
Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin


 ,
,  ,
, 
 ,
,  ,
, 

Abstract
:1. Introduction
2. Results and Discussion
2.1. Cyclooxygenase Inhibition Studies
2.2. Human Serum Albumin Affinity Studies
3. Materials and Methods
3.1. Compounds
3.2. In vitro Cyclooxygenase Inhibition Studies
3.3. Interaction with Human Serum Albumin by Fluorescence Quenching
Assessment of HSA-CDX Binding Parameters
3.4. Computational Studies
3.4.1. Preparation of CDXs, Controls, Decoys, and Macromolecules
3.4.2. Docking
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Tiritan, M.E.; Ribeiro, A.R.; Fernandes, C.; Pinto, M. Chiral pharmaceuticals. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 1–28. [Google Scholar]
- Blaser, H.U. Chirality and its implications for the pharmaceutical industry. Rend. Lincei 2013, 24, 213–216. [Google Scholar] [CrossRef]
- Brocks, D.R. Drug disposition in three dimensions: An update on stereoselectivity in pharmacokinetics. Biopharm. Drug Dispos. 2006, 27, 387–406. [Google Scholar] [CrossRef] [PubMed]
- Mannschreck, A.; Kiesswetter, R.; von Angerer, E. Unequal activities of enantiomers via biological receptors: Examples of chiral drug, pesticide, and fragrance molecules. J. Chem. Educ. 2007, 84, 2012–2017. [Google Scholar] [CrossRef]
- Smith, S.W. Chiral toxicology: It's the same thing only different. Toxicol. Sci. 2009, 110, 4–30. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, B.S. Exploiting the power of stereochemistry in drugs: An overview of racemic and enantiopure drugs. J. Mod. Med. Chem. 2013, 1, 10–36. [Google Scholar] [CrossRef]
- Triggle, D.J. Stereoselectivity of drug action. Drug Discov. Today 1997, 2, 138–147. [Google Scholar] [CrossRef]
- Kasprzyk-Hordern, B. Pharmacologically active compounds in the environment and their chirality. Chem. Soc. Rev. 2010, 39, 4466–4503. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.J.; Calvo, L.; Fernandez, F.; Carganico, G.; Bastida, E.; Bujons, J.; Messeguer, A. Biological activity of ketoprofen and its optical isomers. Eur. J. Pharmacol. 1990, 183, 2263–2264. [Google Scholar] [CrossRef]
- Sánchez, T.; Moreno, J.J. ketoprofen S(+)-enantiomer inhibits prostaglandin production and cell growth in 3T6 fibroblast cultures. Eur. J. Pharmacol. 1999, 370, 63–67. [Google Scholar] [CrossRef]
- Kolluri, S.K.; Corr, M.; James, S.Y.; Bernasconi, M.; Lu, D.; Liu, W.; Cottam, H.B.; Leoni, L.M.; Carson, D.A.; Zhang, X.K. The R-enantiomer of the nonsteroidal antiinflammatory drug etodolac binds retinoid x receptor and induces tumor-selective apoptosis. Proc. Natl. Acad. Sci. USA 2005, 102, 2525–2530. [Google Scholar] [CrossRef] [PubMed]
- Trainor, G.L. The importance of plasma protein binding in drug discovery. Expert Opin. Drug Discov. 2007, 2, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Kerns, E.H.; Di, L. Drug-Like Properties: Concepts, Structure Design and Methods; Elsevier Inc.: Burlington, MA, USA, 2008. [Google Scholar]
- Fasano, M.; Curry, S.; Terreno, E.; Galliano, M.; Fanali, G.; Narciso, P.; Notari, S.; Ascenzi, P. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 2005, 57, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Shen, Q.; Wang, L.; Zhou, H.; Jiang, H.D.; Yu, L.S.; Zeng, S. Stereoselective binding of chiral drugs to plasma proteins. Acta Pharmacol. Sin. 2013, 34, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Goodman, G.; Gilman. The Pharmacological Basis of Therapeutics, 9th ed.; McGraw-Hill: New York, NY, USA, 1996. [Google Scholar]
- Kratochwil, N.A.; Huber, W.; Müller, F.; Kansy, M.; Gerber, P.R. Predicting plasma protein binding of drugs: A new approach. Biochem. Pharmacol. 2002, 64, 1355–1374. [Google Scholar] [CrossRef]
- Oravcova, J.; Bohs, B.; Lindner, W. Drug-protein binding studies—new trends in analytical and experimental methodology. J. Chromatogr. B Biomed. Appl. 1996, 677, 1–28. [Google Scholar] [CrossRef]
- Paal, K.; Shkarupin, A. Paclitaxel binding to the fatty acid-induced conformation of human serum albumin—automated docking studies. Bioorg. Med. Chem. 2007, 15, 7568–7575. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.; Huang, Y.M.; Ma, X.L.; Liao, X.X.; Wang, Q.; Xiong, X.N.; Li, H. Multispectroscopic and docking studies on the binding of chlorogenic acid isomers to human serum albumin: Effects of esteryl position on affinity. Food Chem. 2016, 212, 434–442. [Google Scholar] [CrossRef] [PubMed]
- Pouli, N.; Marakos, P. Fused xanthone derivatives as antiproliferative agents. Anticancer Agents Med. Chem. 2009, 9, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Palmeira, A.; Paiva, A.; Sousa, E.; Seca, H.; Almeida, G.M.; Lima, R.T.; Fernandes, M.X.; Pinto, M.; Vasconcelos, M.H. Insights into the in vitro antitumor mechanism of action of a new pyranoxanthone. Chem. Biol. Drug Des. 2010, 76, 43–58. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.; Paiva, A.; Nazareth, N.; Gales, L.; Damas, A.M.; Nascimento, M.S.J.; Pinto, M. Bromoalkoxyxanthones as promising antitumor agents: Synthesis, crystal structure and effect on human tumor cell lines. Eur. J. Med. Chem. 2009, 44, 3830–3835. [Google Scholar] [CrossRef] [PubMed]
- Panthong, K.; Hutadilok-Towatana, N.; Panthong, A. Cowaxanthone f, a new tetraoxygenated xanthone, and other anti-inflammatory and antioxidant compounds from garcinia cowa. Can. J. Chem. 2009, 87, 1636–1640. [Google Scholar] [CrossRef]
- Nakatani, K.; Nakahata, N.; Arakawa, T.; Yasuda, H.; Ohizumi, Y. Inhibition of cyclooxygenase and prostaglandin E2 synthesis by γ-mangostin, a xanthone derivative in mangosteen, in C6 rat glioma cells. Biochem. Pharmacol. 2002, 63, 73–79. [Google Scholar] [CrossRef]
- Nakatani, K.; Yamakuni, T.; Kondo, N.; Arakawa, T.; Oosawa, K.; Shimura, S.; Inoue, H.; Ohizumi, Y. Γ-mangostin inhibits inhibitor-κB kinase activity and decreases lipopolysaccharide-lnduced cyclooxygenase-2 gene expression in C6 rat glioma cells. Mol. Pharmacol. 2004, 66, 667–674. [Google Scholar] [CrossRef] [PubMed]
- Shagufta; Ahmad, I. Recent insight into the biological activities of synthetic xanthone derivatives. Eur. J. Med. Chem. 2016, 116, 267–280. [Google Scholar]
- Pinto, M.M.M.; Sousa, M.E.; Nascimento, M.S.J. Xanthone derivatives: New insights in biological activities. Curr. Med. Chem. 2005, 12, 2517–2538. [Google Scholar] [CrossRef] [PubMed]
- Vieira, L.M.M.; Kijjoa, A. Naturally-occurring xanthones: Recent developments. Curr. Med. Chem. 2005, 12, 2413–2446. [Google Scholar] [CrossRef] [PubMed]
- Masters, K.S.; Bräse, S. Xanthones from fungi, lichens, and bacteria: The natural products and their synthesis. Chem. Rev. 2012, 112, 3717–3776. [Google Scholar] [CrossRef] [PubMed]
- Pinto, M.M.M.; Castanheiro, R.A.P.; Kijjoa, A. Xanthones from marine-derived microorganisms: Isolation, structure elucidation, and biological activities. In Encyclopedia of Analytical Chemistry; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2014; Vol. 27, pp. 1–21. [Google Scholar]
- Azevedo, C.M.G.; Afonso, C.M.M.; Sousa, D.; Lima, R.T.; Helena Vasconcelos, M.; Pedro, M.; Barbosa, J.; Corrêa, A.G.; Reis, S.; Pinto, M.M.M. Multidimensional optimization of promising antitumor xanthone derivatives. Bioorg. Med. Chem. 2013, 21, 2941–2959. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.E.; Pinto, M.M.M. Synthesis of xanthones: An overview. Curr. Med. Chem. 2005, 12, 2447–2479. [Google Scholar] [CrossRef] [PubMed]
- Waszkielewicz, A.M.; Słoczyńska, K.; Pękala, E.; Żmudzki, P.; Siwek, A.; Gryboś, A.; Marona, H. Design, synthesis, and anticonvulsant activity of some derivatives of xanthone with aminoalkanol moieties. Chem. Biol. Drug Des. 2017, 89, 339–352. [Google Scholar] [CrossRef] [PubMed]
- Szkaradek, N.; Rapacz, A.; Pytka, K.; Filipek, B.; Siwek, A.; Cegła, M.; Marona, H. Synthesis and preliminary evaluation of pharmacological properties of some piperazine derivatives of xanthone. Bioorg. Med. Chem. 2013, 21, 514–522. [Google Scholar] [CrossRef] [PubMed]
- Marona, H.; Pekala, E.; Antkiewicz-Michaluk, L.; Walczak, M.; Szneler, E. Anticonvulsant activity of some xanthone derivatives. Bioorg. Med. Chem. 2008, 16, 7234–7244. [Google Scholar] [CrossRef] [PubMed]
- Sousa, E.P.; Silva, A.M.S.; Pinto, M.M.M.; Pedro, M.M.; Cerqueira, F.A.M.; Nascimento, M.S.J. Isomeric kielcorins and dihydroxyxanthones: Synthesis, structure elucidation, and inhibitory activities of growth of human cancer cell lines and on the proliferation of human lymphocytes in vitro. Hel. Chim. Acta 2002, 85, 2862–2876. [Google Scholar] [CrossRef]
- Rewcastle, G.W.; Atwell, G.J.; Baguley, B.C.; Boyd, M.; Thomsen, L.L.; Zhuang, L.; Denny, W.A. Potential antitumor agents. 63. Structure-activity relationships for side-chain analogues of the colon 38 active agent 9-oxo-9h-xanthene-4-acetic acid. J. Med. Chem. 1991, 34, 2864–2870. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.E.; Tiritan, M.E.; Belaz, K.R.A.; Pedro, M.; Nascimento, M.S.J.; Cass, Q.B.; Pinto, M.M.M. Multimilligram enantioresolution of low-solubility xanthonolignoids on polysaccharide chiral stationary phases using a solid-phase injection system. J. Chromatogr. A 2006, 1120, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Masawang, K.; Tiritan, M.E.; Sousa, E.; De Lima, V.; Afonso, C.; Bousbaa, H.; Sudprasert, W.; Pedro, M.; Pinto, M.M. New chiral derivatives of xanthones: Synthesis and investigation of enantioselectivity as inhibitors of growth of human tumor cell lines. Bioorg. Med. Chem. 2014, 22, 1049–1062. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Oliveira, L.; Tiritan, M.E.; Leitao, L.; Pozzi, A.; Noronha-Matos, J.B.; Correia-De-Sá, P.; Pinto, M.M. Synthesis of new chiral xanthone derivatives acting as nerve conduction blockers in the rat sciatic nerve. Eur. J. Med. Chem. 2012, 55, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Cidade, H.; Rocha, V.; Palmeira, A.; Marques, C.; Tiritan, M.E.; Ferreira, H.; Lobo, J.S.; Almeida, I.F.; Sousa, M.E.; Pinto, M. In silico and in vitro antioxidant and cytotoxicity evaluation of oxygenated xanthone derivatives. Arab. J. Chem. 2017, in press. [Google Scholar] [CrossRef]
- Pereira, D.; Lima, R.T.; Palmeira, A.; Seca, H.; Soares, J.; Gomes, S.; Raimundo, L.; Maciel, C.; Pinto, M.; Sousa, E.; et al. Design and synthesis of new inhibitors of p53-MDM2 interaction with a chalcone scaffold. Arab. J. Chem. 2016, in press. [Google Scholar] [CrossRef]
- Palmeira, A.; Rodrigues, F.; Sousa, E.; Pinto, M.; Vasconcelos, M.H.; Fernandes, M.X. New uses for old drugs: Pharmacophore-based screening for the discovery of p-glycoprotein inhibitors. Chem. Biol. Drug Des. 2011, 78, 57–72. [Google Scholar] [CrossRef] [PubMed]
- Lima, R.T.; Seca, H.; Palmeira, A.; Fernandes, M.X.; Castro, F.; Correia-da-Silva, M.; Nascimento, M.S.J.; Sousa, E.; Pinto, M.; Vasconcelos, M.H. Sulfated small molecules targeting EBV in Burkitt lymphoma: From in silico screening to the evidence of in vitro effect on viral episomal DNA. Chem. Biol. Drug Des. 2013, 81, 631–644. [Google Scholar] [CrossRef] [PubMed]
- Hawkey, C.J. Cox-1 and cox-2 inhibitors. Best Pract. Res. Clin. Gastroenterol. 2001, 15, 801–820. [Google Scholar] [CrossRef] [PubMed]
- Atukorala, I.; Hunter, D.J. Valdecoxib: The rise and fall of a cox-2 inhibitor. Expert Opin. Pharmacother. 2013, 14, 1077–1086. [Google Scholar] [CrossRef] [PubMed]
- Bali, A.; Ohri, R.; Deb, P.K. Synthesis, evaluation and docking studies on 3-alkoxy-4-methanesulfonamido acetophenone derivatives as non-ulcerogenic anti-inflammatory agents. Eur. J. Med. Chem. 2012, 49, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Hegazy, G.H.; Ali, H.I. Design, synthesis, biological evaluation, and comparative cox1 and cox2 docking of p-substituted benzylidenamino phenyl esters of ibuprofenic and mefenamic acids. Bioorg. Med. Chem. 2012, 20, 1259–1270. [Google Scholar] [CrossRef] [PubMed]
- El-Sayed, M.A.; Abdel-Aziz, N.I.; Abdel-Aziz, A.A.; El-Azab, A.S.; ElTahir, K.E. Synthesis, biological evaluation and molecular modeling study of pyrazole and pyrazoline derivatives as selective cox-2 inhibitors and anti-inflammatory agents. Part 2. Bioorg. Med. Chem. 2012, 20, 3306–3316. [Google Scholar] [CrossRef] [PubMed]
- Ryn, J.; Trummlitz, G.; Pairet, M. Cox-2 selectivity and inflammatory processes. Curr. Med. Chem. 2000, 7, 1145–1161. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Aziz, A.A.; ElTahir, K.E.; Asiri, Y.A. Synthesis, anti-inflammatory activity and cox-1/cox-2 inhibition of novel substituted cyclic imides. Part 1: Molecular docking study. Eur. J. Med. Chem. 2011, 46, 1648–1655. [Google Scholar] [CrossRef] [PubMed]
- Rowlinson, S.W.; Kiefer, J.R.; Prusakiewicz, J.J.; Pawlitz, J.L.; Kozak, K.R.; Kalgutkar, A.S.; Stallings, W.C.; Kurumbail, R.G.; Marnett, L.J. A novel mechanism of cyclooxygenase-2 inhibition involving interactions with ser-530 and tyr-385. J. Biol. Chem. 2003, 278, 45763–45769. [Google Scholar] [CrossRef] [PubMed]
- Parikh, H.H.; McElwain, K.; Balasubramanian, V.; Leung, W.; Wong, D.; Morris, M.E.; Ramanathan, M. A rapid spectrofluorimetric technique for determining drug-serum protein binding suitable for high-throughput screening. Pharm. Res. 2000, 17, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Watanabe, H.; Tanase, S.; Nakajou, K.; Maruyama, T.; Kragh-Hansen, U.; Otagiri, M. Role of arg-410 and tyr-411 in human serum albumin for ligand binding and esterase-like activity. Biochem. J. 2000, 349 Pt 3, 813–819. [Google Scholar] [CrossRef] [PubMed]
- Ghuman, J.; Zunszain, P.A.; Petitpas, I.; Bhattacharya, A.A.; Otagiri, M.; Curry, S. Structural basis of the drug-binding specificity of human serum albumin. J. Mol. Biol. 2005, 353, 38–52. [Google Scholar] [CrossRef] [PubMed]
- Zaidi, N.; Ahmad, E.; Rehan, M.; Rabbani, G.; Ajmal, M.R.; Zaidi, Y.; Subbarao, N.; Khan, R.H. Biophysical insight into furosemide binding to human serum albumin: A study to unveil its impaired albumin binding in uremia. J. Phys. Chem. B 2013, 117, 2595–2604. [Google Scholar] [CrossRef] [PubMed]
- Karthikeyan, S.; Bharanidharan, G.; Mani, K.A.; Srinivasan, N.; Kesherwani, M.; Velmurugan, D.; Aruna, P.; Ganesan, S. Determination on the binding of thiadiazole derivative to human serum albumin: A spectroscopy and computational approach. J. Biomol. Struct. Dyn. 2016, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Singh, D.V.; Bharti, S.K.; Agarwal, S.; Roy, R.; Misra, K. Study of interaction of human serum albumin with curcumin by nmr and docking. J. Mol. Model. 2014, 20, 2365. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Brandão, P.; Santos, A.; Tiritan, M.E.; Afonso, C.; Cass, Q.B.; Pinto, M.M. Resolution and determination of enantiomeric purity of new chiral derivatives of xanthones using polysaccharide-based stationary phases. J. Chromatogr. A 2012, 1269, 143–153. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, C.; Tiritan, M.E.; Cass, Q.; Kairys, V.; Fernandes, M.X.; Pinto, M. Enantioseparation and chiral recognition mechanism of new chiral derivatives of xanthones on macrocyclic antibiotic stationary phases. J. Chromatogr. A 2012, 1241, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Coutinho, A.; Prieto, M. Ribonuclease t1 and alcohol dehydrogenase fluorescence quenching by acrylamide—A laboratory experiment for undergraduate students. J. Chem. Edu. 1993, 70, 425–428. [Google Scholar] [CrossRef]
- Casewit, C.J.; Colwell, K.S.; Rappe, A.K. Application of a universal force field to main group compounds. J. Am. Chem. Soc. 1992, 114, 10046–10053. [Google Scholar] [CrossRef]
- Gasteiger, J.; Marsili, M. Iterative partial equalization of orbital electronegativity—A rapid access to atomic charges. Tetrahedron 1980, 36, 3219–3228. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Mysinger, M.M.; Carchia, M.; Irwin, J.J.; Shoichet, B.K. Directory of useful decoys, enhanced (DUD-E): Better ligands and decoys for better benchmarking. J. Med. Chem. 2012, 55, 6582–6594. [Google Scholar] [CrossRef] [PubMed]
- Rose, P.W.; Prlic, A.; Bi, C.; Bluhm, W.F.; Christie, C.H.; Dutta, S.; Green, R.K.; Goodsell, D.S.; Westbrook, J.D.; Woo, J.; et al. The RCSB protein data bank: Views of structural biology for basic and applied research and education. Nucleic Acids Res. 2015, 43, D345–D356. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. Autodock vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Seeliger, D.; de Groot, B.L. Ligand docking and binding site analysis with pymol and autodock/vina. J. Comput. Aided Mol. Des. 2010, 24, 417–422. [Google Scholar] [CrossRef] [PubMed]
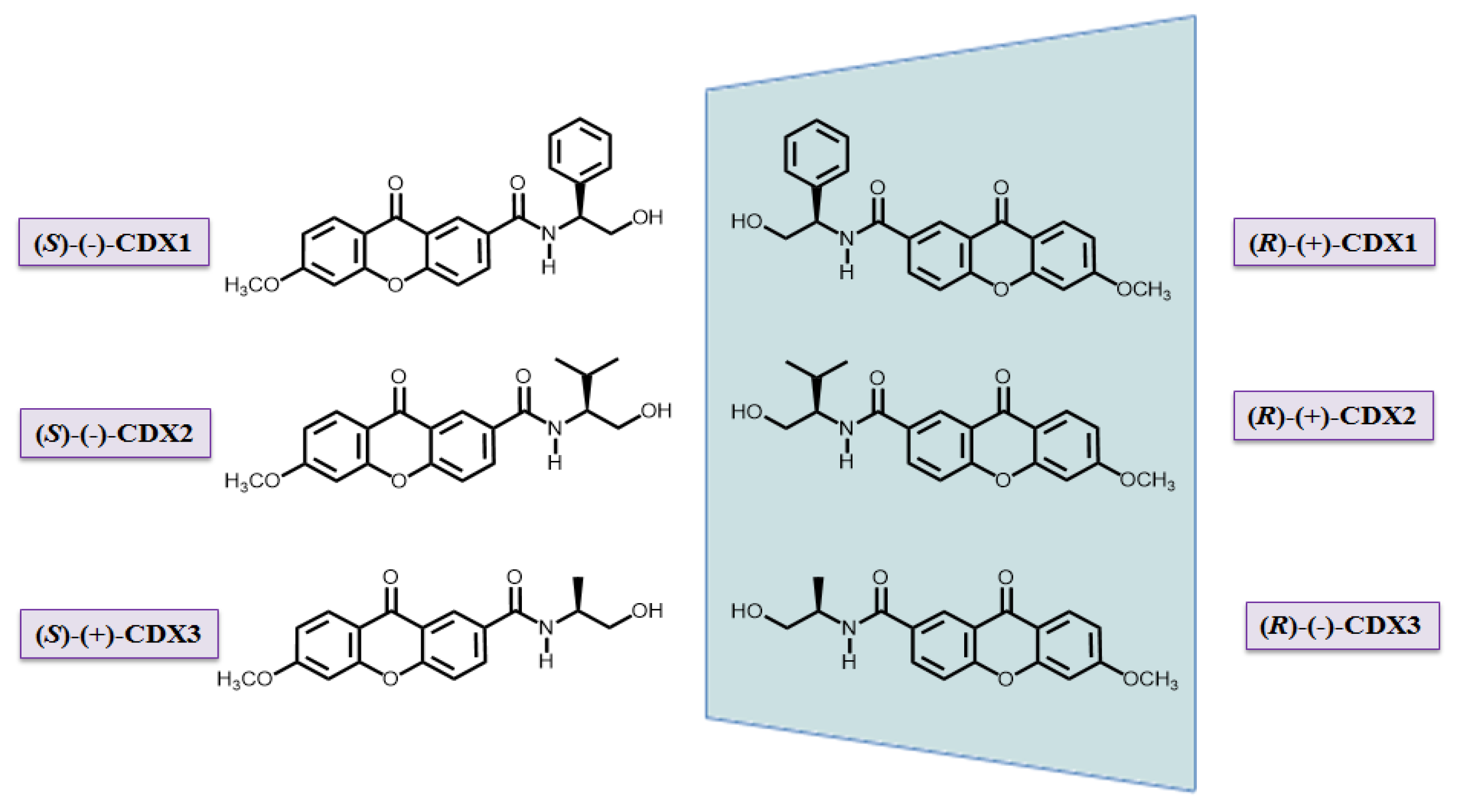
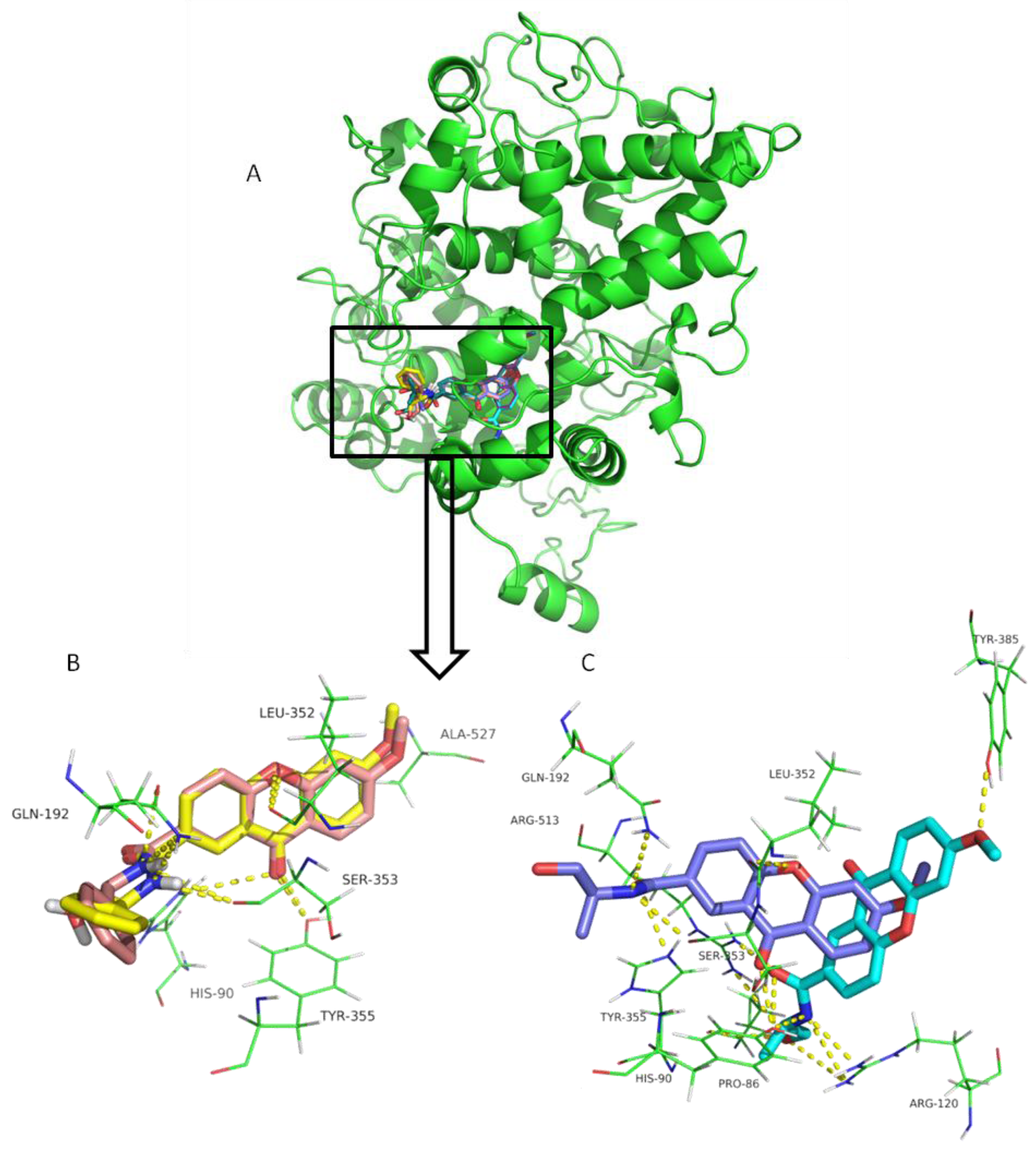
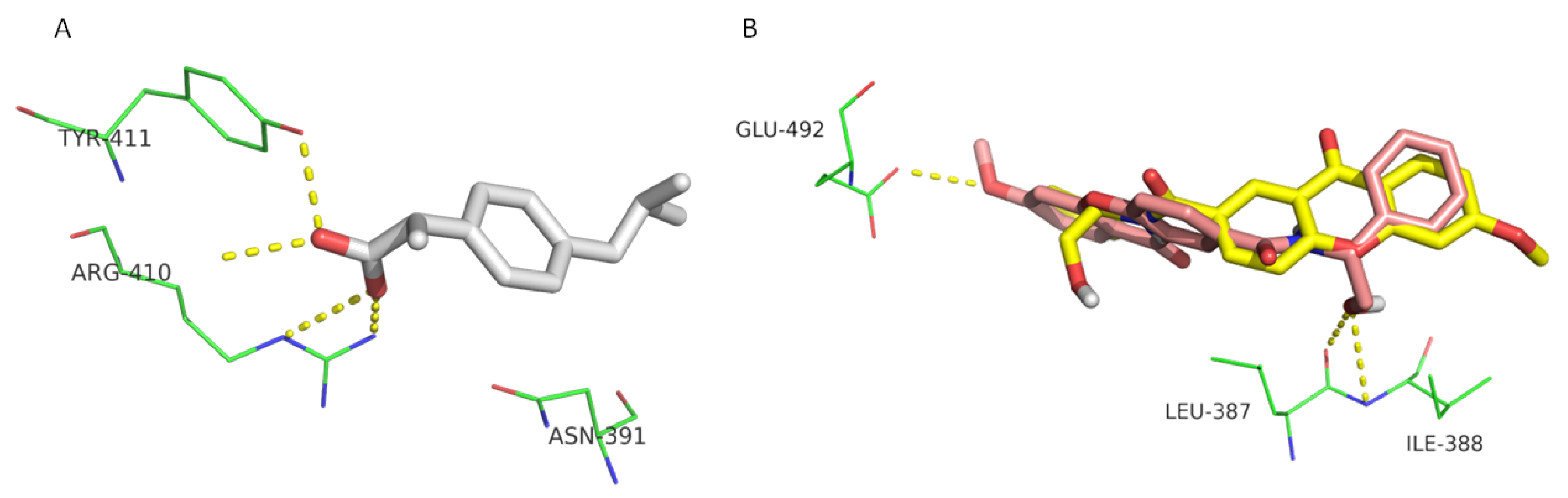

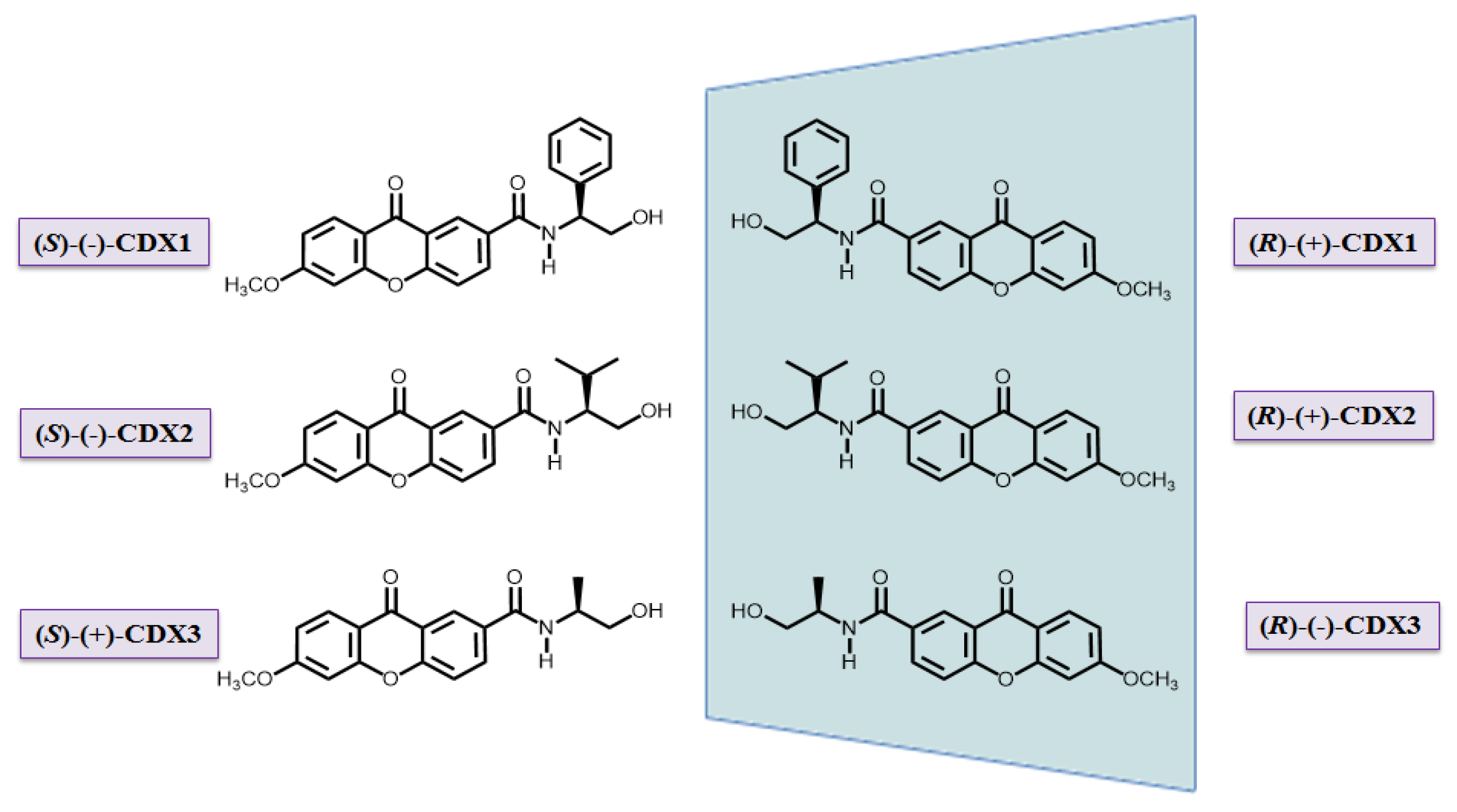{kind=link}
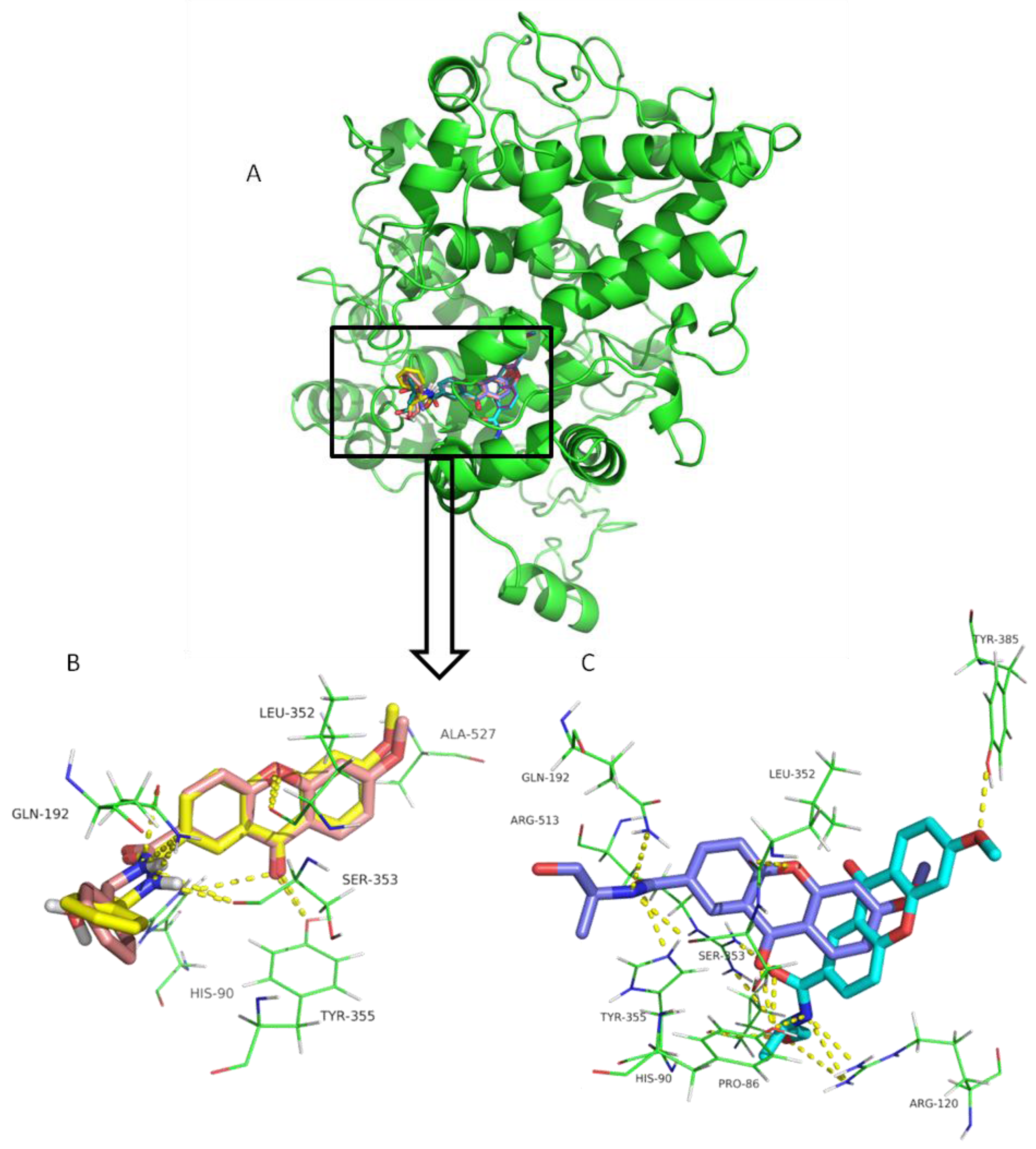{kind=link}
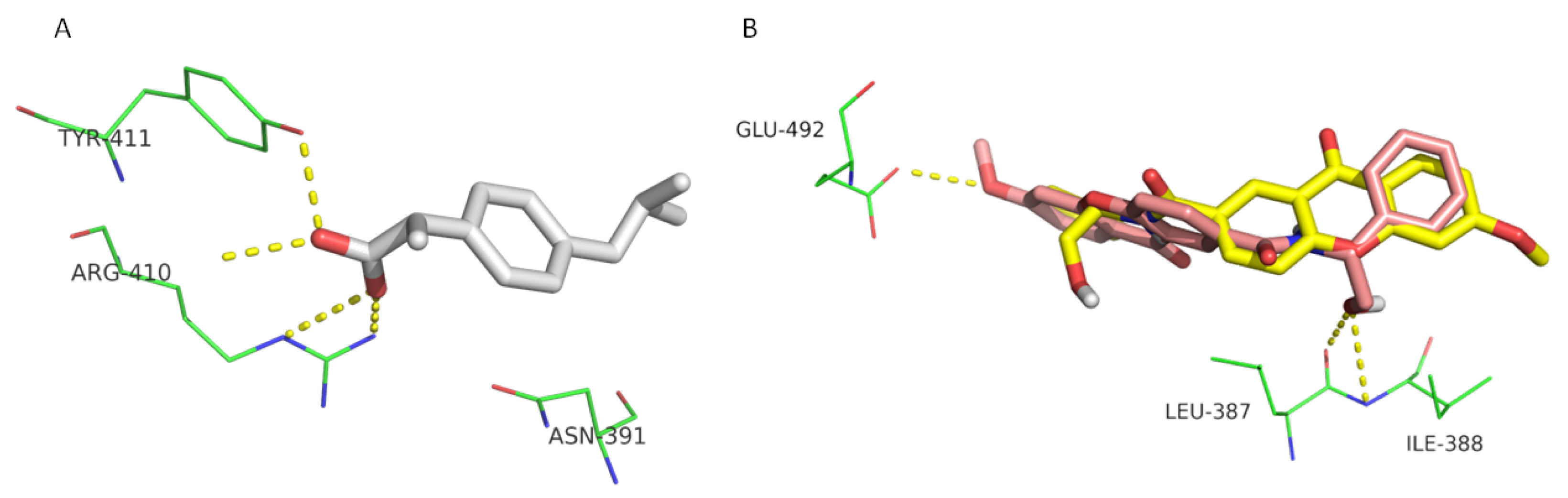{kind=link}
| CDX | COX-1 | COX-2 |
|---|---|---|
| (S)-(−)-CDX1 | 87.6 ± 2.1 | 80.1 ± 12.8 |
| (R)-(+)-CDX1 | 79.6 ± 5.0 | 84.7 ± 5.7 |
| (S)-(−)-CDX2 | 82.9 ± 5.2 | 85.7 ± 4.5 |
| (R)-(+)-CDX2 | 66.8 ± 1.6 | 73.2 ± 0.4 |
| (S)-(+)-CDX3 | 91.7 ± 10.7 | 93.4 ± 11.4 |
| (R)-(−)-CDX3 | 75.2 ± 9.0 | 75.1 ± 7.2 |
| Indomethacin | 83.2 ± 6.4 | 80.7 ± 9.5 |
| Compounds | Docking Score (kcal/mol) | ||
|---|---|---|---|
| COX-1 | COX-2 | ||
| Known ligands | Diclofenac | −6.1 | −7.9 |
| Indomethacin | −5.1 | −7.9 | |
| Naproxen | −7.8 | ||
| Piroxicam | −5.2 | ||
| Celecoxib | −11.5 | ||
| Valdecoxib | −9.5 | ||
| Ligands from database | −7.8 | −9.3 | |
| Decoys from database | −7.3 | −7.6 | |
| (R)-(+)-CDX1 | −4.2 | −7.8 | |
| (S)-(−)-CDX1 | −4.5 | −8.0 | |
| (R)-(+)-CDX2 | −3.4 | −6.5 | |
| (S)-(−)-CDX2 | −5.4 | −7.0 | |
| (R)-(−)-CDX3 | −5.3 | −6.9 | |
| (S)-(+)-CDX3 | −5.6 | −7.5 | |
| Compound | Kd | Ymax | ∆G Binding | Docking Score (kcal/mol) | |
|---|---|---|---|---|---|
| Known ligands | Azaprozone | −5.9 | |||
| Diazepam | −7.1 | ||||
| Fusidic acid | −5.8 | ||||
| (S)-Ibuprofen | −7.3 | ||||
| Iophenoxid acid | −4.4 | ||||
| Naproxen | −7.9 | ||||
| Warfarin | −8.5 | ||||
| (R)-(+)-CDX1 | 61.8 ± 6.5 | 109.6 ± 1.6 | −2.4 ± 0.2 | −7.3 | |
| (S)-(−)-CDX1 | 23.6 ± 0.8 | 105.3 ± 0.4 | −1.9 ± 0.1 | −7.0 | |
| (R)-(+)-CDX2 | 29.2 ± 0.9 | 108.2 ± 0.2 | −2.0 ± 0.1 | −7.2 | |
| (S)-(−)-CDX2 | 24.7 ± 1.1 | 107.4 ± 5.4 | −1.9 ± 0.1 | −7.2 | |
| (R)-(−)-CDX3 | 26.4 ± 1.2 | 113.2 ± 1.4 | −1.9 ± 0.1 | −7.2 | |
| (S)-(+)-CDX3 | 31.4 ± 2.0 | 116.2 ± 0.6 | −2.0 ± 0.2 | −7.0 | |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, C.; Palmeira, A.; Ramos, I.I.; Carneiro, C.; Afonso, C.; Tiritan, M.E.; Cidade, H.; Pinto, P.C.A.G.; Saraiva, M.L.M.F.S.; Reis, S.; et al. Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin. Pharmaceuticals 2017, 10, 50. https://doi.org/10.3390/ph10020050
Fernandes C, Palmeira A, Ramos II, Carneiro C, Afonso C, Tiritan ME, Cidade H, Pinto PCAG, Saraiva MLMFS, Reis S, et al. Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin. Pharmaceuticals. 2017; 10(2):50. https://doi.org/10.3390/ph10020050
Chicago/Turabian StyleFernandes, Carla, Andreia Palmeira, Inês I. Ramos, Carlos Carneiro, Carlos Afonso, Maria Elizabeth Tiritan, Honorina Cidade, Paula C.A.G. Pinto, M. Lúcia M.F.S. Saraiva, Salette Reis, and et al. 2017. "Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin" Pharmaceuticals 10, no. 2: 50. https://doi.org/10.3390/ph10020050
APA StyleFernandes, C., Palmeira, A., Ramos, I. I., Carneiro, C., Afonso, C., Tiritan, M. E., Cidade, H., Pinto, P. C. A. G., Saraiva, M. L. M. F. S., Reis, S., & Pinto, M. M. M. (2017). Chiral Derivatives of Xanthones: Investigation of the Effect of Enantioselectivity on Inhibition of Cyclooxygenases (COX-1 and COX-2) and Binding Interaction with Human Serum Albumin. Pharmaceuticals, 10(2), 50. https://doi.org/10.3390/ph10020050