


Effect of Tryptic Digestion on Sensitivity and Specificity in MALDI-TOF-Based Molecular Diagnostics through Machine Learning

 ,
, 
 , , and
, , and 
Abstract
:1. Introduction
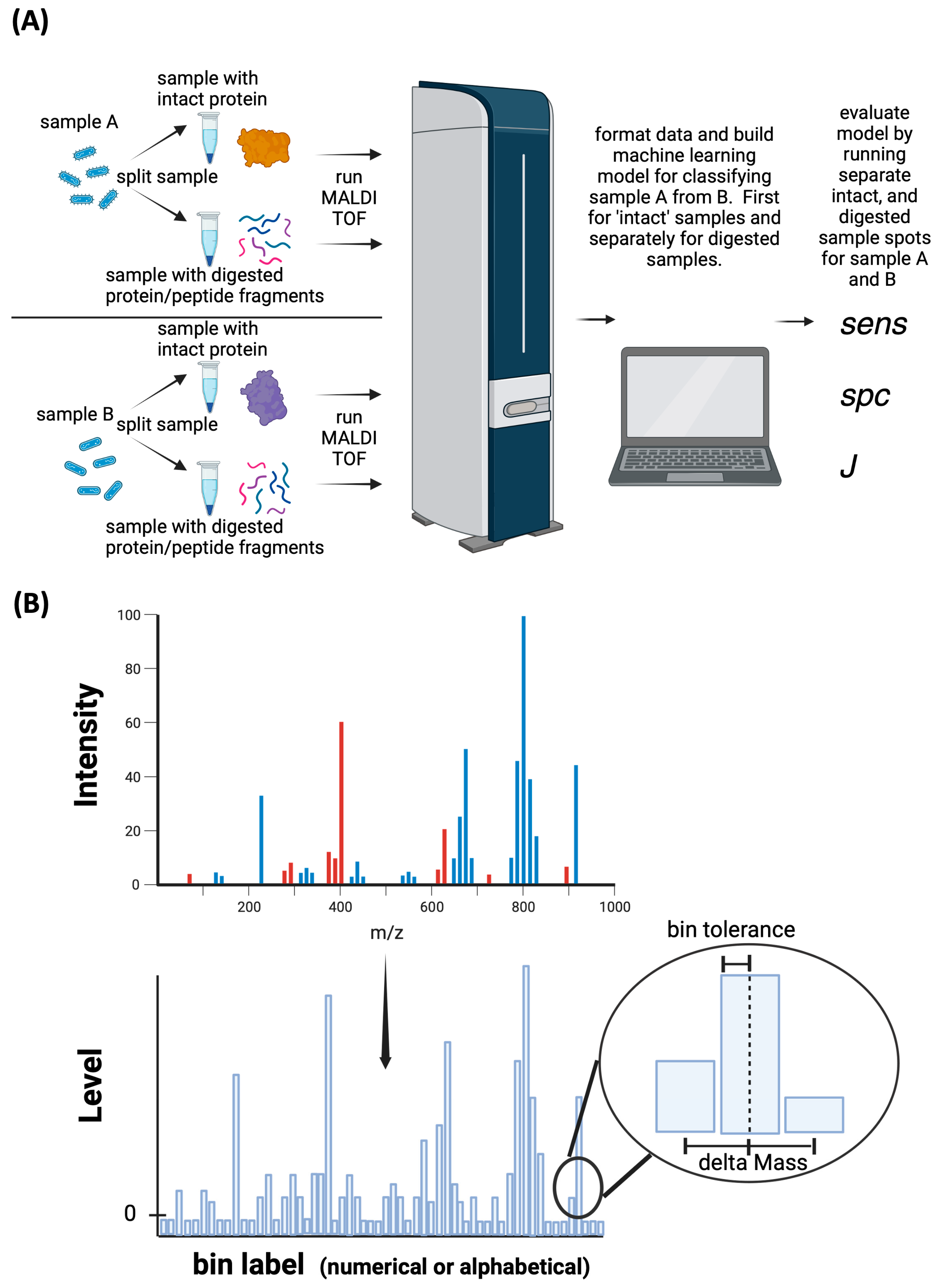
2. Materials and Methods
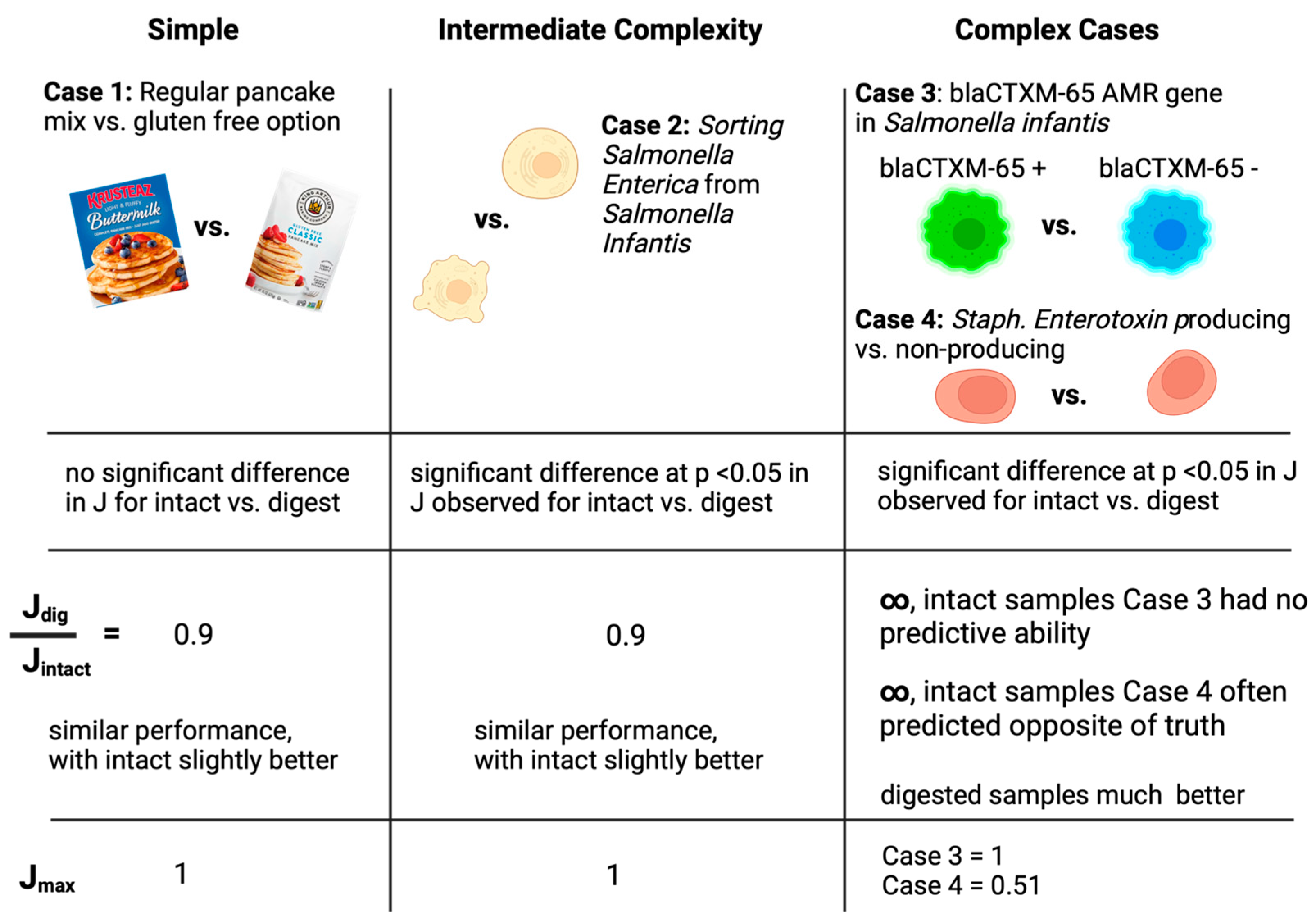
2.1. Case Study 1: Pancake Mix—Gluten-Free vs. Wheat Gluten Mix
Sample Preparation
2.2. Case Study 2: Salmonella enterica ATCC 51741 vs. Salmonella enterica Serovar Infantis
Microbial Culture and Spotting
2.3. Case Study 3: Presence of blaCTX-M-65 AMR Gene in S. enterica Serovar Infantis
2.3.1. Independent Assessment of blaCTX-M-65 AMR Gene Presence
2.3.2. Microbial Culture and Spotting
2.4. Case Study 4: Staphylococcus aureus—Enterotoxin-Positive or -Negative
2.4.1. Independent Assessment of Enterotoxin Presence
2.4.2. Microbial Culture and Spotting
2.5. MALDI-TOF-MS Analysis
2.6. Data Mining
3. Results
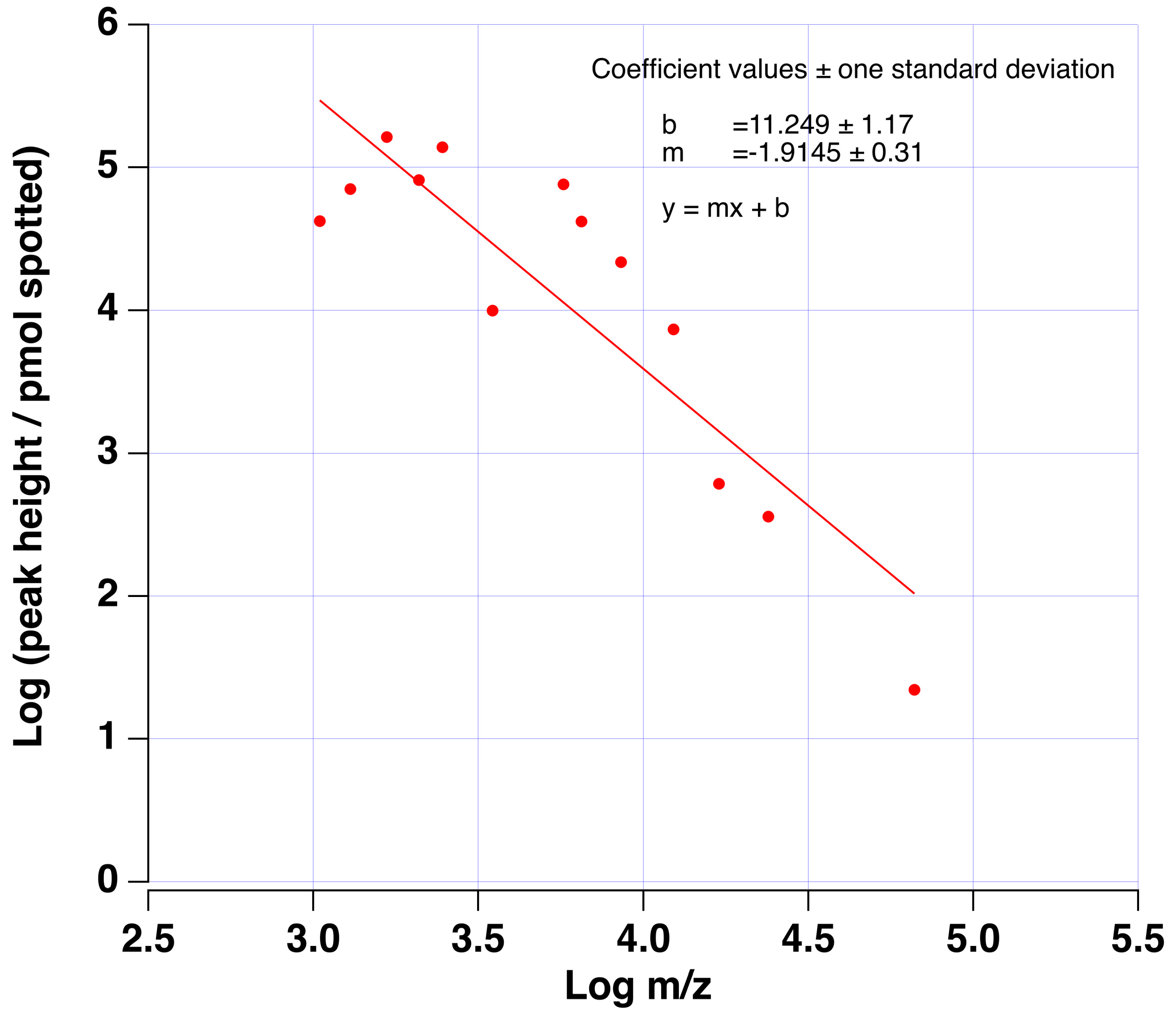
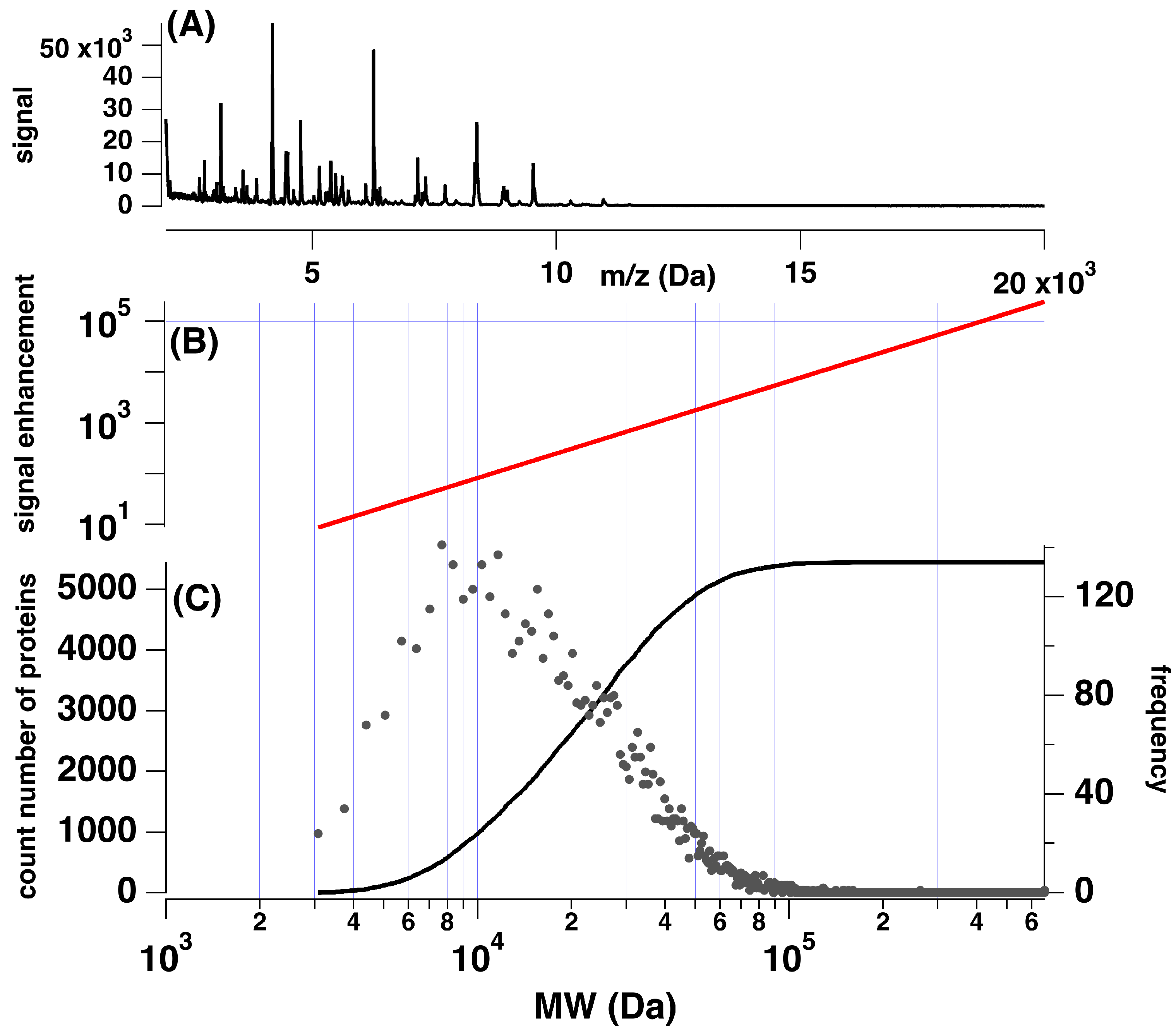
3.1. Estimate of Sensitivity Increase via Tryptic Digestion
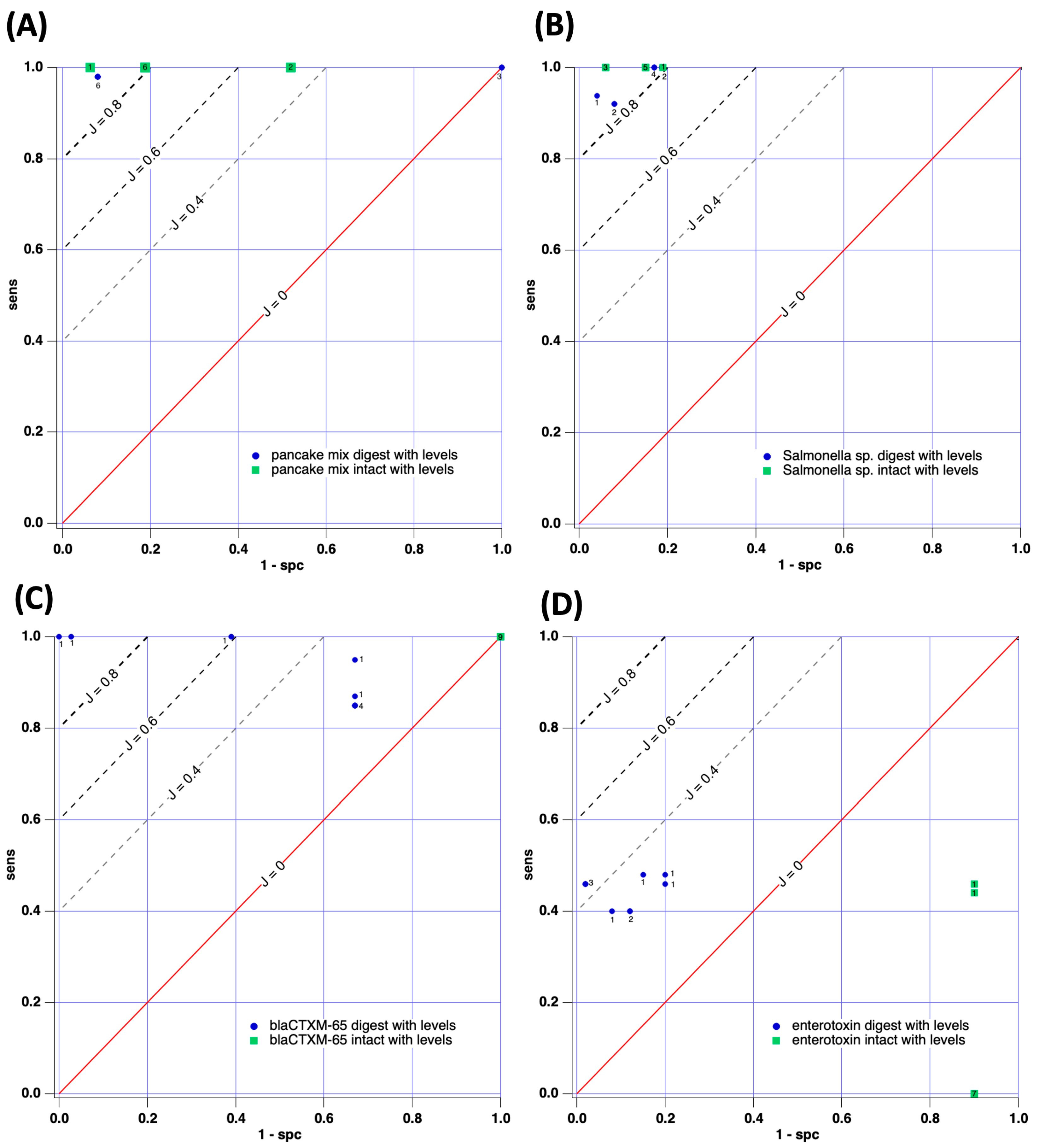
3.2. Results for Case Studies
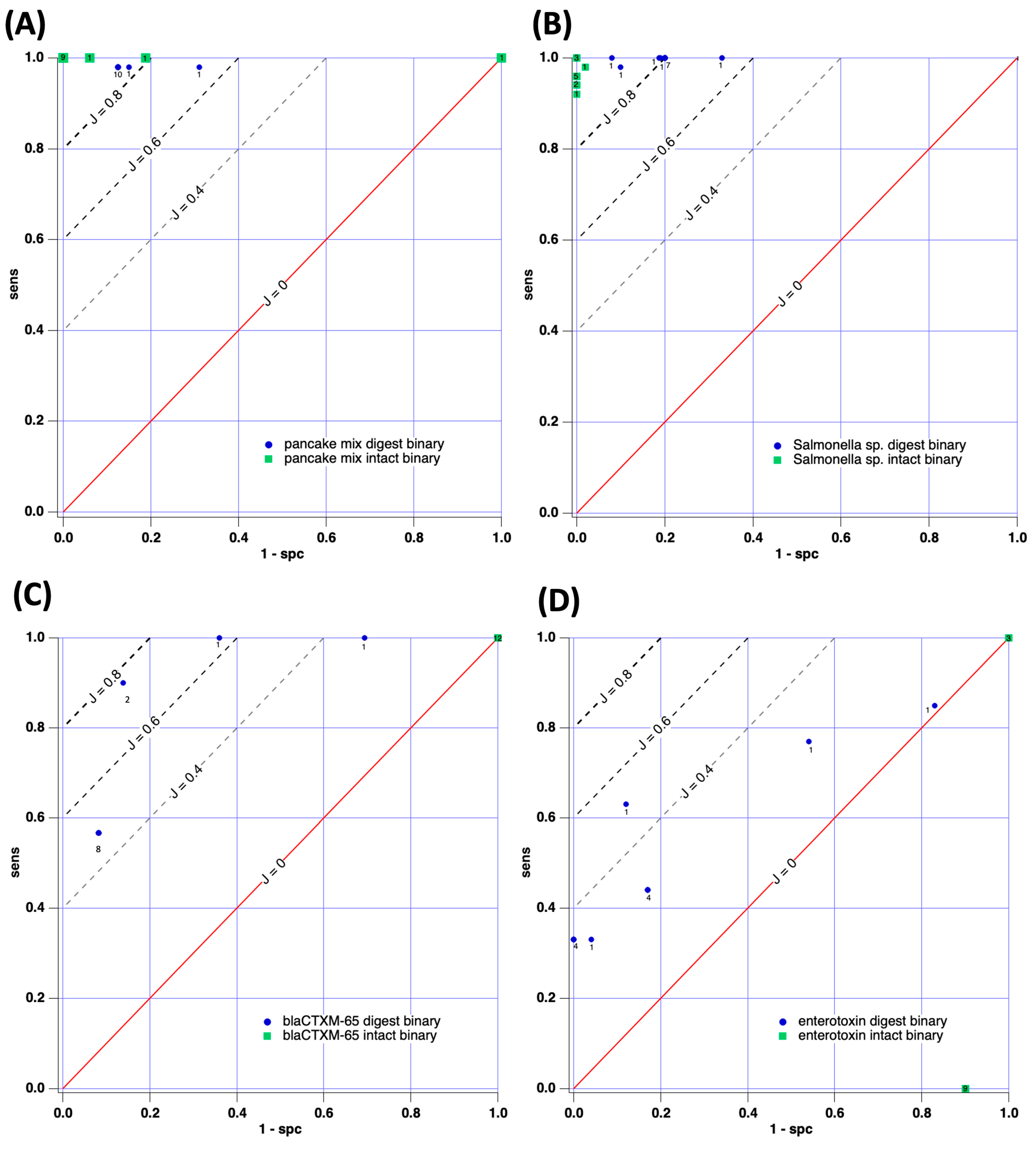
3.2.1. Case Study 1: Pancake Mix—Gluten-Free vs. Wheat Gluten Mix
3.2.2. Case Study 2: Salmonella enterica ATCC 51741 vs. Salmonella enterica Serovar Infantis
3.2.3. Case Study 3: blaCTX-M-65 Gene-Positive or -Negative
3.2.4. Case Study 4: Staphylococcus aureus–Enterotoxin-Positive or -Negative
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Thompson, J.E. Matrix-Assisted Laser Desorption Ionization-Time-of-Flight Mass Spectrometry in Veterinary Medicine: Recent Advances (2019–Present). Vet. World 2022, 15, 2623–2657. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Yi, J.; Han, G.; Qiao, L. MALDI-TOF Mass Spectrometry in Clinical Analysis and Research. ACS Meas. Sci. Au 2022, 2, 385–404. [Google Scholar] [CrossRef] [PubMed]
- Feucherolles, M.; Poppert, S.; Utzinger, J.; Becker, S.L. MALDI-TOF Mass Spectrometry as a Diagnostic Tool in Human and Veterinary Helminthology: A Systematic Review. Parasites Vectors 2019, 12, 245. [Google Scholar] [CrossRef] [PubMed]
- Do, T.; Guran, R.; Adam, V.; Zitka, O. Use of MALDI-TOF Mass Spectrometry for Virus Identification: A Review. Analyst 2022, 147, 3131–3154. [Google Scholar] [CrossRef] [PubMed]
- Singhal, N.; Kumar, M.; Kanaujia, P.K.; Virdi, J.S. MALDI-TOF Mass Spectrometry: An Emerging Technology for Microbial Identification and Diagnosis. Front. Microbiol. 2015, 6, 144398. [Google Scholar] [CrossRef]
- Santos, T.; Capelo, J.L.; Santos, H.M.; Oliveira, I.; Marinho, C.; Gonçalves, A.; Araújo, J.E.; Poeta, P.; Igrejas, G. Use of MALDI-TOF Mass Spectrometry Fingerprinting to Characterize Enterococcus Spp. and Escherichia Coli Isolates. J. Proteom. 2015, 127 Pt B, 321–331. [Google Scholar] [CrossRef]
- Raharimalala, F.N.; Andrianinarivomanana, T.M.; Rakotondrasoa, A.; Collard, J.M.; Boyer, S. Usefulness and Accuracy of MALDI-TOF Mass Spectrometry as a Supplementary Tool to Identify Mosquito Vector Species and to Invest in Development of International Database. Med. Vet. Entomol. 2017, 31, 289–298. [Google Scholar] [CrossRef]
- Fernández-Álvarez, C.; Torres-Corral, Y.; Saltos-Rosero, N.; Santos, Y. MALDI-TOF Mass Spectrometry for Rapid Differentiation of Tenacibaculum Species Pathogenic for Fish. Appl. Microbiol. Biotechnol. 2017, 101, 5377–5390. [Google Scholar] [CrossRef]
- Fernández-Álvarez, C.; Torres-Corral, Y.; Santos, Y. Use of Ribosomal Proteins as Biomarkers for Identification of Flavobacterium Psychrophilum by MALDI-TOF Mass Spectrometry. J. Proteom. 2018, 170, 59–69. [Google Scholar] [CrossRef]
- Yolanda, T.C.; Clara, F.Á.; Ysabel, S. Proteomic and Molecular Fingerprinting for Identification and Tracking of Fish Pathogenic Streptococcus. Aquaculture 2019, 498, 322–334. [Google Scholar] [CrossRef]
- Kyritsi, M.A.; Kristo, I.; Hadjichristodoulou, C. Serotyping and Detection of Pathogenecity Loci of Environmental Isolates of Legionella Pneumophila Using MALDI-TOF MS. Int. J. Hyg. Environ. Health 2020, 224, 113441. [Google Scholar] [CrossRef] [PubMed]
- Christoforidou, S.; Kyritsi, M.; Boukouvala, E.; Ekateriniadou, L.; Zdragas, A.; Samouris, G.; Hadjichristodoulou, C. Identification of Brucella Spp. Isolates and Discrimination from the Vaccine Strain Rev.1 by MALDI-TOF Mass Spectrometry. Mol. Cell Probes 2020, 51, 101533. [Google Scholar] [CrossRef] [PubMed]
- Hamidi, H.; Bagheri Nejad, R.; Es-Haghi, A.; Ghassempour, A. A Combination of MALDI-TOF MS Proteomics and Species-Unique Biomarkers’ Discovery for Rapid Screening of Brucellosis. J. Am. Soc. Mass. Spectrom. 2022, 33, 1530–1540. [Google Scholar] [CrossRef]
- Chen, M.; Wei, X.; Zhang, J.; Zhou, H.; Chen, N.; Wang, J.; Feng, Y.; Yu, S.; Zhang, J.; Wu, S.; et al. Differentiation of Bacillus Cereus and Bacillus Thuringiensis Using Genome-Guided MALDI-TOF MS Based on Variations in Ribosomal Proteins. Microorganisms 2022, 10, 918. [Google Scholar] [CrossRef]
- Araújo, J.E.; Santos, T.; Jorge, S.; Pereira, T.M.; Reboiro-Jato, M.; Pavón, R.; Magriço, R.; Teixeira-Costa, F.; Ramos, A.; Santos, H.M. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry-Based Profiling as a Step Forward in the Characterization of Peritoneal Dialysis Effluent. Anal. Methods 2015, 7, 7467–7473. [Google Scholar] [CrossRef]
- Araújo, J.E.; Jorge, S.; Magriço, R.; Costa, T.E.; Ramos, A.; Reboiro-Jato, M.; Riverola, F.; Lodeiro, C.; Capelo, J.L.; Santos, H.M. Classifying Patients in Peritoneal Dialysis by Mass Spectrometry-Based Profiling. Talanta 2016, 152, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Fatou, B.; Saudemont, P.; Leblanc, E.; Vinatier, D.; Mesdag, V.; Wisztorski, M.; Focsa, C.; Salzet, M.; Ziskind, M.; Fournier, I. In Vivo Real-Time Mass Spectrometry for Guided Surgery Application. Sci. Rep. 2016, 6, 25919. [Google Scholar] [CrossRef]
- Araújo, J.E.; López-Fernández, H.; Diniz, M.S.; Baltazar, P.M.; Pinheiro, L.C.; da Silva, F.C.; Carrascal, M.; Videira, P.; Santos, H.M.; Capelo, J.L. Dithiothreitol-Based Protein Equalization Technology to Unravel Biomarkers for Bladder Cancer. Talanta 2018, 180, 36–46. [Google Scholar] [CrossRef]
- Torres, I.; Giménez, E.; Vinuesa, V.; Pascual, T.; Moya, J.M.; Alberola, J.; Martínez-Sapiña, A.; Navarro, D. Matrix-Assisted Laser Desorption/Ionization Time-of-Flight Mass Spectrometry (MALDI-TOF-MS) Proteomic Profiling of Cerebrospinal Fluid in the Diagnosis of Enteroviral Meningitis: A Proof-of-Principle Study. Eur. J. Clin. Microbiol. Infect. Dis. 2018, 37, 2331–2339. [Google Scholar] [CrossRef]
- Thompson, J.; Nunn, S.L.E.; Sarkar, S.; Clayton, B. Diagnostic Screening of Bovine Mastitis Using MALDI-TOF MS Direct-Spotting of Milk and Machine Learning. Vet. Sci. 2023, 10, 101. [Google Scholar] [CrossRef]
- Feucherolles, M.; Nennig, M.; Becker, S.L.; Martiny, D.; Losch, S.; Penny, C.; Cauchie, H.M.; Ragimbeau, C. Combination of MALDI-TOF Mass Spectrometry and Machine Learning for Rapid Antimicrobial Resistance Screening: The Case of Campylobacter Spp. Front. Microbiol. 2022, 12, 804484. [Google Scholar] [CrossRef] [PubMed]
- Tran, N.K.; Howard, T.; Walsh, R.; Pepper, J.; Loegering, J.; Phinney, B.; Salemi, M.R.; Rashidi, H.H. Novel Application of Automated Machine Learning with MALDI-TOF-MS for Rapid High-Throughput Screening of COVID-19: A Proof of Concept. Sci. Rep. 2021, 11, 8219. [Google Scholar] [CrossRef] [PubMed]
- Weis, C.V.; Jutzeler, C.R.; Borgwardt, K. Machine Learning for Microbial Identification and Antimicrobial Susceptibility Testing on MALDI-TOF Mass Spectra: A Systematic Review. Clin. Microbiol. Infect. 2020, 26, 1310–1317. [Google Scholar] [CrossRef]
- Lazari, L.C.; Rosa-Fernandes, L.; Palmisano, G. Machine Learning Approaches to Analyze MALDI-TOF Mass Spectrometry Protein Profiles. Methods Mol. Biol. 2022, 2511, 375–394. [Google Scholar] [CrossRef] [PubMed]
- Courtiol, P.; Maussion, C.; Moarii, M.; Pronier, E.; Pilcer, S.; Sefta, M.; Manceron, P.; Toldo, S.; Zaslavskiy, M.; Le Stang, N.; et al. Deep Learning-Based Classification of Mesothelioma Improves Prediction of Patient Outcome. Nat. Med. 2019, 25, 1519–1525. [Google Scholar] [CrossRef]
- Kleppe, A.; Skrede, O.J.; De Raedt, S.; Liestøl, K.; Kerr, D.J.; Danielsen, H.E. Designing Deep Learning Studies in Cancer Diagnostics. Nat. Rev. Cancer 2021, 21, 199–211. [Google Scholar] [CrossRef]
- Ludwig, J.A.; Weinstein, J.N. Biomarkers in Cancer Staging, Prognosis and Treatment Selection. Nat. Rev. Cancer 2005, 5, 845–856. [Google Scholar] [CrossRef] [PubMed]
- Kather, J.N.; Calderaro, J. Development of AI-Based Pathology Biomarkers in Gastrointestinal and Liver Cancer. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 591–592. [Google Scholar] [CrossRef]
- Liang, J.; Zhang, W.; Yang, J.; Wu, M.; Dai, Q.; Yin, H.; Xiao, Y.; Kong, L. Deep Learning Supported Discovery of Biomarkers for Clinical Prognosis of Liver Cancer. Nat. Mach. Intell. 2023, 5, 408–420. [Google Scholar] [CrossRef]
- Zhang, X.; Jonassen, I.; Goksøyr, A. Machine Learning Approaches for Biomarker Discovery Using Gene Expression Data. In Bioinformatics; Exon: Brisbane, Australia, 2021; pp. 53–64. [Google Scholar] [CrossRef]
- Gonçalves, J.P.L.; Bollwein, C.; Schwamborn, K. Mass Spectrometry Imaging Spatial Tissue Analysis toward Personalized Medicine. Life 2022, 12, 1037. [Google Scholar] [CrossRef]
- Rapid Trypsin Digestion of Complex Protein Mixtures for Proteomics Analysis. Available online: https://www.sigmaaldrich.com/US/en/technical-documents/technical-article/protein-biology/protein-mass-spectrometry/rapid-trypsin-digestion (accessed on 14 September 2023).
- Thermo Fisher Scientific-US. Protein Digestion for Mass Spectrometry. Available online: https://www.thermofisher.com/us/en/home/life-science/protein-biology/protein-mass-spectrometry-analysis/sample-prep-mass-spectrometry/protein-digestion-mass-spectrometry.html (accessed on 14 September 2023).
- Huynh, M.L.; Russell, P.; Walsh, B. Tryptic digestion of in-gel proteins for mass spectrometry analysis. Methods Mol Biol. 2009, 519, 507–513. [Google Scholar] [CrossRef] [PubMed]
- Merefith, O.B.; Wren, J.J. Determination of Molecular-Weight Distribution in Wheat-Flour Proteins by Extraction and Gel Filtration in a Dissociating Medium. Cereal Chem. 1966, 43, 169–186. Available online: https://www.cerealsgrains.org/publications/cc/backissues/1966/Documents/CC1966a16.html (accessed on 29 July 2023).
- Perdomo, A.; Webb, H.E.; Bugarel, M.; Friedman, C.R.; Francois Watkins, L.K.; Loneragan, G.H.; Calle, A. First Known Report of Mcr-Harboring Enterobacteriaceae in the Dominican Republic. Int. J. Environ. Res. Public Health 2023, 20, 5123. [Google Scholar] [CrossRef] [PubMed]
- Josefsen, M.H.; Andersen, S.C.; Christensen, J.; Hoorfar, J. Microbial Food Safety: Potential of DNA Extraction Methods for Use in Diagnostic Metagenomics. J. Microbiol. Methods. 2015, 114, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Tolosi, R.; Apostolakos, I.; Laconi, A.; Carraro, L.; Grilli, G.; Cagnardi, P.; Piccirillo, A. Rapid Detection and Quantification of Plasmid-mediated Colistin Resistance Genes (Mcr-1 to Mcr-5) by Real-time PCR in Bacterial and Environmental Samples. J. Appl. Microbiol. 2020, 129, 1523–1529. [Google Scholar] [CrossRef]
- Calle, A.; Fernandez, M.; Montoya, B.; Schmidt, M.; Thompson, J. Uv-c led irradiation reduces salmonella on chicken and food contact surfaces. Foods 2021, 10, 1459. [Google Scholar] [CrossRef]
- Jechorek, R.P.; Johnston, R.L. Evaluation of the VIDAS Staph Enterotoxin II (SET 2) Immunoassay Method for the Detection of Staphylococcal Enterotoxins in Selected Foods: Collaborative study. J. AOAC Int. 2008, 91, 164–173. Available online: https://pubmed.ncbi.nlm.nih.gov/18376599/ (accessed on 31 July 2023). [CrossRef]
- UniProt. proteome:UP000248731 in UniProtKB Search (5445). Available online: https://www.uniprot.org/uniprotkb?dir=descend&query=proteome%3AUP000248731&sort=length (accessed on 28 August 2023).
- Protein Digest Protocols. Protease Digestion for Mass Spectrometry. Available online: https://www.promega.com/resources/guides/protein-analysis/protease-digestion-for-mass-spec/ (accessed on 28 August 2023).
- Ortega, E.; Abriouel, H.; Lucas, R.; Gálvez, A. Multiple Roles of Staphylococcus Aureus Enterotoxins: Pathogenicity, Superantigenic Activity, and Correlation to Antibiotic Resistance. Toxins 2010, 2, 2117. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Case Study | Trial | Sens (Avg.) | Spc (Avg.) | Javg | Jmax |
|---|---|---|---|---|---|
| Case Study 1 | Intact binary | 1 | 0.90 | 0.90 | 1 |
| Intact w/signals | 1 | 0.75 | 0.75 | 0.94 | |
| Digest binary Digest w/signals | 0.98 0.99 | 0.86 0.61 | 0.84 0.60 | 0.86 0.90 | |
| Case Study 2 | Intact binary | 0.97 | 0.99 | 0.96 | 1 |
| Intact w/signals | 1 | 0.88 | 0.88 | 0.94 | |
| Digest binary | 0.99 | 0.81 | 0.81 | 0.92 | |
| Digest w/signals | 0.98 | 0.86 | 0.84 | 0.90 | |
| Case Study 3 | Intact binary | 1 | 0 | 0 | 0 |
| Intact w/signals | 1 | 0 | 0 | 0 | |
| Digest binary | 0.70 | 0.82 | 0.51 | 0.78 | |
| Digest w/signals | 0.91 | 0.51 | 0.42 | 1 | |
| Case Study 4 | Intact binary | 0.33 | 0.07 | −0.60 | 0 |
| Intact w/signals | 0.1 | 0.1 | −0.80 | −0.4 | |
| Digest binary | 0.47 | 0.82 | 0.29 | 0.51 | |
| Digest w/signals | 0.44 | 0.90 | 0.34 | 0.44 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sarkar, S.; Squire, A.; Diab, H.; Rahman, M.K.; Perdomo, A.; Awosile, B.; Calle, A.; Thompson, J. Effect of Tryptic Digestion on Sensitivity and Specificity in MALDI-TOF-Based Molecular Diagnostics through Machine Learning. Sensors 2023, 23, 8042. https://doi.org/10.3390/s23198042
Sarkar S, Squire A, Diab H, Rahman MK, Perdomo A, Awosile B, Calle A, Thompson J. Effect of Tryptic Digestion on Sensitivity and Specificity in MALDI-TOF-Based Molecular Diagnostics through Machine Learning. Sensors. 2023; 23(19):8042. https://doi.org/10.3390/s23198042
Chicago/Turabian StyleSarkar, Sumon, Abigail Squire, Hanin Diab, Md. Kaisar Rahman, Angela Perdomo, Babafela Awosile, Alexandra Calle, and Jonathan Thompson. 2023. "Effect of Tryptic Digestion on Sensitivity and Specificity in MALDI-TOF-Based Molecular Diagnostics through Machine Learning" Sensors 23, no. 19: 8042. https://doi.org/10.3390/s23198042
APA StyleSarkar, S., Squire, A., Diab, H., Rahman, M. K., Perdomo, A., Awosile, B., Calle, A., & Thompson, J. (2023). Effect of Tryptic Digestion on Sensitivity and Specificity in MALDI-TOF-Based Molecular Diagnostics through Machine Learning. Sensors, 23(19), 8042. https://doi.org/10.3390/s23198042