
Advances in Drug Discovery of New Antitubercular Multidrug-Resistant Compounds



Abstract
:1. Introduction
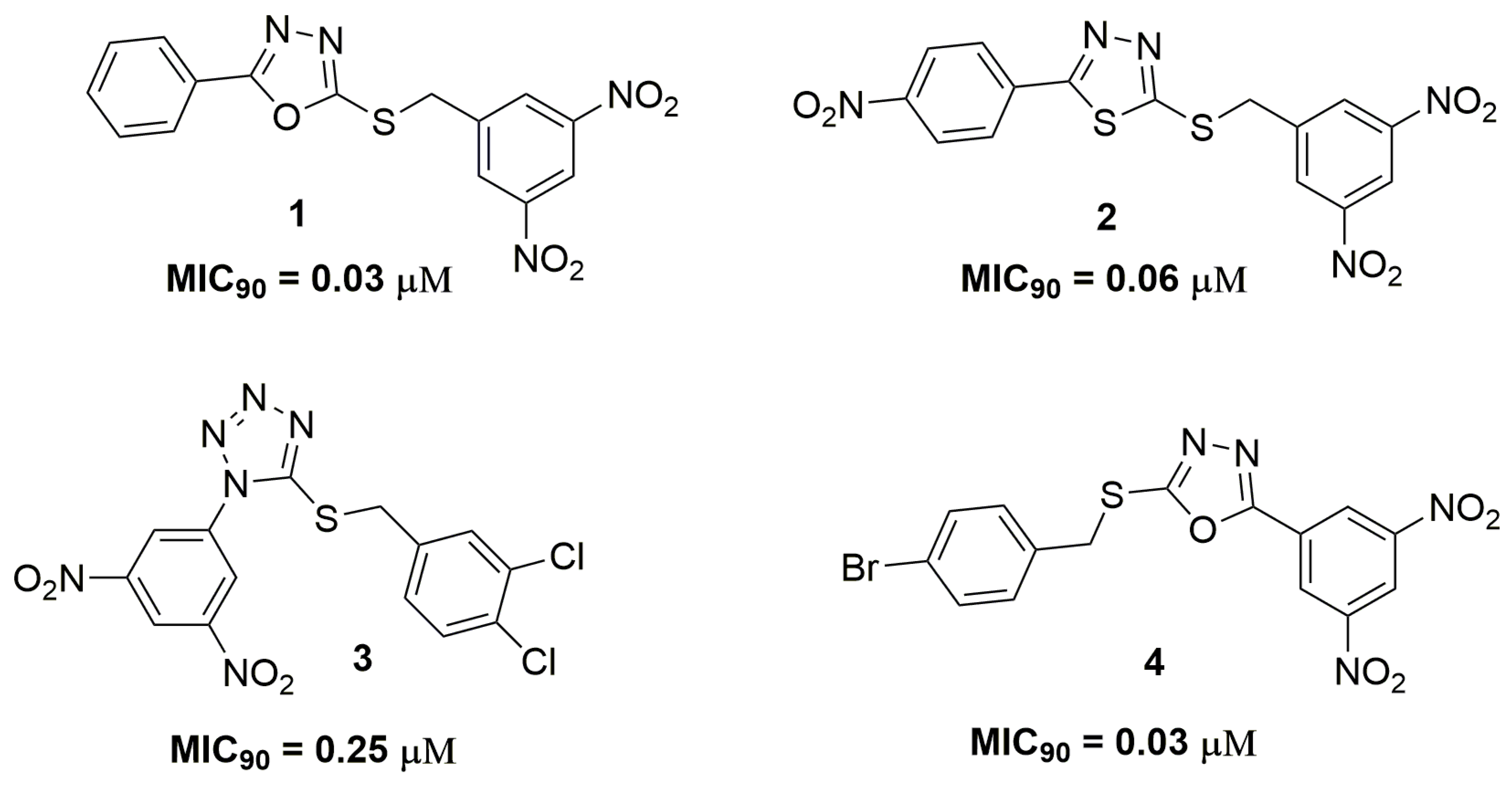
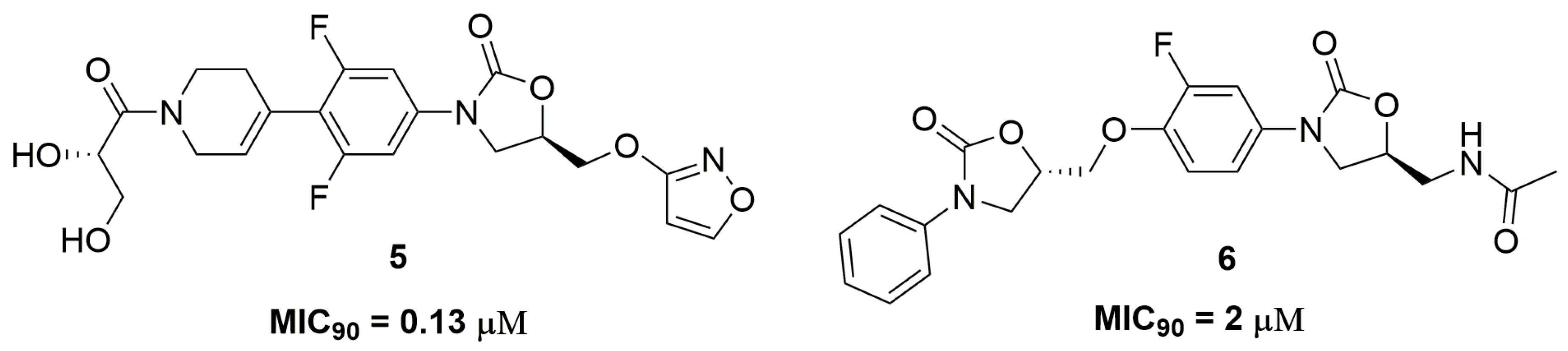
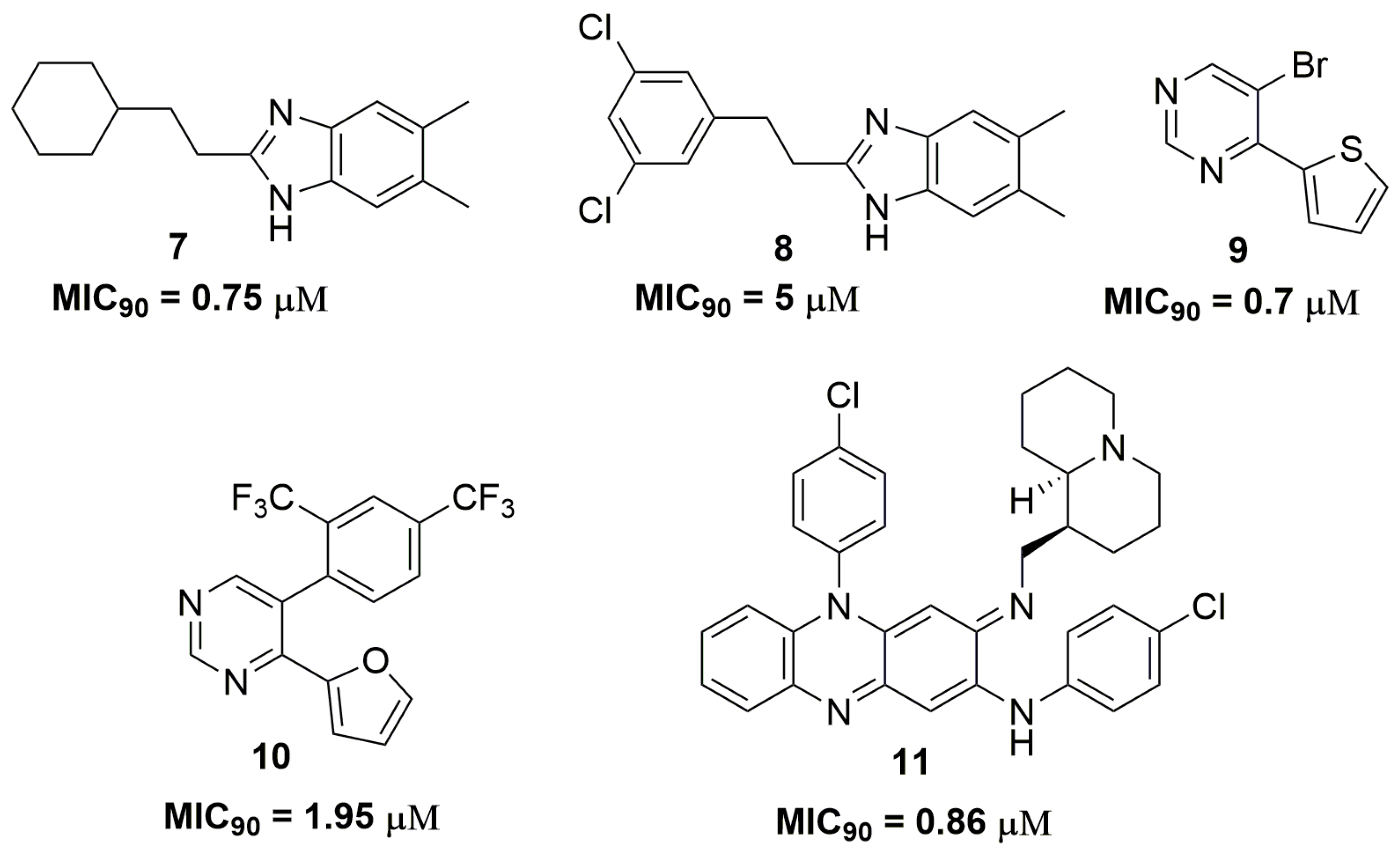
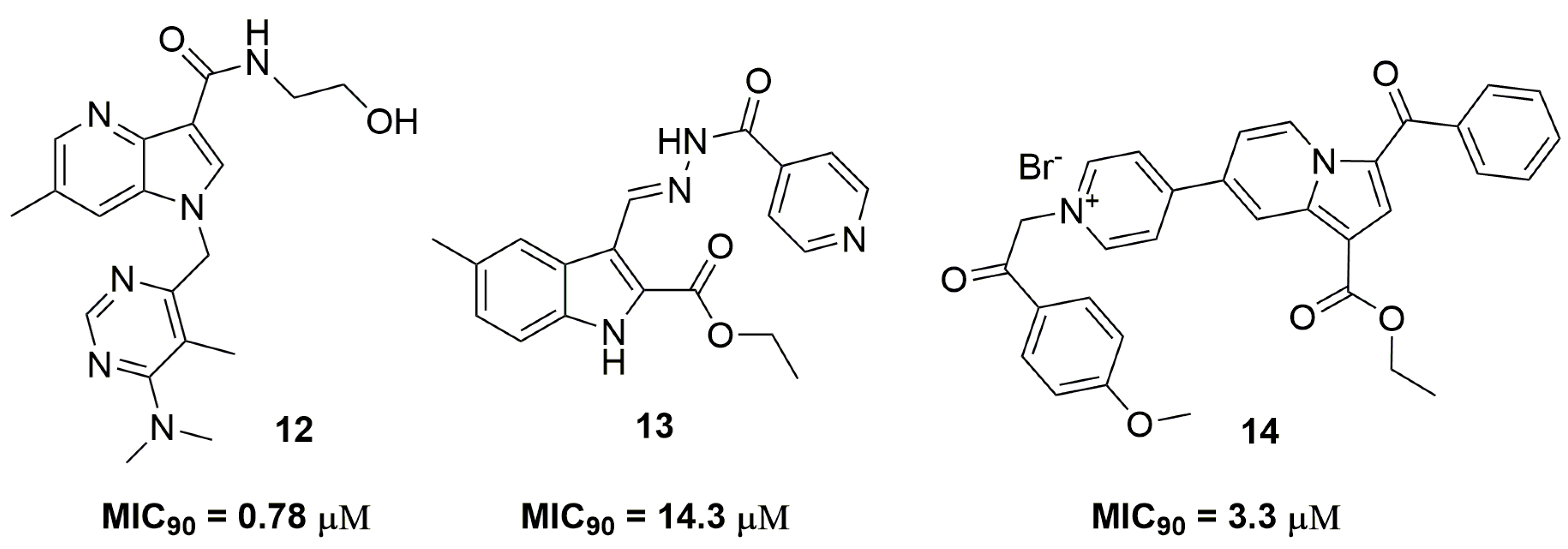
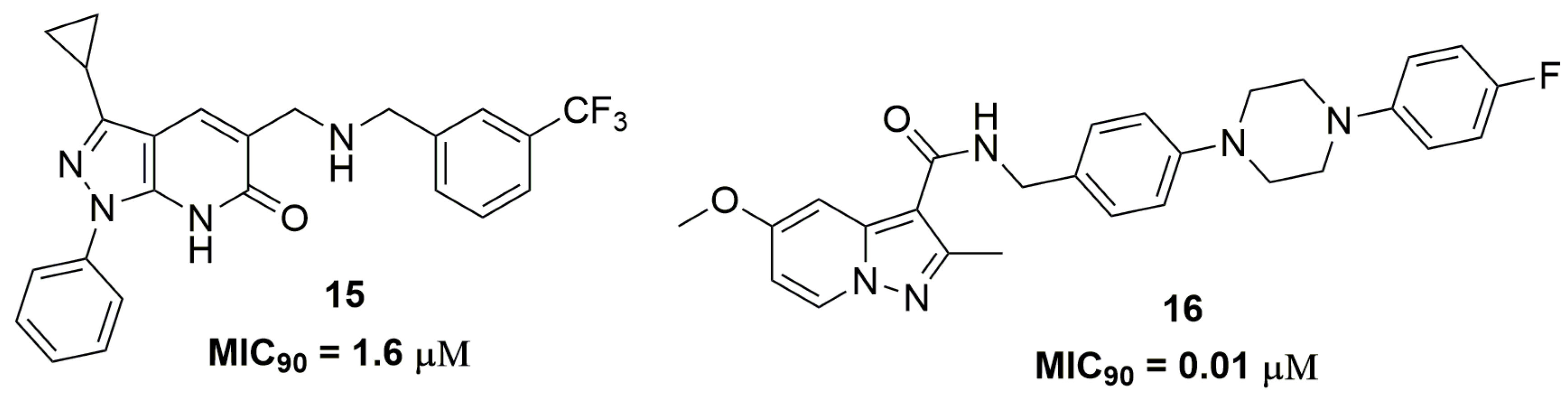
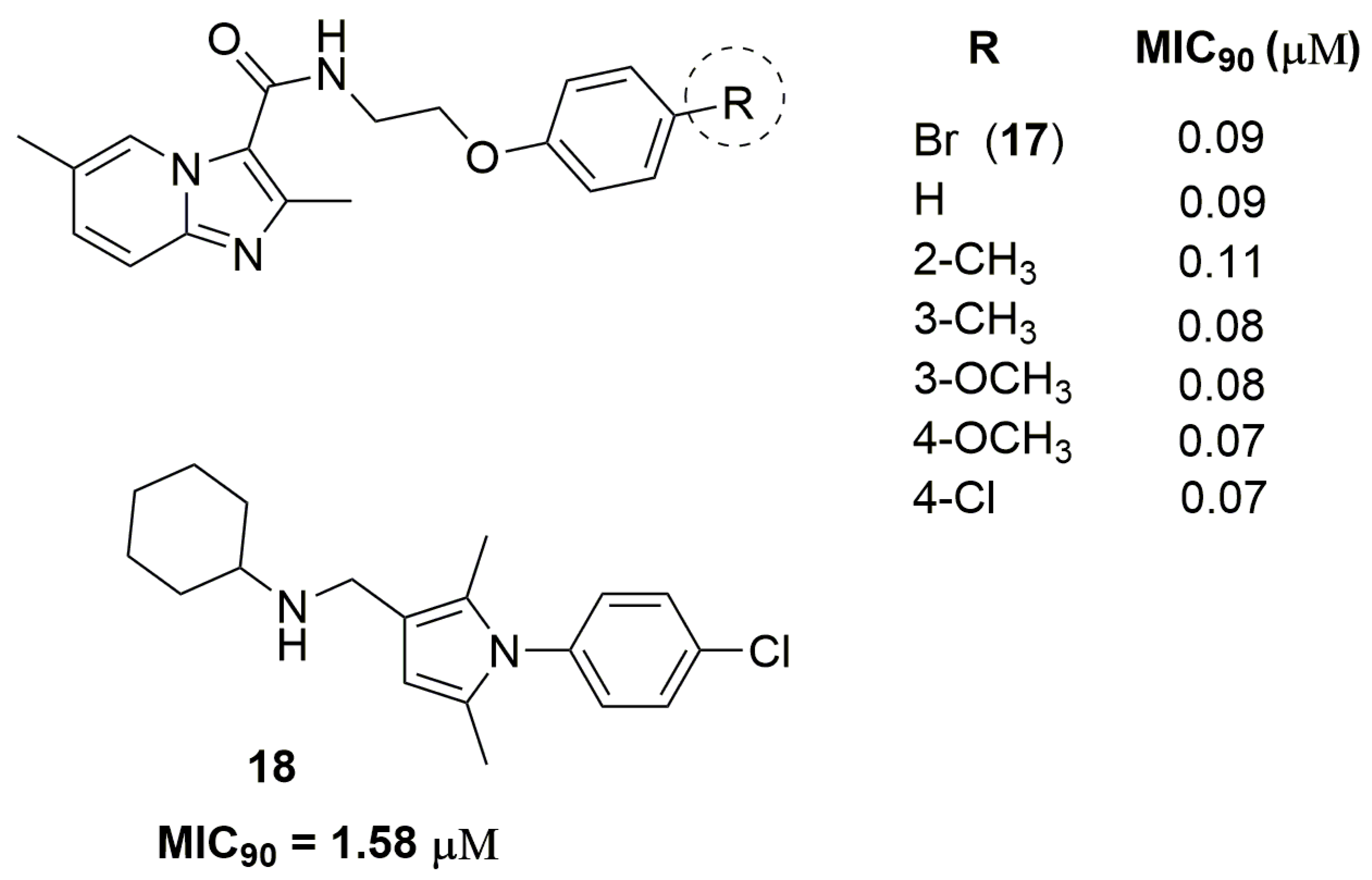
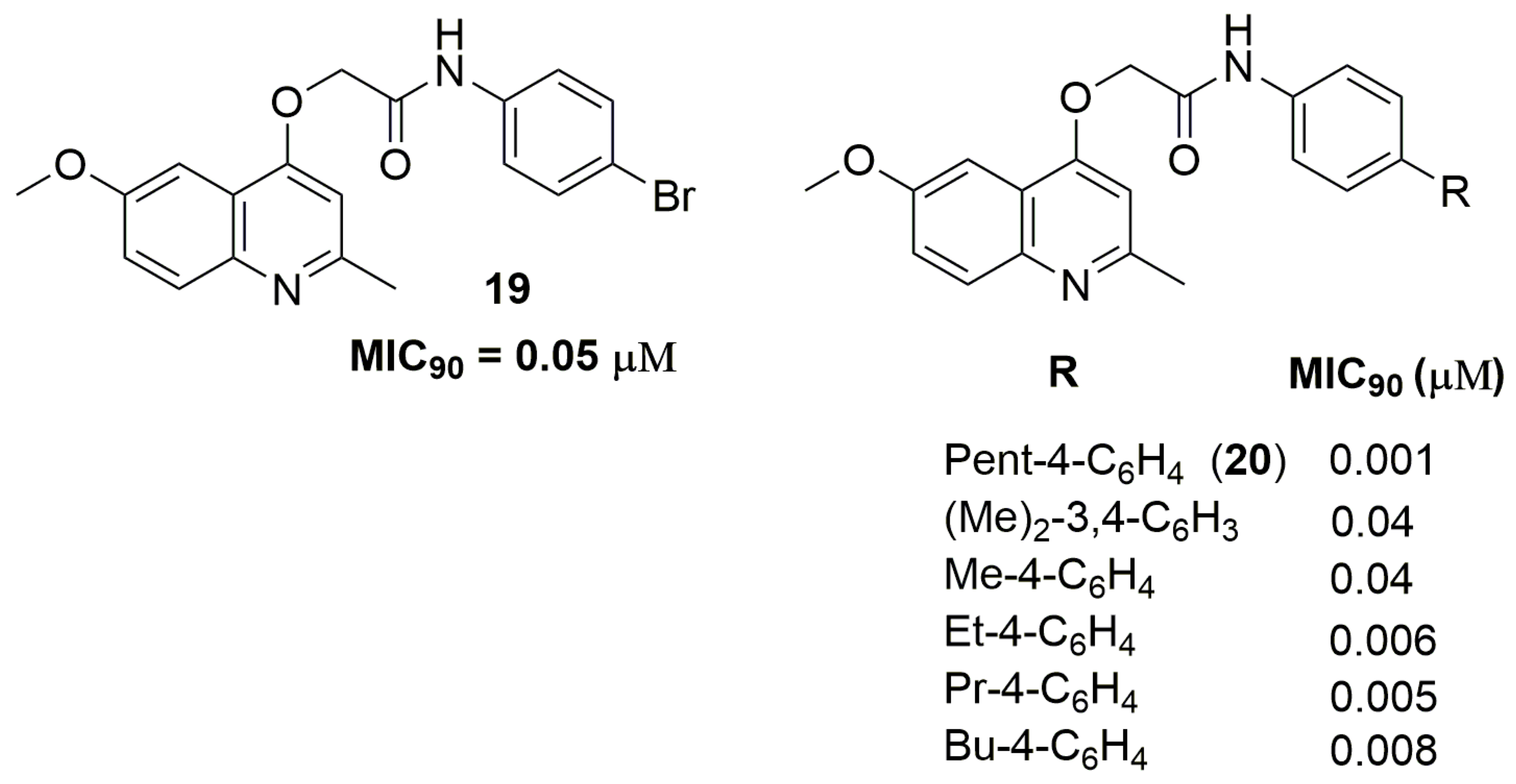
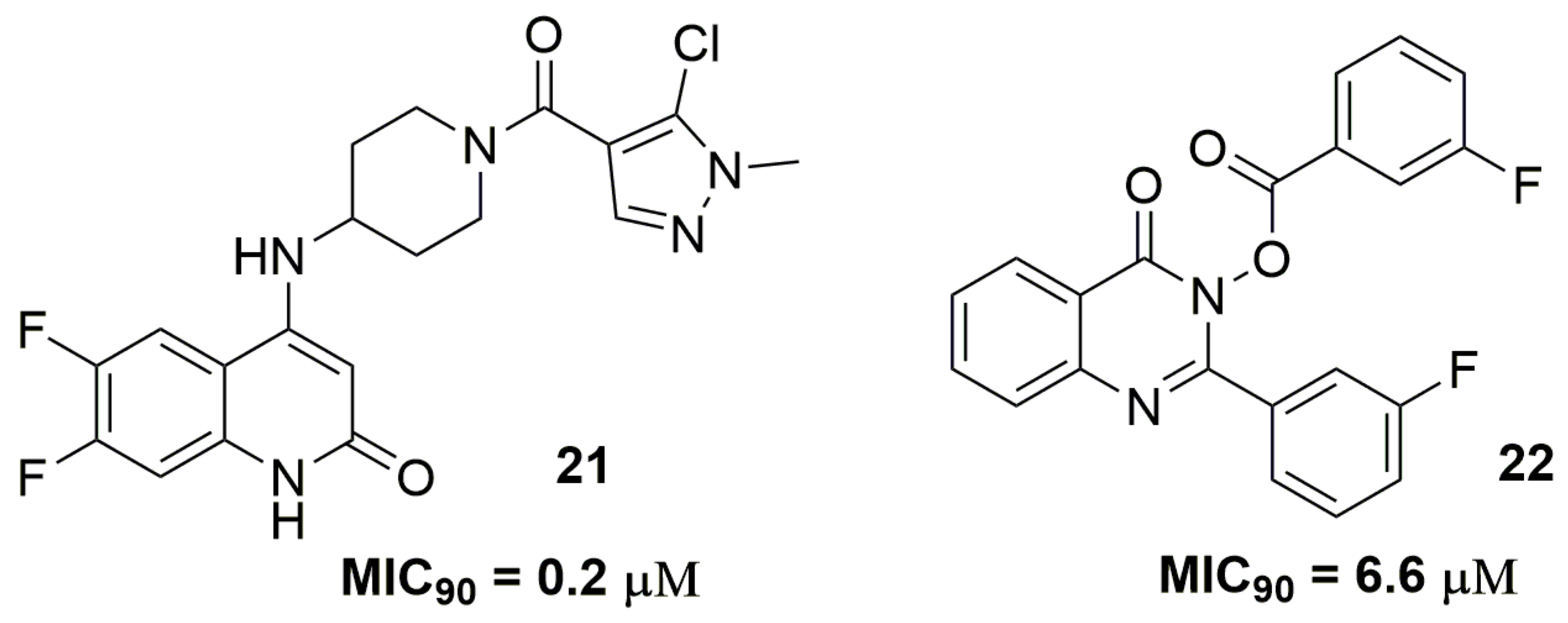
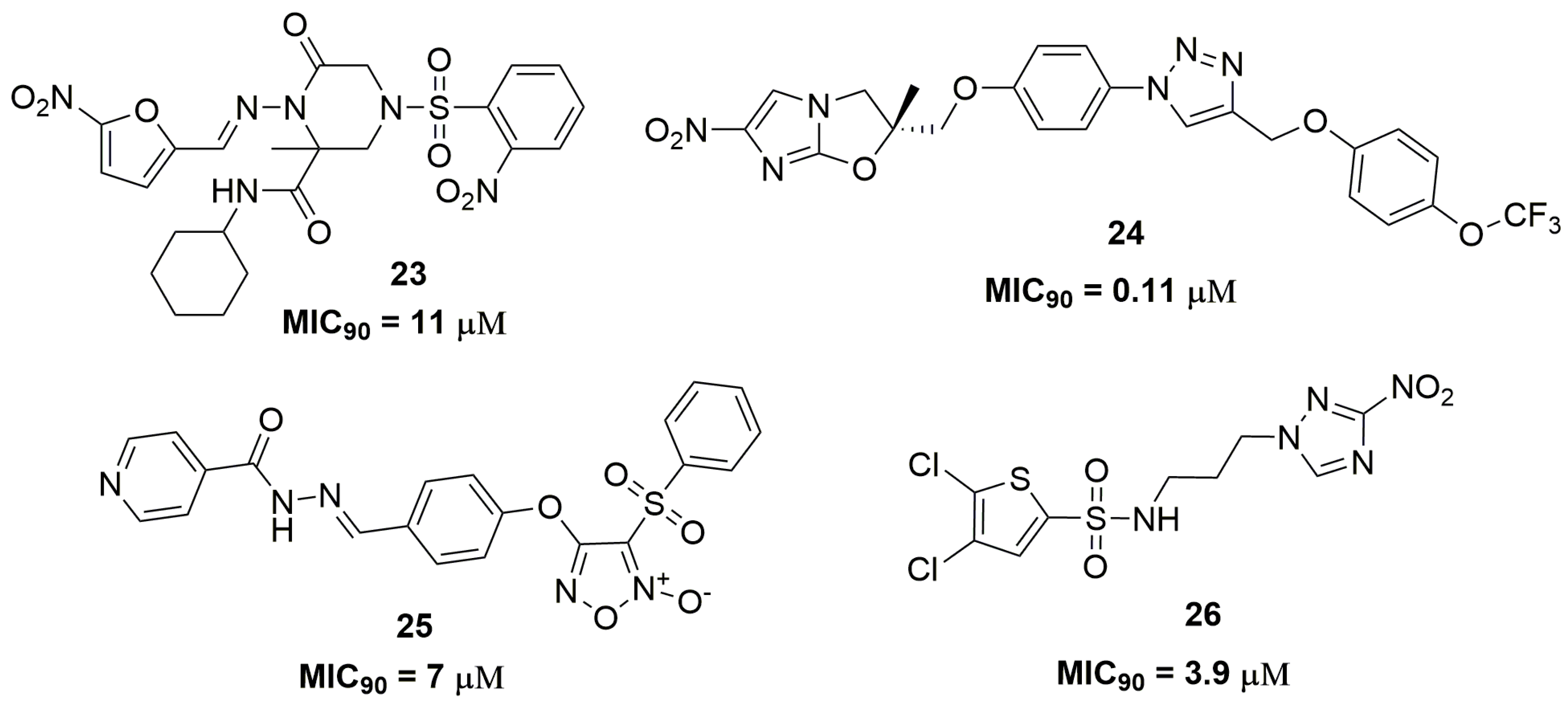
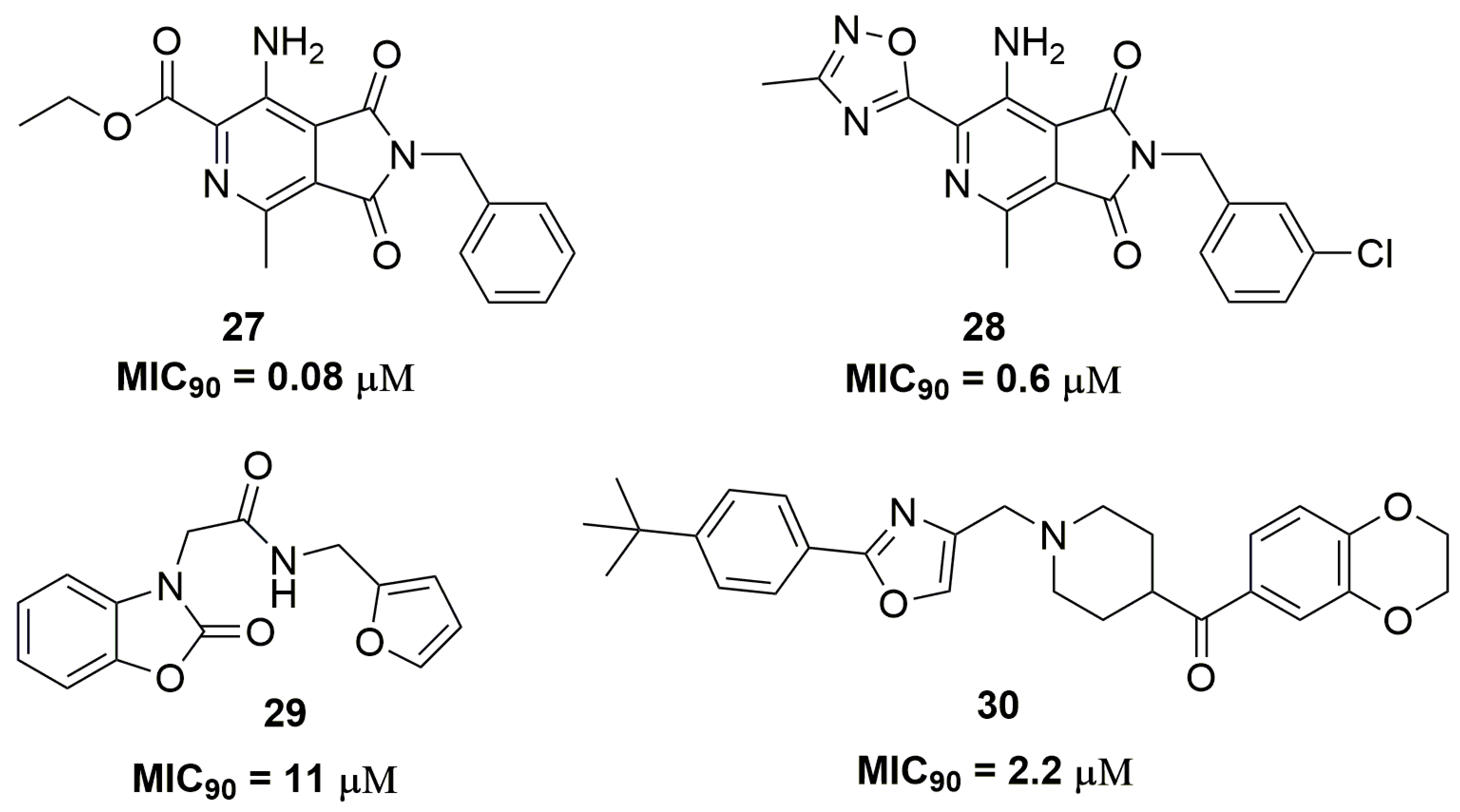
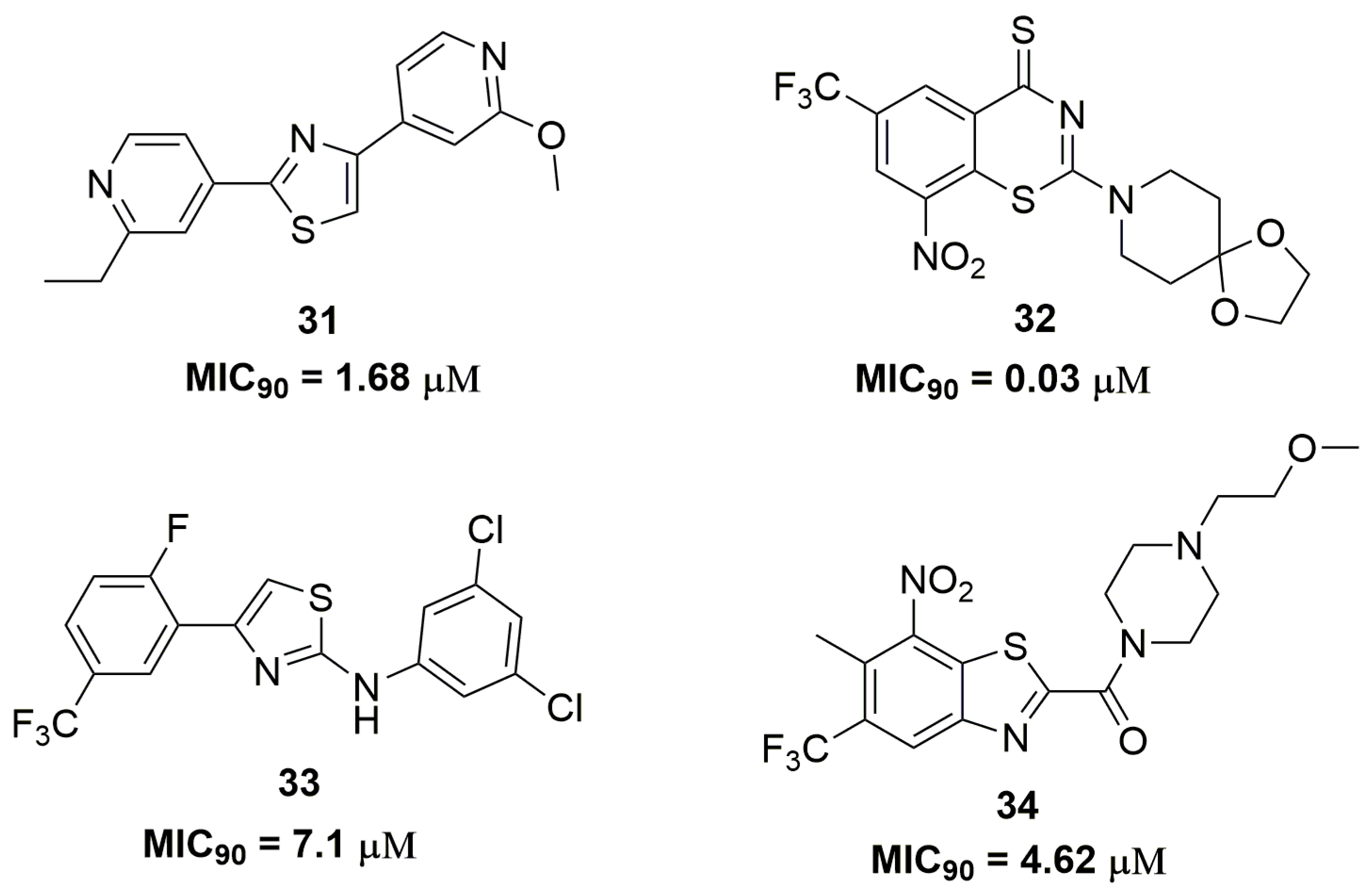
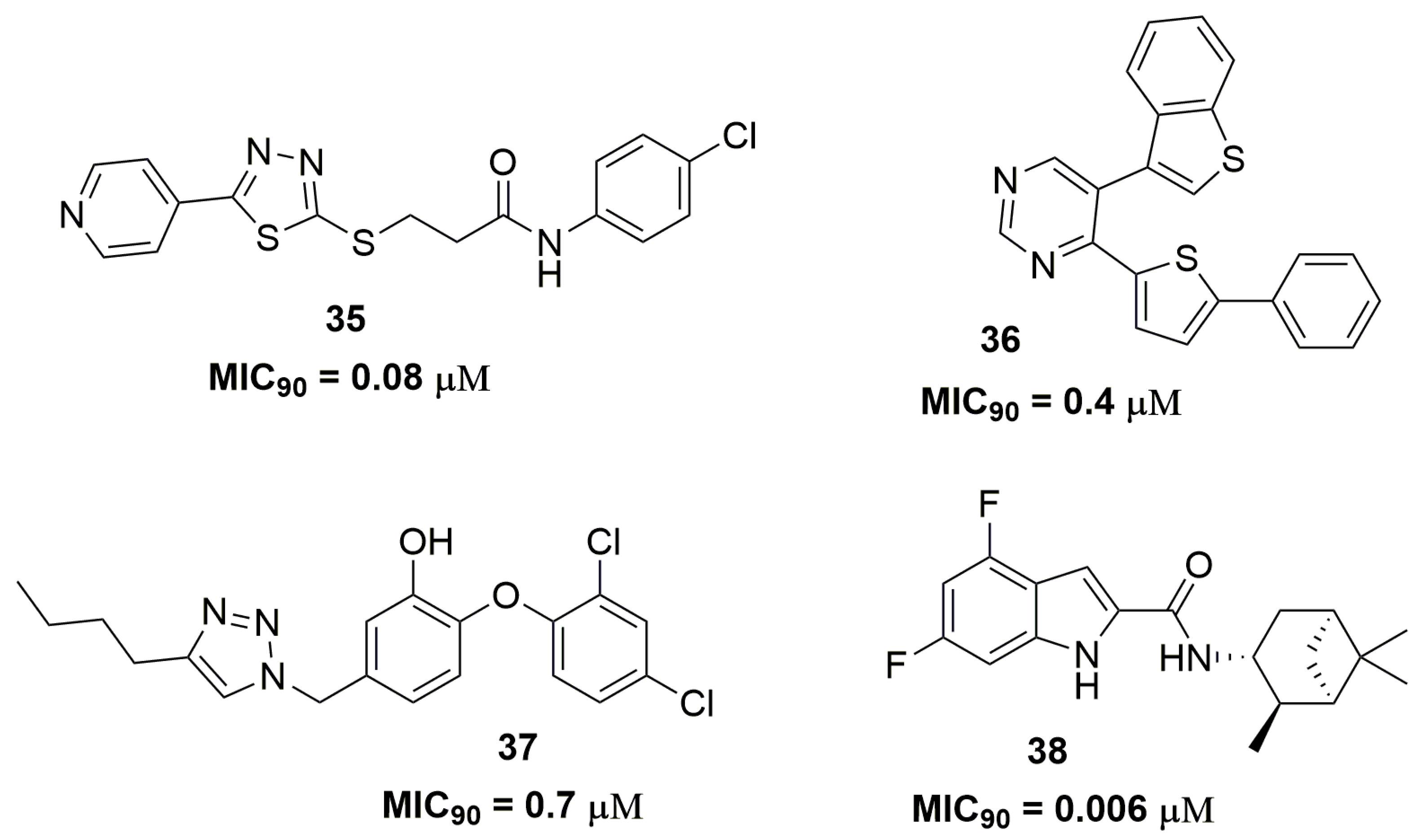
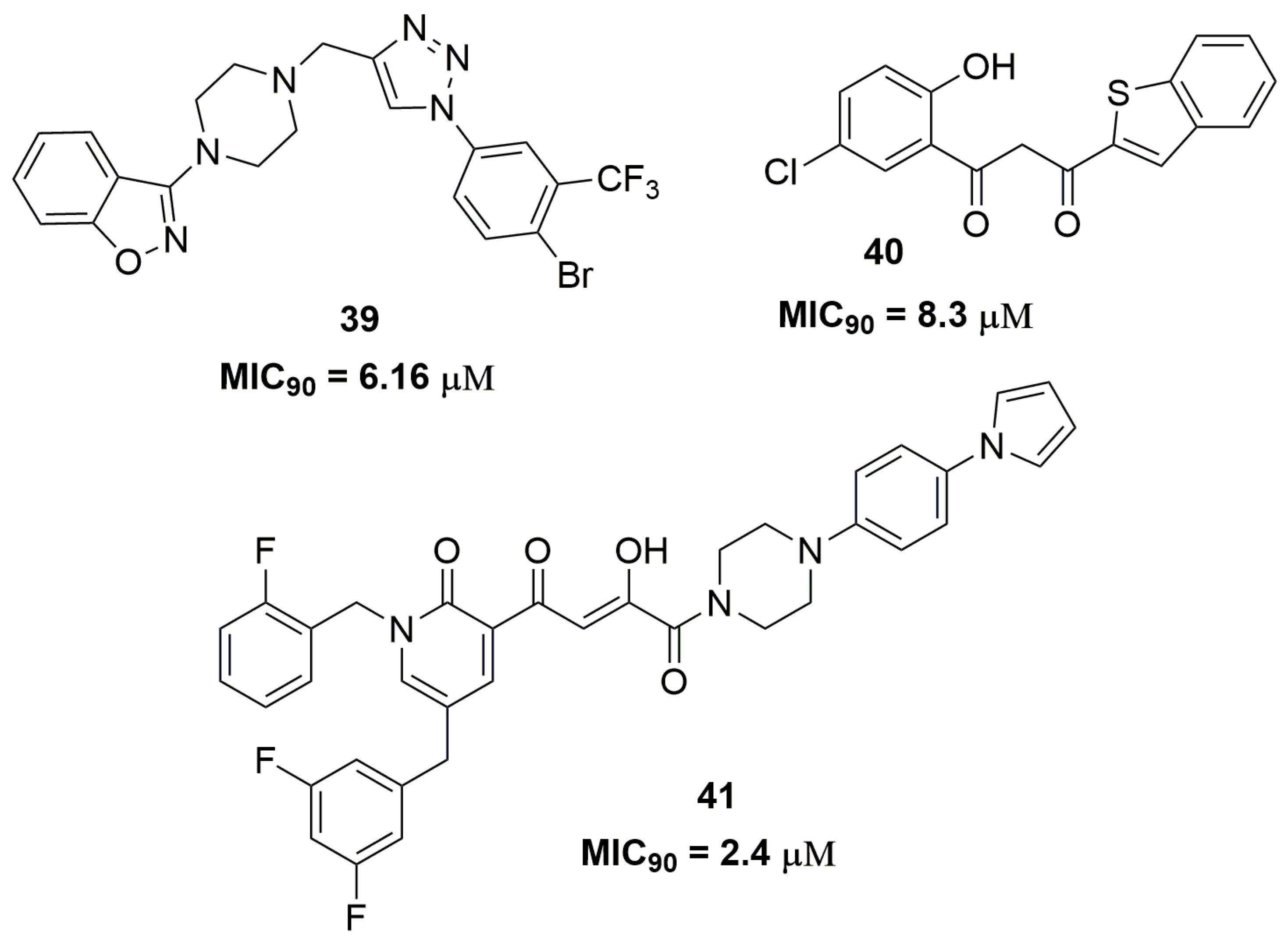
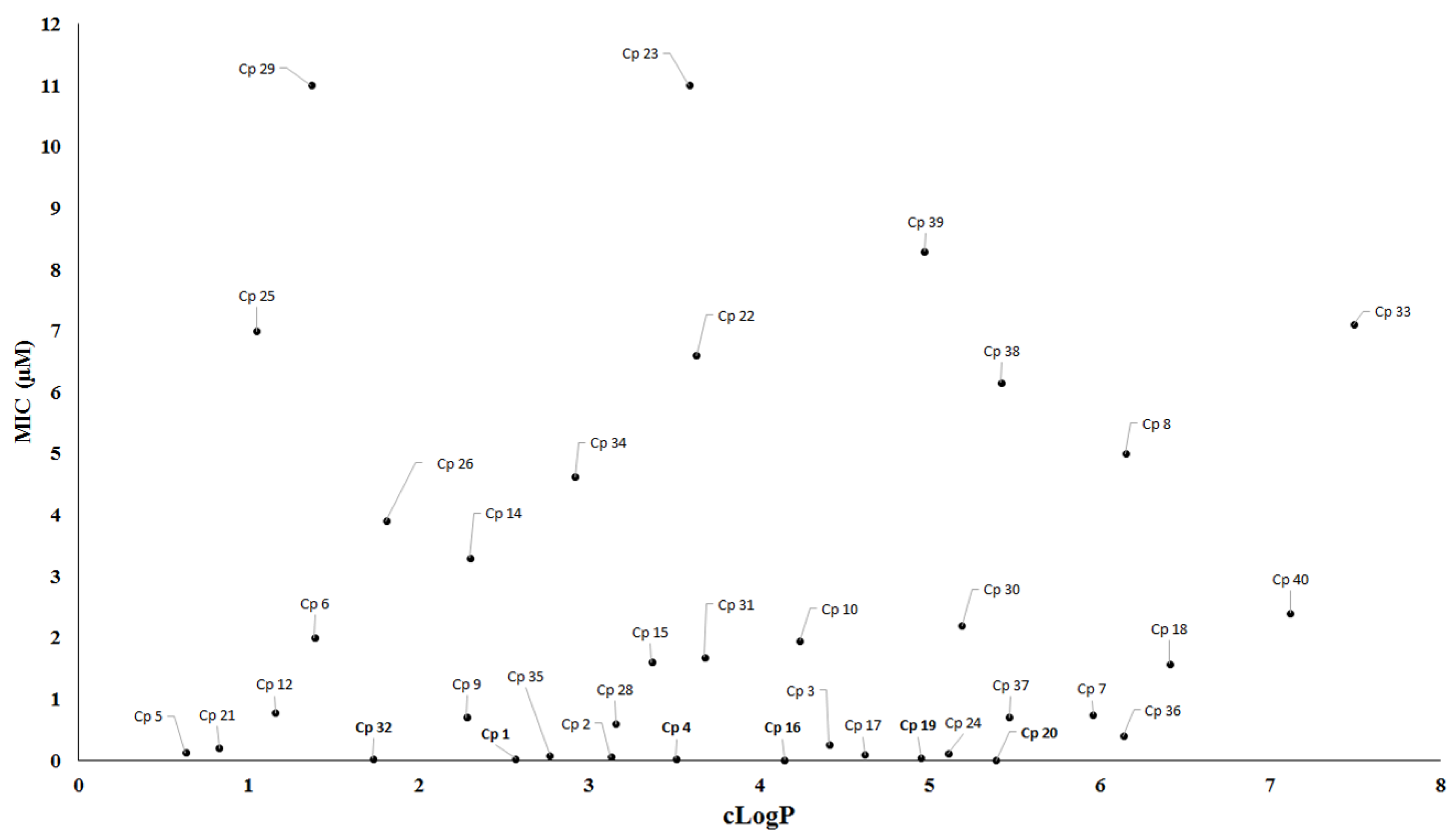
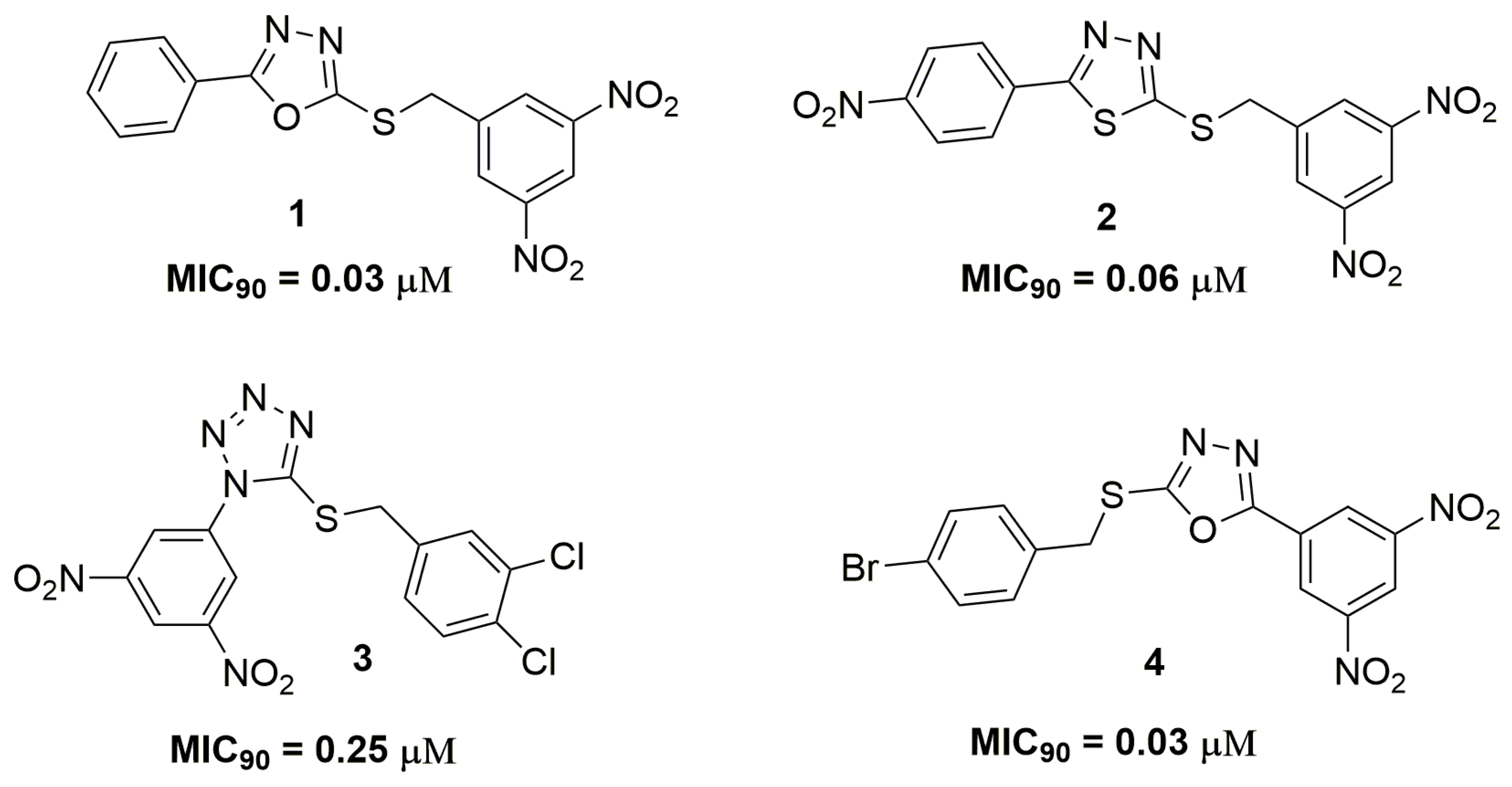
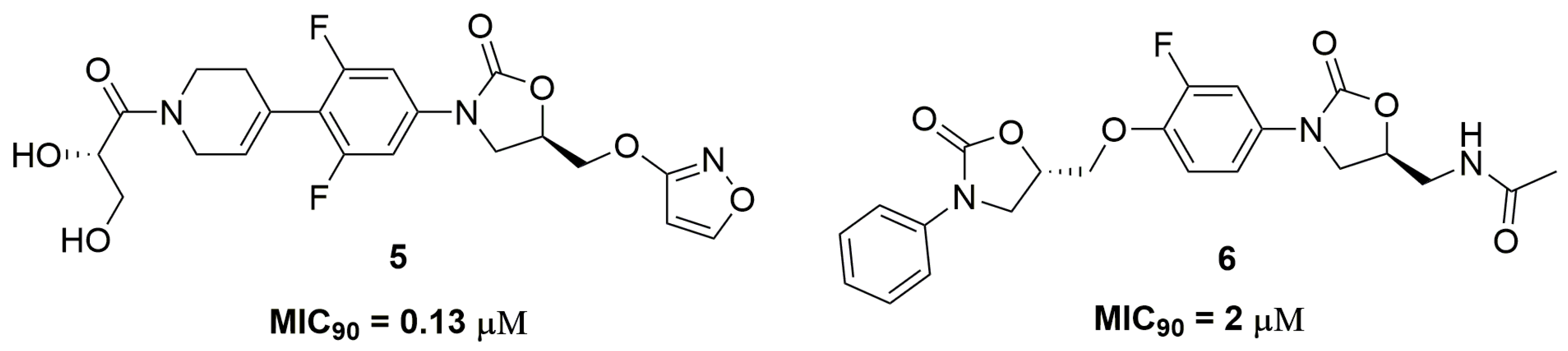
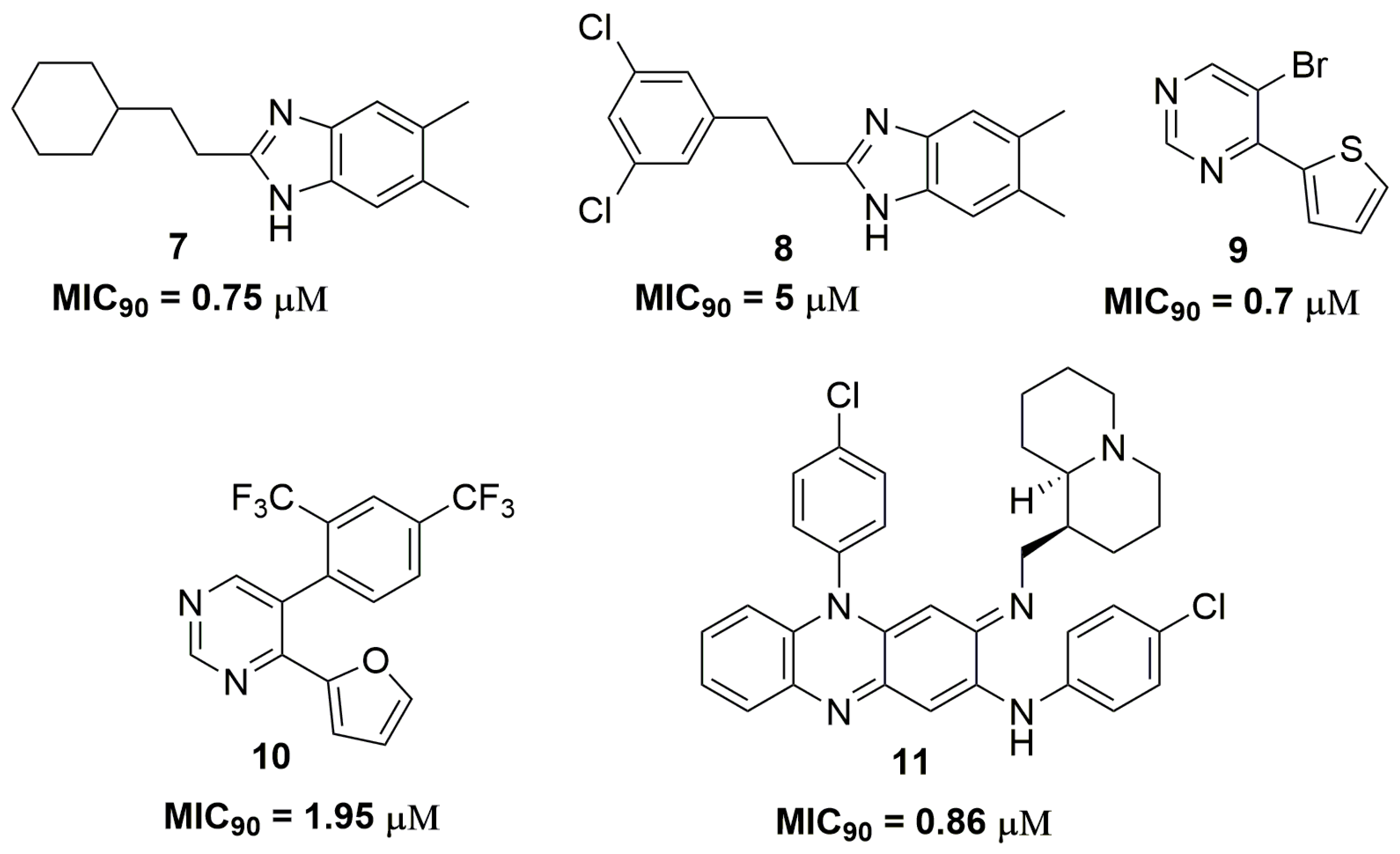
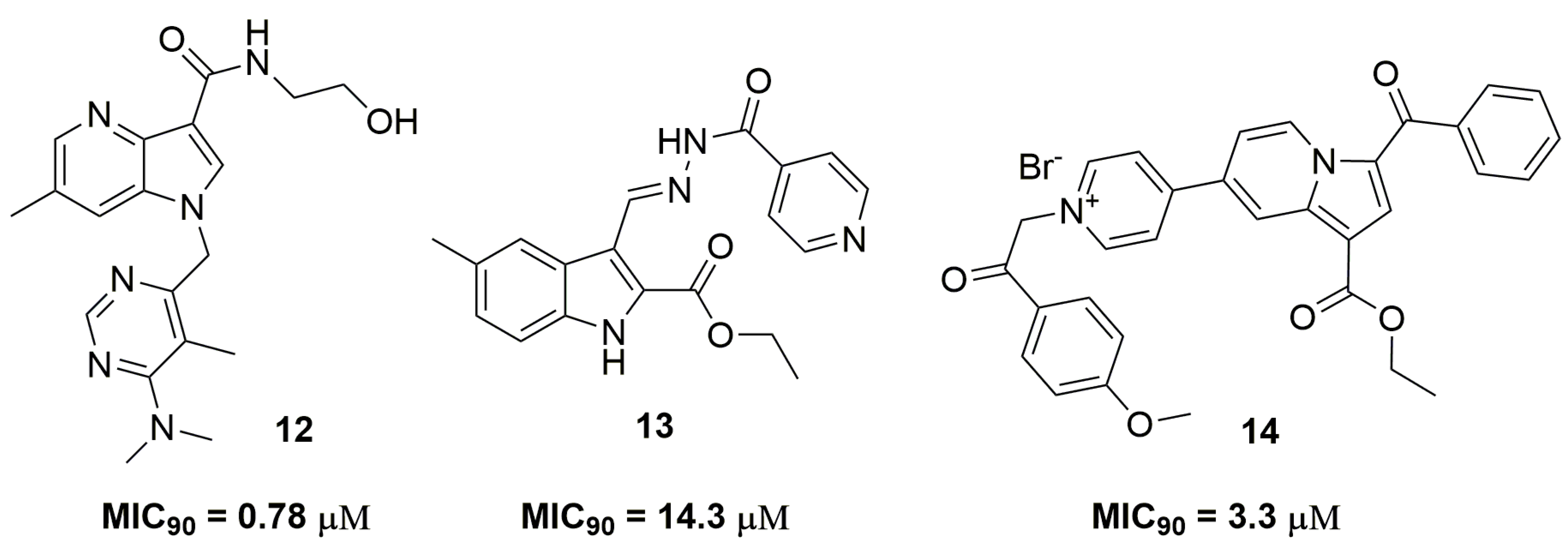
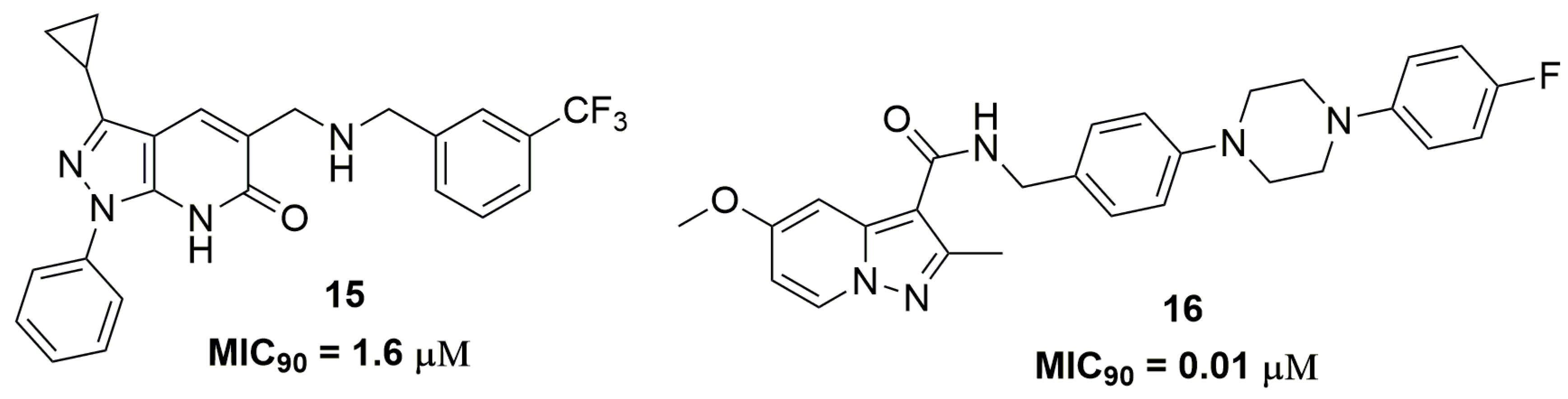
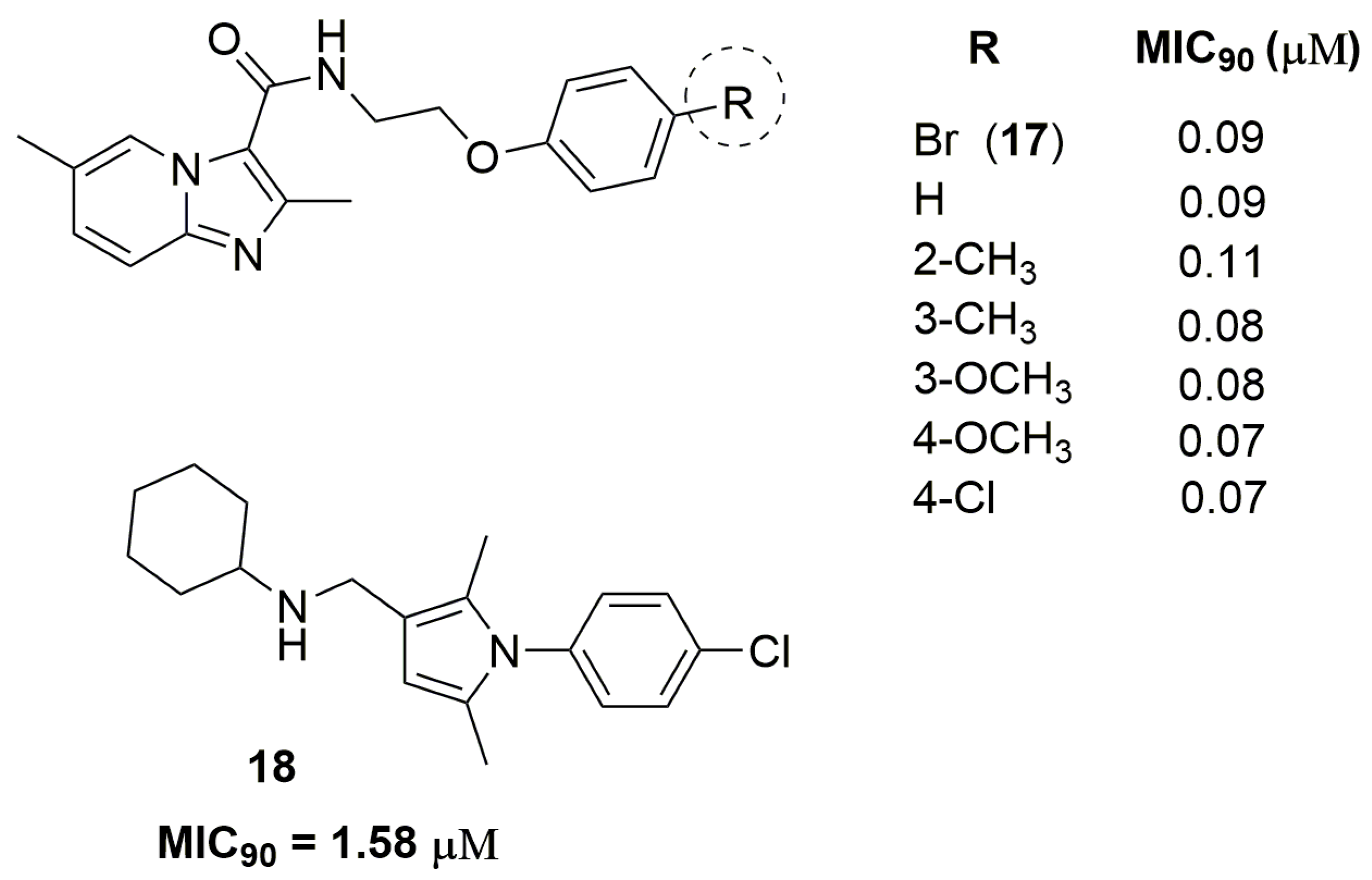
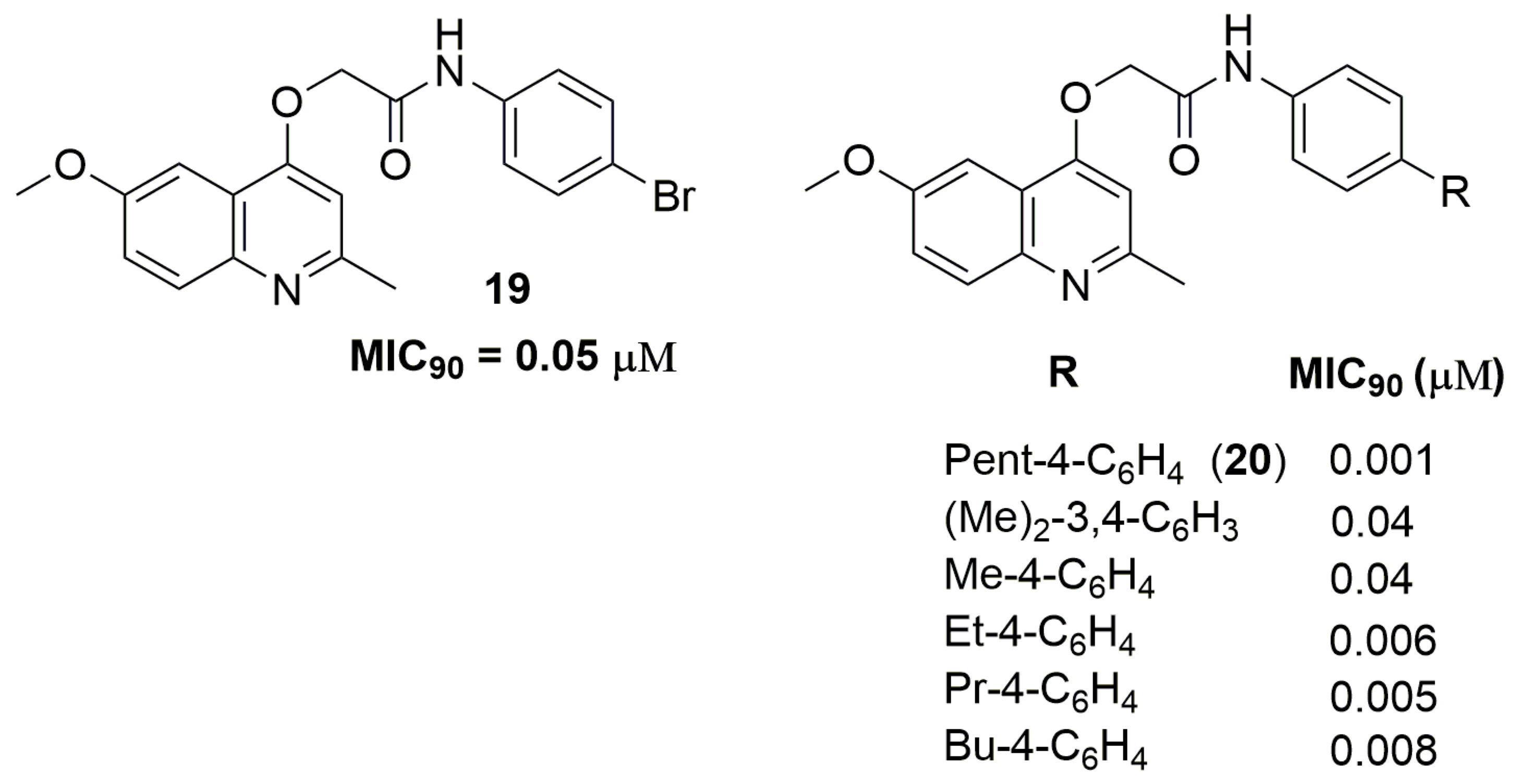
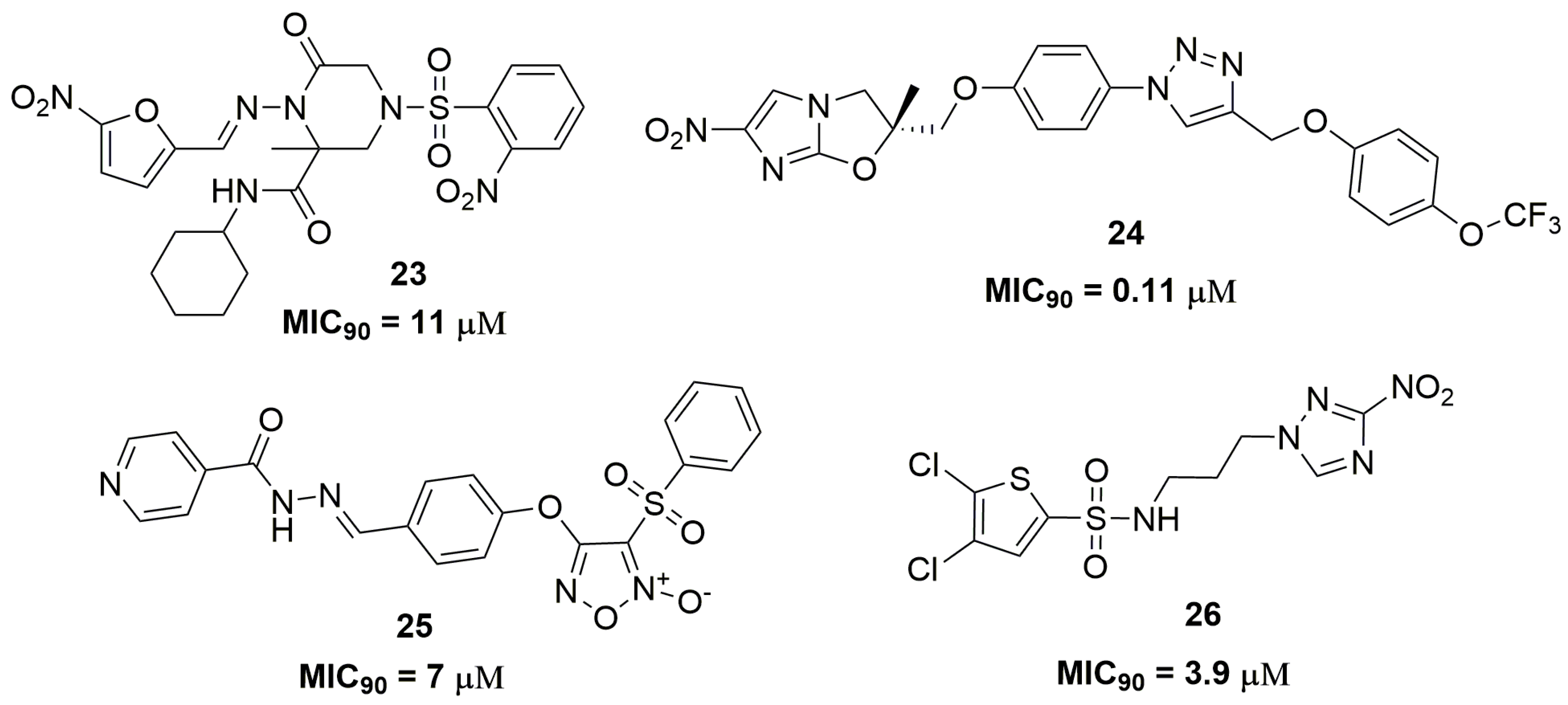
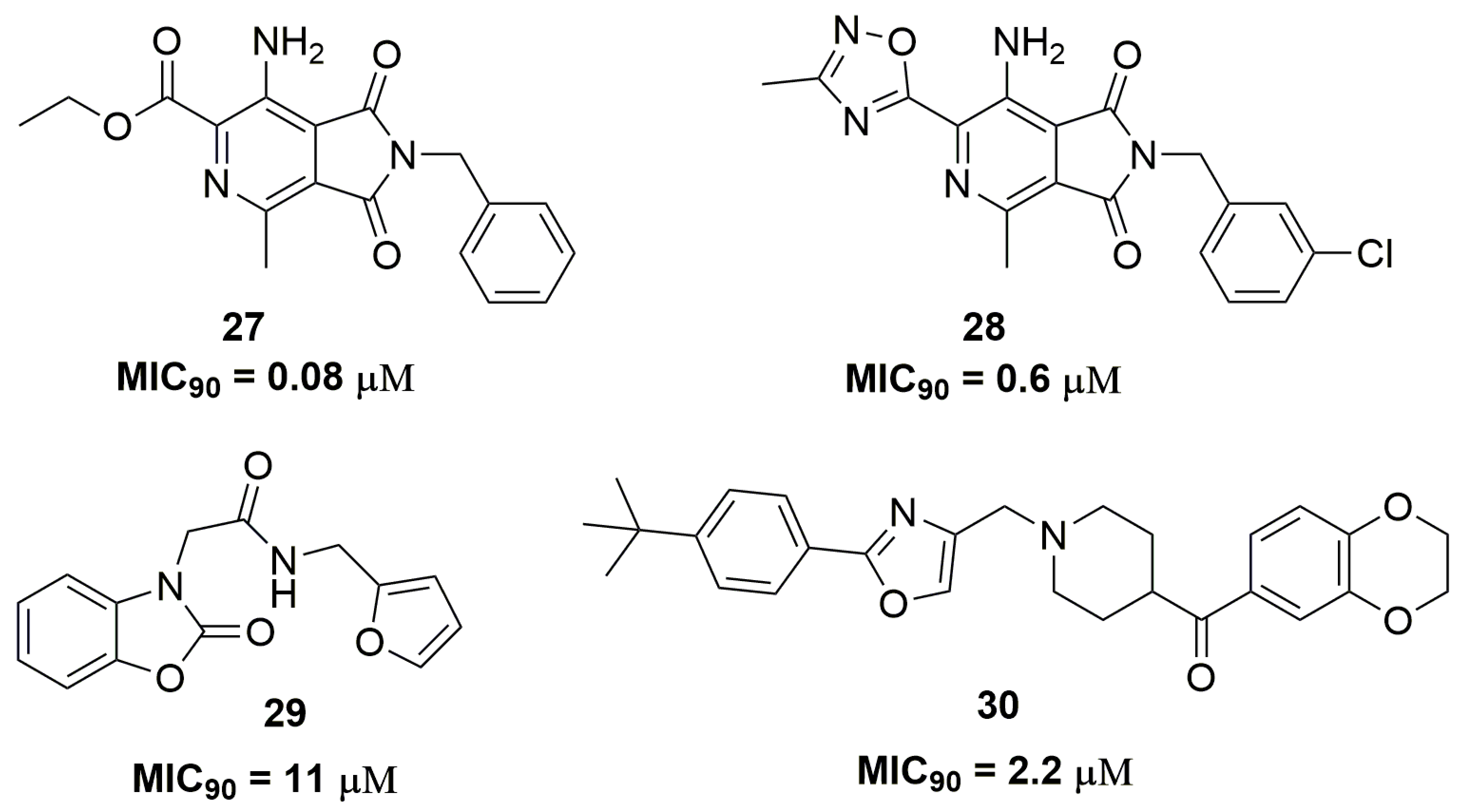
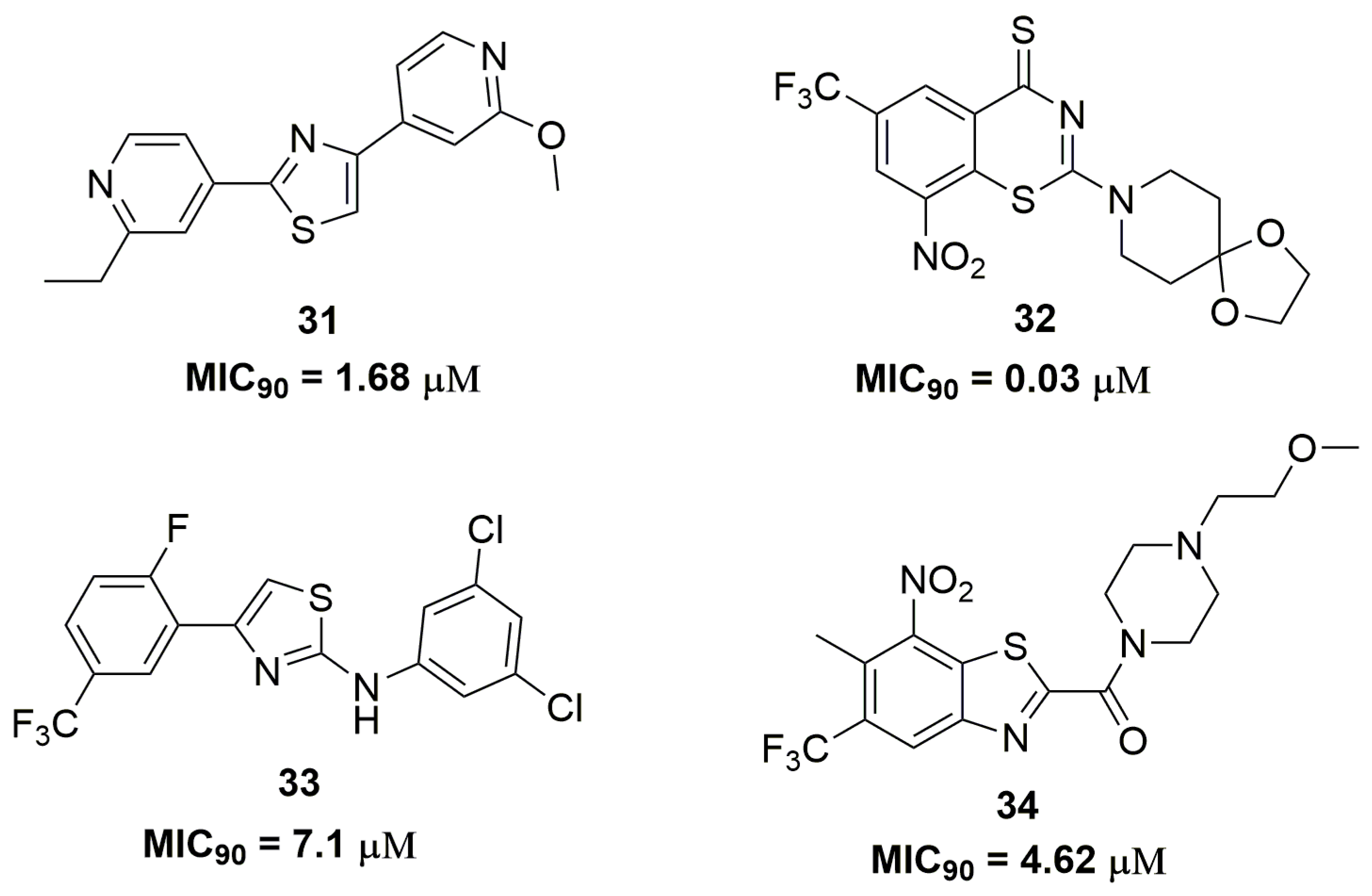
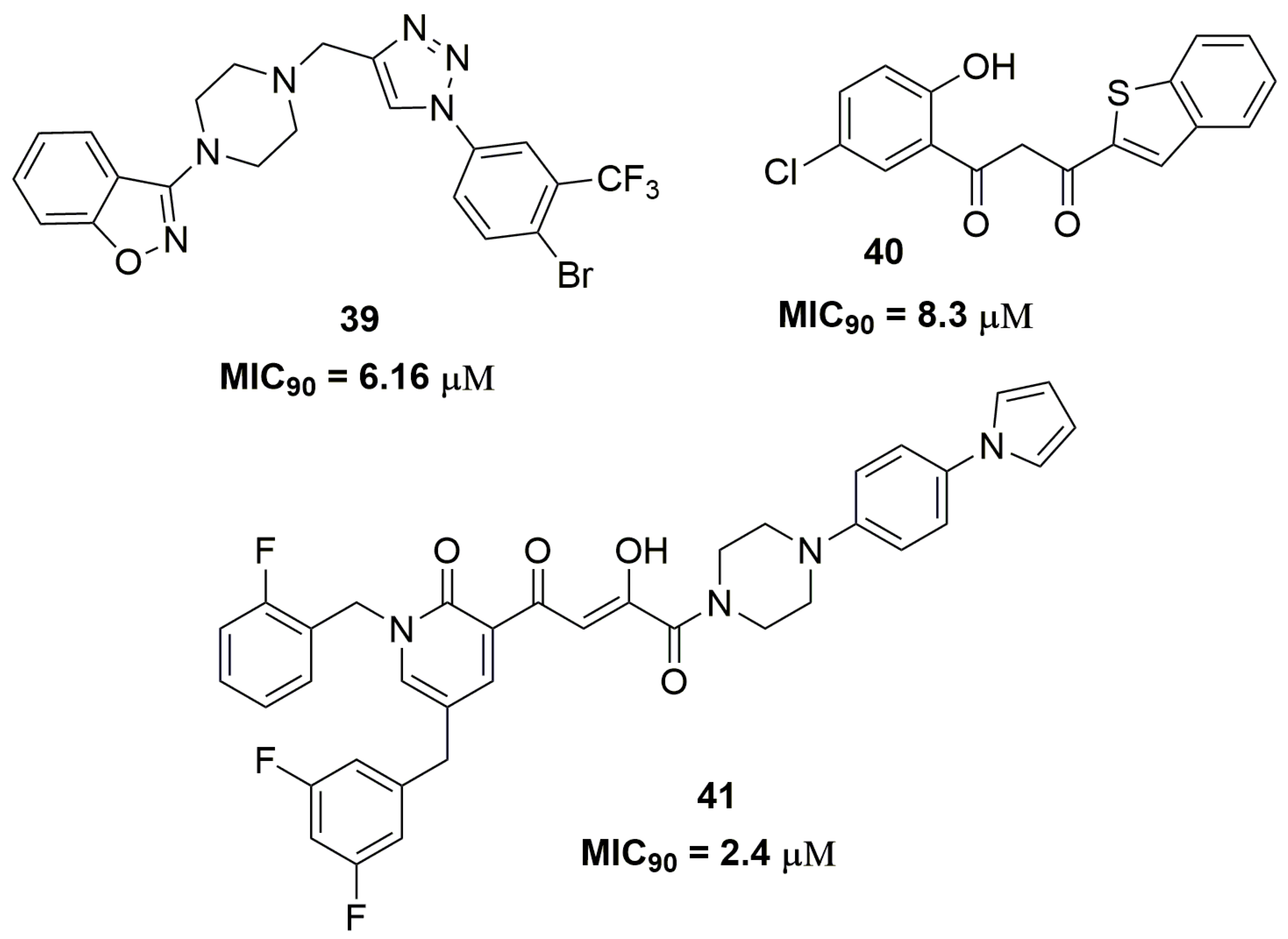
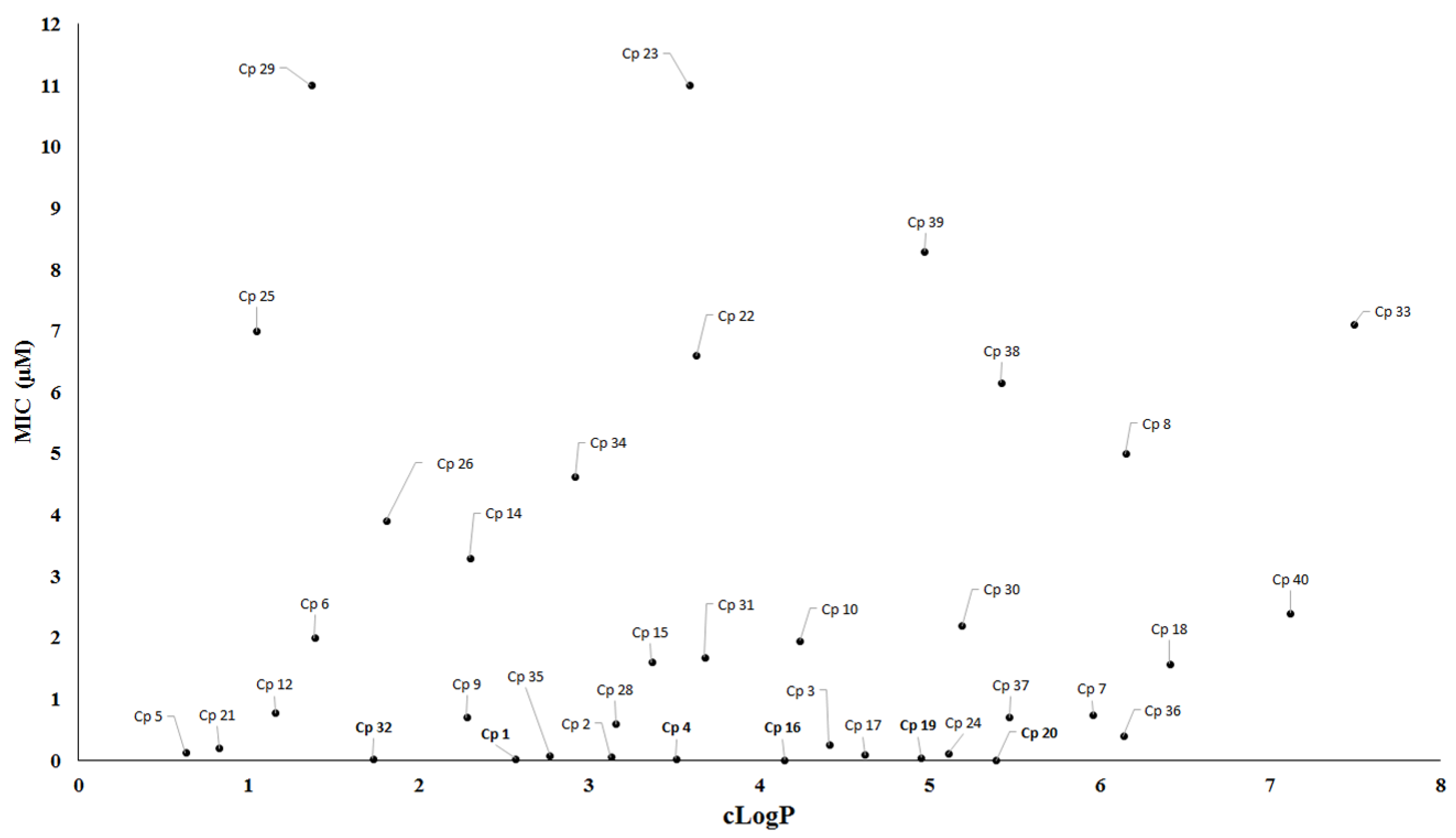
2. Antituberculosis Compounds
3. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- World Health Organization. Global Tuberculosis Report; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- World Health Organization. Global Tuberculosis Report 2014 (WHO/HTM/TB/2014.08); World Health Organization: Geneva, Switzerland, 2014. [Google Scholar]
- Zumla, A.; Nahid, P.; Cole, S.T. Advances in the development of new tuberculosis drugs and treatment regimens. Nat. Rev. Drug Discov. 2013, 12, 388–404. [Google Scholar] [CrossRef] [PubMed]
- Klopper, M.; Warren, R.M.; Hayes, C.; van Pittius, N.C.G.; Streicher, E.M.; Muller, B.; Sirgel, F.A.; Chabula-Nxiweni, M.; Hoosain, E.; Coetzee, G.; et al. Emergence and spread of extensively and totally drug-resistant tuberculosis, South Africa. Emerg. Infect. Dis. 2013, 19, 449–455. [Google Scholar] [CrossRef] [PubMed]
- Slomski, A. South Africa Warns of Emergence of “Totally” Drug-Resistant Tuberculosis. J. Am. Med. Assoc. 2013, 309, 1097–1098. [Google Scholar] [CrossRef] [PubMed]
- Koul, A.; Arnoult, E.; Lounis, N.; Guillemont, J.; Andries, K. The challenge of new drug discovery for tuberculosis. Nature 2011, 469, 483–490. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.S.; Jornada, D.H.; Souza, P.C.; Man Chin, C.; Pavan, F.R.; Santos, J.L. Current Advances in Antitubercular Drug Discovery: Potent Prototypes and New Targets. Curr. Med. Chem. 2015, 22, 3133–3161. [Google Scholar] [CrossRef]
- Pai, M.; Behr, M.A.; Dowdy, D.; Dheda, K.; Divangahi, M.; Boehme, C.C.; Ginsberg, A.; Swaminathan, S.; Spigelman, M.; Getahun, H.; et al. Tuberculosis. Nat. Rev. Dis. Prim. 2016, 2, 1–23. [Google Scholar] [CrossRef] [PubMed]
- Wallis, R.S.; Maeurer, M.; Mwaba, P.; Chakaya, J.; Rustomjee, R.; Migliori, G.B.; Marais, B.; Schito, M.; Churchyard, G.; Swaminathan, S.; et al. Tuberculosis—Advances in development of new drugs, treatment regimens, host-directed therapies, and biomarkers. Lancet Infect. Dis. 2016, 16, e34–e46. [Google Scholar] [CrossRef]
- Bloemberg, G.V.; Keller, P.M.; Stucki, D.; Trauner, A.; Borrell, S.; Latshang, T.; Coscolla, M.; Rothe, T.; Hömke, R.; Ritter, C.; et al. Acquired Resistance to Bedaquiline and Delamanid in Therapy for Tuberculosis. N. Engl. J. Med. 2015, 373, 1986–1988. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Chen, J.; Cui, P.; Shi, W.; Shi, X.; Niu, H.; Chan, D.; Yew, W.W.; Zhang, W.; Zhang, Y. Mycobacterium tuberculosis Mutations Associated with Reduced Susceptibility to Linezolid. Antimicrob. Agents Chemother. 2016, 60, 2542–2544. [Google Scholar] [CrossRef] [PubMed]
- Segala, E.; Sougakoff, W.; Nevejans-Chauffour, A.; Jarlier, V.; Petrella, S. New mutations in the mycobacterial ATP synthase: New insights into the binding of the diarylquinoline TMC207 to the ATP synthase C-Ring structure. Antimicrob. Agents Chemother. 2012, 56, 2326–2334. [Google Scholar] [CrossRef] [PubMed]
- Karabanovich, G.; Zemanova, J.; Smutny, T.; Szekely, R.; Sarkan, M.; Centarova, I.; Vocat, A.; Pavkova, I.; Conka, P.; Nemecek, J.; et al. Development of 3,5-Dinitrobenzylsulfanyl-1,3,4-oxadiazoles and Thiadiazoles as Selective Antitubercular Agents Active Against Replicating and Nonreplicating Mycobacterium tuberculosis. J. Med. Chem. 2016, 59, 2362–2380. [Google Scholar] [CrossRef] [PubMed]
- Karabanovich, G.; Němeček, J.; Valášková, L.; Carazo, A.; Konečná, K.; Stolaříková, J.; Hrabálek, A.; Pavliš, O.; Pávek, P.; Vávrová, K.; et al. S-substituted 3,5-dinitrophenyl 1,3,4-oxadiazole-2-thiols and tetrazole-5-thiols as highly efficient antitubercular agents. Eur. J. Med. Chem. 2017, 126, 369–383. [Google Scholar] [CrossRef] [PubMed]
- Zurenko, G.E.; Gibson, J.K.; Shinabarger, D.L.; Aristoff, P.A.; Ford, C.W.; Tarpley, W.G. Oxazolidinones: A new class of antibacterials. Curr. Opin. Pharmacol. 2001, 1, 470–476. [Google Scholar] [CrossRef]
- Wilson, D.N.; Schluenzen, F.; Harms, J.M.; Starosta, A.L.; Connell, S.R.; Fucini, P. The oxazolidinone antibiotics perturb the ribosomal peptidyl-transferase center and effect tRNA positioning. Proc. Natl. Acad. Sci. USA 2008, 105, 13339–13344. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, V.; Solapure, S.; Iyer, H.; Ghosh, A.; Sharma, S.; Kaur, P.; Deepthi, R.; Subbulakshmi, V.; Ramya, V.; Ramachandran, V.; et al. Bactericidal activity and mechanism of action of AZD5847, a novel oxazolidinone for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 495–502. [Google Scholar] [CrossRef] [PubMed]
- Furin, J.J.; Bois, D.; Van Brakel, E.; Chheng, P.; Venter, A.; Peloquin, C.A.; Alsultan, A.; Thiel, B.A.; Debanne, S.M.; Boom, W.H.; et al. Early Bactericidal Activity of AZD5847 in Patients with Pulmonary Tuberculosis. Antimicrob. Agents Chemother. 2016, 60, 6591–6599. [Google Scholar] [CrossRef] [PubMed]
- Ang, W.; Ye, W.; Sang, Z.; Liu, Y.; Yang, T.; Deng, Y.; Luo, Y.; Wei, Y. Discovery of novel bis-oxazolidinone compounds as potential potent and selective antitubercular agents. Bioorg. Med. Chem. Lett. 2014, 24, 1496–1501. [Google Scholar] [CrossRef] [PubMed]
- Gobis, K.; Foks, H.; Serocki, M.; Augustynowicz-Kopec, E.; Napiorkowska, A. Synthesis and evaluation of in vitro antimycobacterial activity of novel 1H-benzo[d]imidazole derivatives and analogues. Eur. J. Med. Chem. 2014, 89, 13–20. [Google Scholar] [CrossRef] [PubMed]
- Gobis, K.; Foks, H.; Suchan, K.; Augustynowicz-Kopec, E.; Napiorkowska, A.; Bojanowski, K. Novel 2-(2-phenalkyl)-1H-benzo[d]imidazoles as antitubercular agents. Synthesis, biological evaluation and structure-activity relationship. Bioorg. Med. Chem. 2015, 23, 2112–2120. [Google Scholar] [CrossRef] [PubMed]
- Kravchenko, M.A.; Verbitskiy, E.V.; Medvinskiy, I.D.; Rusinov, G.L.; Charushin, V.N. Synthesis and antituberculosis activity of novel 5-styryl-4-(hetero)aryl- pyrimidines via combination of the Pd-catalyzed Suzuki cross-coupling and S NH reactions. Bioorg. Med. Chem. Lett. 2014, 24, 3118–3120. [Google Scholar] [CrossRef] [PubMed]
- Verbitskiy, E.V.; Baskakova, S.A.; Kravchenko, M.A.; Skornyakov, S.N.; Rusinov, G.L.; Chupakhin, O.N.; Charushin, V.N. Synthesis and evaluation of antitubercular activity of fluorinated 5-aryl-4-(hetero)aryl substituted pyrimidines. Bioorg. Med. Chem. 2016, 24, 3771–3780. [Google Scholar] [CrossRef] [PubMed]
- Tonelli, M.; Novelli, F.; Tasso, B.; Sparatore, A.; Boido, V.; Sparatore, F.; Cannas, S.; Molicotti, P.; Zanetti, S.; Parapini, S.; et al. Antitubercular activity of quinolizidinyl/pyrrolizidinylalkyliminophenazines. Bioorg. Med. Chem. 2014, 22, 6837–6845. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, M.; Shandil, R.; Manjunatha, M.R.; Solapure, S.; Ramachandran, V.; Kumar, N.; Saralaya, R.; Panduga, V.; Reddy, J.; Prabhakar, K.R.; et al. 1,4-azaindole, A potential drug candidate for treatment of tuberculosis. Antimicrob. Agents Chemother. 2014, 58, 5325–5331. [Google Scholar] [CrossRef] [PubMed]
- Shirude, P.S.; Shandil, R.; Sadler, C.; Naik, M.; Hosagrahara, V.; Hammed, S.; Shinde, V.; Bathula, C.; Humnabadkar, V.; Kumar, N.; et al. Azaindoles: Noncovalent DprE1 Inhibitors from Scaffold Morphing Efforts, Kill Mycobacterium tuberculosis and Are Efficacious in Vivo. J. Med. Chem. 2013, 56, 9701–9708. [Google Scholar] [CrossRef] [PubMed]
- Velezheva, V.; Brennan, P.; Ivanov, P.; Kornienko, A.; Lyubimov, S.; Kazarian, K.; Nikonenko, B.; Majorov, K.; Apt, A. Synthesis and antituberculosis activity of indole-pyridine derived hydrazides, hydrazide-hydrazones, and thiosemicarbazones. Bioorg. Med. Chem. Lett. 2016, 26, 978–985. [Google Scholar] [CrossRef] [PubMed]
- Danac, R.; Mangalagiu, I.I. Antimycobacterial activity of nitrogen heterocycles derivatives: Bipyridine derivatives. Part III. Eur. J. Med. Chem. 2014, 74, 664–670. [Google Scholar] [CrossRef] [PubMed]
- Panda, M.; Ramachandran, S.; Ramachandran, V.; Shirude, P.S.; Humnabadkar, V.; Nagalapur, K.; Sharma, S.; Kaur, P.; Guptha, S.; Narayan, A.; et al. Discovery of pyrazolopyridones as a novel class of noncovalent DprE1 inhibitor with potent anti-mycobacterial activity. J. Med. Chem. 2014, 57, 4761–4771. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, G.; Pasca, M.R.; Chiarelli, L.R.; Manina, G.; Mattevi, A.; Binda, C. The DprE1 enzyme, one of the most vulnerable targets of Mycobacterium tuberculosis. Appl. Microbiol. Biotechnol. 2013, 97, 8841–8848. [Google Scholar] [CrossRef] [PubMed]
- Brecik, M.; Centárová, I.; Mukherjee, R.; Kolly, G.S.; Huszár, S.; Bobovská, A.; Kilacsková, E.; Mokošová, V.; Svetlíková, Z.; Šarkan, M.; et al. DprE1 Is a Vulnerable Tuberculosis Drug Target Due to Its Cell Wall Localization. ACS Chem. Biol. 2015, 10, 1631–1636. [Google Scholar] [CrossRef] [PubMed]
- Tang, J.; Wang, B.; Wu, T.; Wan, J.; Tu, Z.; Njire, M.; Wan, B.; Franzblauc, S.G.; Zhang, T.; Lu, X.; et al. Design, Synthesis, and Biological Evaluation of Pyrazolo[1,5-a]pyridine-3-carboxamides as Novel Antitubercular Agents. ACS Med. Chem. Lett. 2015, 6, 814–818. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Lu, Y.; Li, L.; Zhao, R.; Wang, B.; Lv, K.; Liu, M.; You, X. Identification of N -(2-Phenoxyethyl)imidazo[1,2-a]pyridine-3-carboxamides as New Antituberculosis Agents. ACS Med. Chem. Lett. 2016, 7, 1130–1133. [Google Scholar] [CrossRef] [PubMed]
- Bhakta, S.; Scalacci, N.; Maitra, A.; Brown, A.K.; Dasugari, S.; Evangelopoulos, D.; McHugh, T.D.; Mortazavi, P.N.; Twist, A.; Petricci, E.; et al. Design and Synthesis of 1-((1,5-Bis(4-chlorophenyl)-2-methyl-1H-pyrrol-3-yl)methyl)-4-methylpiperazine (BM212) and N-Adamantan-2-yl-N′-((E)-3,7-dimethylocta-2,6-dienyl)ethane-1,2-diamine (SQ109) Pyrrole Hybrid Derivatives: Discovery of Potent Antituberc. J. Med. Chem. 2016, 59, 2780–2793. [Google Scholar] [CrossRef] [PubMed]
- Singh, S.; Kaur, G.; Mangla, V.; Gupta, M.K. Quinoline and quinolones: Promising scaffolds for future antimycobacterial agents. J. Enzym. Inhib. Med. Chem. 2015, 30, 492–504. [Google Scholar] [CrossRef] [PubMed]
- Pissinate, K.; Villela, A.D.; Rodrigues, V.; Giacobbo, B.C.; Grams, E.S.; Abbadi, B.L.; Trindade, R.V.; Roesler Nery, L.; Bonan, C.D.; Back, D.F.; et al. 2-(Quinolin-4-yloxy)acetamides Are Active against Drug-Susceptible and Drug-Resistant Mycobacterium tuberculosis Strains. ACS Med. Chem. Lett. 2016, 7, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Giacobbo, B.C.; Pissinate, K.; Rodrigues-Junior, V.; Villela, A.D.; Grams, E.S.; Abbadi, B.L.; Subtil, F.T.; Sperotto, N.; Trindade, R.V.; Back, D.F.; et al. New insights into the SAR and drug combination synergy of 2-(quinolin-4-yloxy)acetamides against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2017, 126, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.; Humnabadkar, V.; Tantry, S.J.; Panda, M.; Narayan, A.; Guptha, S.; Panduga, V.; Manjrekar, P.; Jena, L.K.; Koushik, K.; et al. 4-Aminoquinolone piperidine amides: Noncovalent inhibitors of DprE1 with long residence time and potent antimycobacterial activity. J. Med. Chem. 2014, 57, 5419–5434. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Baig, I.A.; Sun, H.J.; Cui, C.J.; Guo, R.; Jung, I.P.; Wang, D.; Dong, M.; Yoon, M.Y.; Wang, J.G. Synthesis, crystal structure and biological evaluation of substituted quinazolinone benzoates as novel antituberculosis agents targeting acetohydroxyacid synthase. Eur. J. Med. Chem. 2015, 94, 298–305. [Google Scholar] [CrossRef] [PubMed]
- Gokhale, K.; Tilak, B. Mechanisms of bacterial acetohydroxyacid synthase (AHAS) and specific inhibitors of Mycobacterium tuberculosis AHAS as potential drug candidates against tuberculosis. Curr. Drug Targets 2015, 16, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, T.; Boshoff, H. Nitroimidazoles for the treatment of TB: Past, present and future. Futur. Med. Chem. 2011, 3, 1427–1454. [Google Scholar] [CrossRef] [PubMed]
- Rakesha, D.B.; Madhura, D.B.; Maddox, M.; Lee, R.B.; Trivedi, A.; Yang, L.; Scherman, M.S.; Gilliland, J.C.; Gruppo, V.; McNeil, M.R.; et al. Antitubercular nitrofuran isoxazolines with improved pharmacokinetic properties. Bioorg. Med. Chem. 2012, 20, 6063–6072. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.; Rojas, R.; Gilman, R.H.; Sauvain, M.; Lima, L.M.; Barreiro, E.J.; González, M.; Cerecetto, H. Hybrid furoxanyl N-acylhydrazone derivatives as hits for the development of neglected diseases drug candidates. Eur. J. Med. Chem. 2013, 59, 64–74. [Google Scholar] [CrossRef] [PubMed]
- Krasavin, M.; Parchinsky, V.; Kantin, G.; Manicheva, O.; Dogonadze, M.; Vinogradova, T.; Karge, B.; Brönstrup, M. New nitrofurans amenable by isocyanide multicomponent chemistry are active against multidrug-resistant and poly-resistant Mycobacterium tuberculosis. Bioorg. Med. Chem. 2017, 25, 1867–1874. [Google Scholar] [CrossRef] [PubMed]
- Lewis, J.M.; Sloan, D.J. The role of delamanid in the treatment of drug-resistant tuberculosis. Ther. Clin. Risk Manag. 2015, 11, 779–791. [Google Scholar] [PubMed]
- Xavier, A.S.; Lakshmanan, M. Delamanid: A new armor in combating drug-resistant tuberculosis. J. Pharmacol. Pharmacother. 2014, 5, 222–224. [Google Scholar] [CrossRef] [PubMed]
- Munagala, G.; Yempalla, K.R.; Singh, S.; Sharma, S.; Kalia, N.P.; Rajput, V.S.; Kumar, S.; Sawant, S.D.; Khan, I.A.; Vishwakarma, R.A.; et al. Synthesis of new generation triazolyl- and isoxazolyl-containing 6-nitro-2,3-dihydroimidazooxazoles as anti-TB agents: In vitro, structure-activity relationship, pharmacokinetics and in vivo evaluation. Org. Biomol. Chem. 2015, 13, 3610–3624. [Google Scholar] [CrossRef] [PubMed]
- Fernandes, G.F.; de Souza, P.C.; Marino, L.B.; Chegaev, K.; Gugliemo, S.; Lazzarato, L.; Fruttero, R.; Chung, M.C.; Pavan, F.R.; Dos Santos, J.L. Synthesis and biological activity of furoxan derivatives against Mycobacterium tuberculosis. Eur. J. Med. Chem. 2016, 123, 523–531. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulou, M.V.; Bloomer, W.D.; Rosenzweig, H.S.; Arena, A.; Arrieta, F.; Rebolledo, J.C.J.; Smith, D.K. Nitrotriazole- and imidazole-based amides and sulfonamides as antitubercular agents. Antimicrob. Agents Chemother. 2014, 58, 6828–6836. [Google Scholar] [CrossRef] [PubMed]
- Van Der Westhuyzen, R.; Winks, S.; Wilson, C.R.; Boyle, G.A.; Gessner, R.K.; Soares De Melo, C.; Taylor, D.; De Kock, C.; Njoroge, M.; Brunschwig, C.; et al. Pyrrolo[3,4-c]pyridine-1,3(2H)-diones: A novel antimycobacterial class targeting mycobacterial respiration. J. Med. Chem. 2015, 58, 9371–9381. [Google Scholar] [CrossRef] [PubMed]
- Pedgaonkar, G.S.; Sridevi, J.P.; Jeankumar, V.U.; Saxena, S.; Devi, P.B.; Renuka, J.; Yogeeswari, P.; Sriram, D. Development of benzo[d]oxazol-2(3H)-ones derivatives as novel inhibitors of Mycobacterium tuberculosis InhA. Bioorganic Med. Chem. 2014, 22, 6134–6145. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Gao, N.; Zhu, N.; Lin, Y.; Li, Y.; Chen, M.; You, X.; Lu, Y.; Wan, K.; Jiang, J.D.; et al. Discovery of the disubstituted oxazole analogues as a novel class anti-tuberculotic agents against MDR- and XDR-MTB. Bioorg. Med. Chem. Lett. 2015, 25, 5178–5181. [Google Scholar] [CrossRef] [PubMed]
- Krátký, M.; Vinsova, J. Sulphur-Containing Heterocycles as Antimycobacterial Agents: Recent Advances in Thiophene and Thiadiazole Derivatives. Curr. Top. Med. Chem. 2016, 16, 2921–2952. [Google Scholar] [CrossRef] [PubMed]
- Bellale, E.; Naik, M.; Vb, V.; Ambady, A.; Narayan, A.; Ravishankar, S.; Ramachandran, V.; Kaur, P.; McLaughlin, R.; Whiteaker, J.; et al. Diarylthiazole: An antimycobacterial scaffold potentially targeting PrrB-PrrA two-component system. J. Med. Chem. 2014, 57, 6572–6582. [Google Scholar] [CrossRef] [PubMed]
- Haydel, S.E.; Malhotra, V.; Cornelison, G.L.; Clark-Curtiss, J.E. Transient requirement of the PrrA-PrrB two-component system for early intracellular multiplication of Mycobacterium tuberculosis. J. Bacteriol. 2012, 194, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Ewann, F.; Jackson, M.; Pethe, K.; Cooper, A.; Mielcarek, N.; Ensergueix, D.; Gicquel, B.; Locht, C.; Supply, P. Transient Requirement of the PrrA-PrrB Two-Component System for Early Intracellular Multiplication of Mycobacterium tuberculosis. Infect. Immun. 2002, 70, 2256–2263. [Google Scholar] [CrossRef] [PubMed]
- Gao, C.; Peng, C.; Shi, Y.; You, X.; Ran, K.; Xiong, L.; Ye, T.; Zhang, L.; Wang, N.; Zhu, Y.; et al. Benzothiazinethione is a potent preclinical candidate for the treatment of drug-resistant tuberculosis. Sci. Rep. 2016, 6, 29717. [Google Scholar] [CrossRef] [PubMed]
- Makarov, V.; Manina, G.; Mikusova, K.; Möllmann, U.; Ryabova, O.; Saint-Joanis, B.; Dhar, N.; Pasca, M.R.; Buroni, S.; Lucarelli, A.P.; et al. Benzothiazinones kill Mycobacterium tuberculosis by blocking arabinan synthesis. Science 2009, 324, 801–804. [Google Scholar] [CrossRef] [PubMed]
- Pieroni, M.; Wan, B.; Cho, S.; Franzblau, S.G.; Costantino, G. Design, synthesis and investigation on the structure-activity relationships of N-substituted 2-aminothiazole derivatives as antitubercular agents. Eur. J. Med. Chem. 2014, 72, 26–34. [Google Scholar] [CrossRef] [PubMed]
- Landge, S.; Mullick, A.B.; Nagalapur, K.; Neres, J.; Subbulakshmi, V.; Murugan, K.; Ghosh, A.; Sadler, C.; Fellows, M.D.; Humnabadkar, V.; et al. Discovery of benzothiazoles as antimycobacterial agents: Synthesis, structure-activity relationships and binding studies with Mycobacterium tuberculosis decaprenylphosphoryl-B-D-ribose 2’-oxidase. Bioorg. Med. Chem. 2015, 23, 7694–7710. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, N.S.; Dhawale, S.C. Linked pyridinyl-thiadiazoles: Design and synthesis as potential candidate for treatment of XDR and MDR tuberculosis. Eur. J. Med. Chem. 2015, 102, 243–248. [Google Scholar] [CrossRef] [PubMed]
- Verbitskiy, E.V.; Cheprakova, E.M.; Slepukhin, P.A.; Kravchenko, M.A.; Skornyakov, S.N.; Rusinov, G.L.; Chupakhin, O.N.; Charushin, V.N. Synthesis, and structure-activity relationship for C(4) and/or C(5) thienyl substituted pyrimidines, as a new family of antimycobacterial compounds. Eur. J. Med. Chem. 2015, 97, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Stec, J.; Vilchèze, C.; Lun, S.; Perryman, A.L.; Wang, X.; Freundlich, J.S.; Bishai, W.; Jacobs, W.R.; Kozikowski, A.P. Biological evaluation of potent triclosan-derived inhibitors of the enoyl-acyl carrier protein reductase InhA in drug-sensitive and drug-resistant strains of Mycobacterium tuberculosis. ChemMedChem 2014, 9, 2528–2537. [Google Scholar] [CrossRef] [PubMed]
- Stec, J.; Onajole, O.K.; Lun, S.; Guo, H.; Merenbloom, B.; Vistoli, G.; Bishai, W.R.; Kozikowski, A.P. Indole-2-carboxamide-based MmpL3 Inhibitors Show Exceptional Antitubercular Activity in an Animal Model of Tuberculosis Infection. J. Med. Chem. 2016, 59, 6232–6247. [Google Scholar] [CrossRef] [PubMed]
- Naidu, K.M.; Srinivasarao, S.; Agnieszka, N.; Ewa, A.K.; Kumar, M.M.K.; Chandra Sekhar, K.V.G. Seeking potent anti-tubercular agents: Design, synthesis, anti-tubercular activity and docking study of various ((triazoles/indole)-piperazin-1-yl/1,4-diazepan-1-yl)benzo[d]isoxazole derivatives. Bioorg. Med. Chem. Lett. 2016, 26, 2245–2250. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, P.S.; Nikam, M.D.; Nawale, L.U.; Khedkar, V.M.; Sarkar, D.; Gill, C.H. Synthesis and Antitubercular Activity of New Benzo[b]thiophenes. ACS Med. Chem. Lett. 2016, 7, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Nair, V.; Okello, M.O.; Mangu, N.K.; Seo, B.I.; Gund, M.G. A novel molecule with notable activity against multi-drug resistant tuberculosis. Bioorg. Med. Chem. Lett. 2015, 25, 1269–1273. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.; Hu, X.; Liu, Z.; Zhang, T.; Wang, R.; Wan, B.; Franzblau, S.; You, Q. Benzylsulfanyl benzo-heterocycle amides and hydrazones as new agents against drug susceptible and resistant Mycobacterium tuberculosis. Med. Chem. Commun. 2017. [Google Scholar] [CrossRef]
- Zheng, R.; Blanchard, J.S. Steady-State and Pre-Steady-State Kinetic Analysis of Mycobacterium tuberculosis Pantothenate Synthetase. Biochemistry 2001, 40, 12904–12912. [Google Scholar] [CrossRef] [PubMed]
- Leeson, P.D.; Springthorpe, B. The influence of drug-like concepts on decision-making in medicinal chemistry. Nat. Rev. Drug Discov. 2007, 6, 881–890. [Google Scholar] [CrossRef] [PubMed]
- Curatolo, W. Physical chemical properties of oral drug candidates in the discovery and exploratory development settings. Pharm. Sci. Technol. Today 1998, 1, 387–393. [Google Scholar] [CrossRef]
- Mdluli, K.; Kaneko, T.; Upton, A. Tuberculosis drug discovery and emerging targets. Ann. N. Y. Acad. Sci. 2014, 1323, 56–75. [Google Scholar] [CrossRef] [PubMed]
- Wenlock, M.C.; Barton, P. In Silico Physicochemical Parameter Predictions. Mol. Pharm. 2013, 10, 1224–1235. [Google Scholar] [CrossRef] [PubMed]
- Tetko, I.V. Computing chemistry on the web. Drug Discov. Today 2005, 10, 1497–1500. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and developmental settings. Adv. Drug Deliv. Rev. 1997, 23, 3–25. [Google Scholar] [CrossRef]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Organic Chemistry Portal Fragment Based Druglikeness. Available online: http://www.organic-chemistry.org/prog/peo/druglike (accessed on 29 May 2017).
- Zuniga, E.S.; Early, J.; Parish, T. The future for early-stage tuberculosis drug discovery. Futur. Microbiol. 2015, 10, 217–229. [Google Scholar] [CrossRef] [PubMed]
- Manjunatha, U.H.; Smith, P.W. Perspective: Challenges and opportunities in TB drug discovery from phenotypic screening. Bioorg. Med. Chem. 2015, 23, 5087–5097. [Google Scholar] [CrossRef] [PubMed]
- Waring, M.J. Lipophilicity in drug discovery. Expert Opin. Drug Discov. 2010, 5, 235–248. [Google Scholar] [CrossRef] [PubMed]
- Chen, M.; Borlak, J.; Tong, W. High lipophilicity and high daily dose of oral medications are associated with significant risk for drug-induced liver injury. Hepatology 2013, 58, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Tarcsay, A.; Keserű, G.M. Contributions of molecular properties to drug promiscuity. J. Med. Chem. 2013, 56, 1789–1795. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | MIC90 (μM) 2 | cLogP 3 | H bond Donors | H bond Acceptors | Molecular Weight | Drug-Likeness |
|---|---|---|---|---|---|---|
| 1 | 0.03 | 1.84 | 0 | 9 | 358.33 | −4.43 |
| 2 | 0.06 | 1.22 | 0 | 11 | 419.39 | −3.14 |
| 4 | 0.03 | 2.57 | 0 | 9 | 437.22 | −6.22 |
| 16 | 0.01 | 2.83 | 1 | 7 | 473.55 | 7.03 |
| 17 | 0.09 | 2.98 | 1 | 5 | 388.26 | 1.53 |
| 19 | 0.05 | 3.75 | 1 | 5 | 401.25 | −1.04 |
| 20 | 0.001 | 4.23 | 1 | 5 | 364.44 | −1.44 |
| 32 | 0.03 | 1.23 | 0 | 7 | 433.43 | −14.4 |
| 35 | 0.08 | 3.44 | 1 | 5 | 376.89 | 3.49 |
| 38 | 0.006 | 3.72 | 2 | 3 | 332.39 | −1.2 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fernandes, G.F.d.S.; Man Chin, C.; Dos Santos, J.L. Advances in Drug Discovery of New Antitubercular Multidrug-Resistant Compounds . Pharmaceuticals 2017, 10, 51. https://doi.org/10.3390/ph10020051
Fernandes GFdS, Man Chin C, Dos Santos JL. Advances in Drug Discovery of New Antitubercular Multidrug-Resistant Compounds . Pharmaceuticals. 2017; 10(2):51. https://doi.org/10.3390/ph10020051
Chicago/Turabian StyleFernandes, Guilherme Felipe dos Santos, Chung Man Chin, and Jean Leandro Dos Santos. 2017. "Advances in Drug Discovery of New Antitubercular Multidrug-Resistant Compounds " Pharmaceuticals 10, no. 2: 51. https://doi.org/10.3390/ph10020051
APA StyleFernandes, G. F. d. S., Man Chin, C., & Dos Santos, J. L. (2017). Advances in Drug Discovery of New Antitubercular Multidrug-Resistant Compounds . Pharmaceuticals, 10(2), 51. https://doi.org/10.3390/ph10020051