAR Studies on Tetrahydroisoquinoline Derivatives: The Role of Flexibility and Bioisosterism To Raise Potency and Selectivity toward P-glycoprotein. J. Med. Chem. 2014, 57, 9983–9994. [Google Scholar] [CrossRef] [PubMed]
- Goos, J.A.C.M.; Verbeek, J.; Geldof, A.A.; Hiemstra, A.C.; van de Wiel, M.A.; Adamzek, K.A.; Delis-Van Diemen, P.M.; Stroud, S.G.; Bradley, D.P.; Meijer, G.A.; et al. Molecular imaging of aurora kinase A (AURKA) expression: Synthesis and preclinical evaluation of radiolabeled alisertib (MLN8237). Nucl. Med. Biol. 2016, 43, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Verbeek, J.; Syvänen, S.; Schuit, R.C.; Eriksson, J.; de Lange, E.C.; Windhorst, A.D.; Luurtsema, G.; Lammertsma, A.A. Synthesis and preclinical evaluation of [11C]D617, a metabolite of (R)-[11C]verapamil. Nucl. Med. Biol. 2012, 39, 530–539. [Google Scholar] [CrossRef] [PubMed]
- Adachi, Y.; Suzuki, H.; Sugiyama, Y. Comparative studies on in vitro methods for evaluating in vivo function of MDR1 P-glycoprotein. Pharm. Res. 2001, 18, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Neway, W.E.; Ohe, T.; Chen, I.; Rowe, J.F.; Hochman, J.H.; Chiba, M.; Lin, J.H. In vitro substrate identification studies for p-glycoprotein-mediated transport: species difference and predictability of in vivo results. J. Pharmacol. Exp. Ther. 2001, 296, 723–735. [Google Scholar] [PubMed]
- Schwab, D.; Fischer, H.; Tabatabaei, A.; Poli, S.; Huwyler, J. Comparison of in vitro P-glycoprotein screening assays: recommendations for their use in drug discovery. J. Med. Chem. 2003, 46, 1716–1725. [Google Scholar] [CrossRef] [PubMed]
- Bentz, J.; O’Connor, M.P.; Bednarczyk, D.; Coleman, J.; Lee, C.; Palm, J.; Pak, Y.A.; Perloff, E.S.; Reyner, E.; Balimane, P.; et al. Variability in P-glycoprotein inhibitory potency (IC₅₀) using various in vitro experimental systems: implications for universal digoxin drug-drug interaction risk assessment decision criteria. Drug Metab. Dispos. 2013, 41, 1347–1366. [Google Scholar] [CrossRef] [PubMed]
- Pavek, P.; Merino, G.; Wagenaar, E.; Bolscher, E.; Novotna, M.; Jonker, J.W.; Schinkel, A.H. Human breast cancer resistance protein: interactions with steroid drugs, hormones, the dietary carcinogen 2-amino-1-methyl-6-phenylimidazo(4,5-b)pyridine, and transport of cimetidine. J. Pharmacol. Exp. Ther. 2005, 312, 144–152. [Google Scholar] [CrossRef] [PubMed]
- Evers, R.; Kool, M.; Smith, A.J.; van Deemter, L.; de Haas, M.; Borst, P. Inhibitory effect of the reversal agents V-104, GF120918 and Pluronic L61 on MDR1 Pgp-, MRP1- and MRP2-mediated transport. Br. J. Cancer 2000, 83, 366–374. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: a tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Lagas, J.S.; Kruijssen, C.M.M.; van der Wetering, K.; van de Beijnen, J.H.; Schinkel, A.H. Transport of Diclofenac by Breast Cancer Resistance Protein (ABCG2) and Stimulation of Multidrug Resistance Protein 2 (ABCC2) -Mediated Drug Transport by Diclofenac and Benzbromarone. Drug Metab. Dispos. 2009, 37, 129–136. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Contino, M.; Cantore, M.; Capparelli, E.; Grazia, M.; Cassano, G.; Gasparre, G.; Leopoldo, M.; Berardi, F.; Perrone, R. Naphthalenyl derivatives for hitting P-gp/MRP1/BCRP transporters. Bioorg. Med. Chem. 2013, 21, 1324–1332. [Google Scholar] [CrossRef] [PubMed]
- Colabufo, N.A.; Berardi, F.; Perrone, R.; Rapposelli, S.; Digiacomo, M.; Vanni, M. 2-[(3-Methoxyphenylethyl)phenoxy]-Based ABCB1 Inhibitors: Effect of Different Basic Side-Chains on Their Biological Properties. J. Med. Chem. 2008, 51, 7602–7613. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Hof, P.R.; Grant, S.C.; Blackband, S.J.; Bennett, R.; Slatest, L.; McGuigan, M.D.; Benveniste, H. A three-dimensional digital atlas database of the adult C57BL/6J mouse brain by magnetic resonance microscopy. Neuroscience 2005, 135, 1203–1215. [Google Scholar] [CrossRef] [PubMed]















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| In Vitro | In Vivo | |||||||
|---|---|---|---|---|---|---|---|---|
| MDCKII-MDR1 Cells | ||||||||
| Compound | ER 0.01 µM | Calcein-AM EC50 (µM) | Substrate In Vitro | Brain-to-Plasma Knockout or Treated/Control | AUC Whole Brain Knockout or Treated/Control | Substrate In Vivo | ||
| Mice | Rats | Mice | Rats | |||||
| Verapamil | 5.5 | 1.1 | + | 7.1 2 | 6.2 1 3.7 2 | 7.1 3 | + | |
| N-FeVer | 2.8 | 1.1 | + | 5.8 3 | 1.2 1 | 3.7 3 | + | |
| O-FeVer | 37 | 2.4 | + | 6.51 | 5.9 1 | 2.4 3 | + | |
| MC224 | 1.8 | 0.54 | − | 1.42 | 1.1 2 | − | ||
| MC225 | 2.4 | 0.35 | + | 4.3 1 3.2 2 | 1.9 4 | 1.8 1 2.02 | 3.7 4 | + |
| MC198 | 2.1 | 0.27 | + | 2.2 2 | 1.72 | + | ||
| KE64 | 1.9 | 0.014 | − | 2.1 2 | 1.62 | + | ||
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Raaphorst, R.M.; Savolainen, H.; Cantore, M.; Van de Steeg, E.; Van Waarde, A.; Colabufo, N.A.; Elsinga, P.H.; Lammertsma, A.A.; Windhorst, A.D.; Luurtsema, G. Comparison of In Vitro Assays in Selecting Radiotracers for In Vivo P-Glycoprotein PET Imaging. Pharmaceuticals 2017, 10, 76. https://doi.org/10.3390/ph10030076
Raaphorst RM, Savolainen H, Cantore M, Van de Steeg E, Van Waarde A, Colabufo NA, Elsinga PH, Lammertsma AA, Windhorst AD, Luurtsema G. Comparison of In Vitro Assays in Selecting Radiotracers for In Vivo P-Glycoprotein PET Imaging. Pharmaceuticals. 2017; 10(3):76. https://doi.org/10.3390/ph10030076
Chicago/Turabian StyleRaaphorst, Renske M., Heli Savolainen, Mariangela Cantore, Evita Van de Steeg, Aren Van Waarde, Nicola A. Colabufo, Philip H. Elsinga, Adriaan A. Lammertsma, Albert D. Windhorst, and Gert Luurtsema. 2017. "Comparison of In Vitro Assays in Selecting Radiotracers for In Vivo P-Glycoprotein PET Imaging" Pharmaceuticals 10, no. 3: 76. https://doi.org/10.3390/ph10030076
APA StyleRaaphorst, R. M., Savolainen, H., Cantore, M., Van de Steeg, E., Van Waarde, A., Colabufo, N. A., Elsinga, P. H., Lammertsma, A. A., Windhorst, A. D., & Luurtsema, G. (2017). Comparison of In Vitro Assays in Selecting Radiotracers for In Vivo P-Glycoprotein PET Imaging. Pharmaceuticals, 10(3), 76. https://doi.org/10.3390/ph10030076