
Dielectric Spectroscopy Based Detection of Specific and Nonspecific Cellular Mechanisms


Abstract
1. Introduction
2. Materials and Methods
2.1. Yeast Sample Preparation
2.2. Measurement Techniques
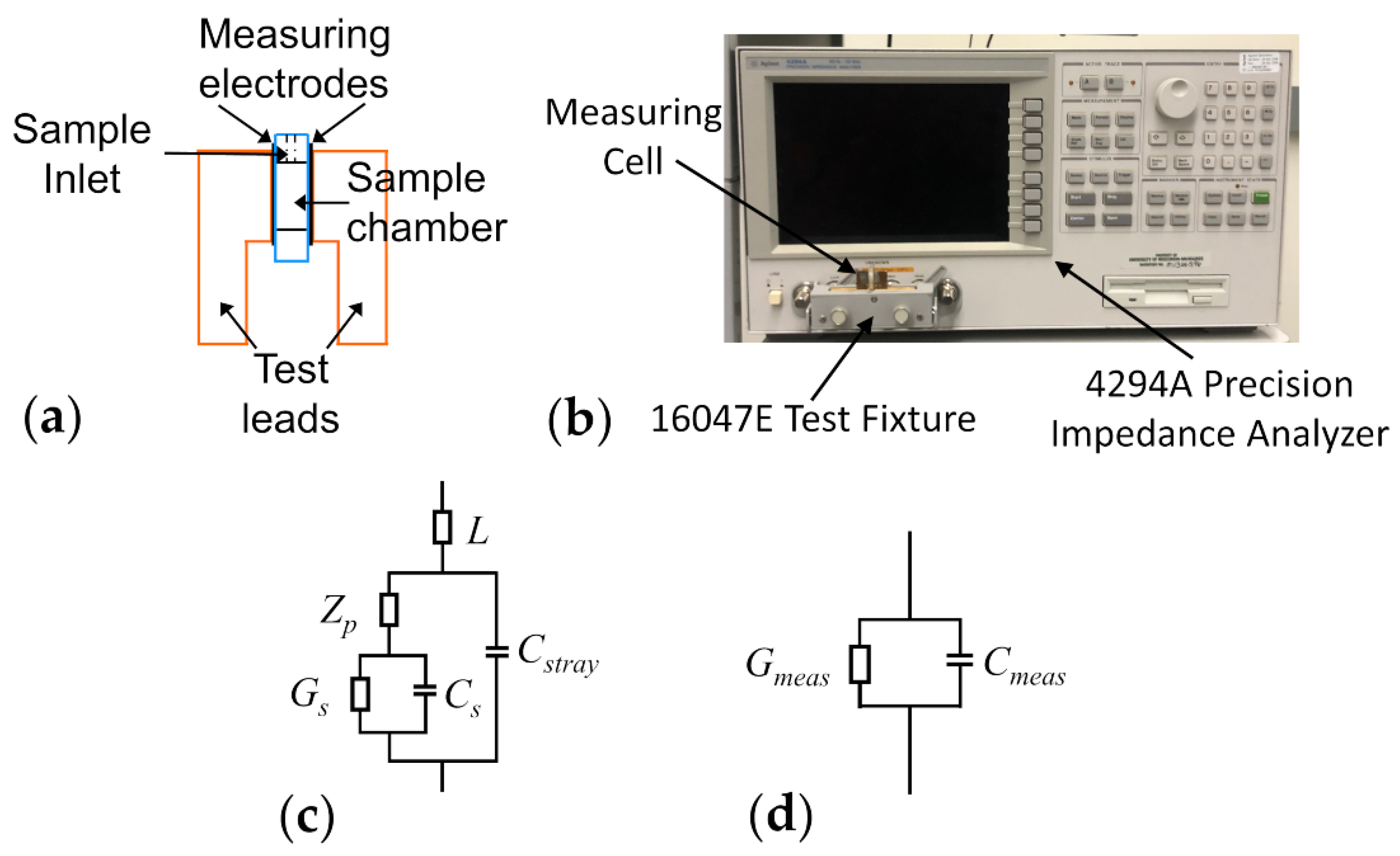
2.2.1. Dielectric Measurements
2.2.2. Fluorescence Microspectroscopy
2.2.3. Differential Interference Contrast Measurements
2.3. Simulation of the Measured Dielectric Data
2.3.1. Electrical Model of Yeast Cells
2.3.2. Correction for Electrode Polarization
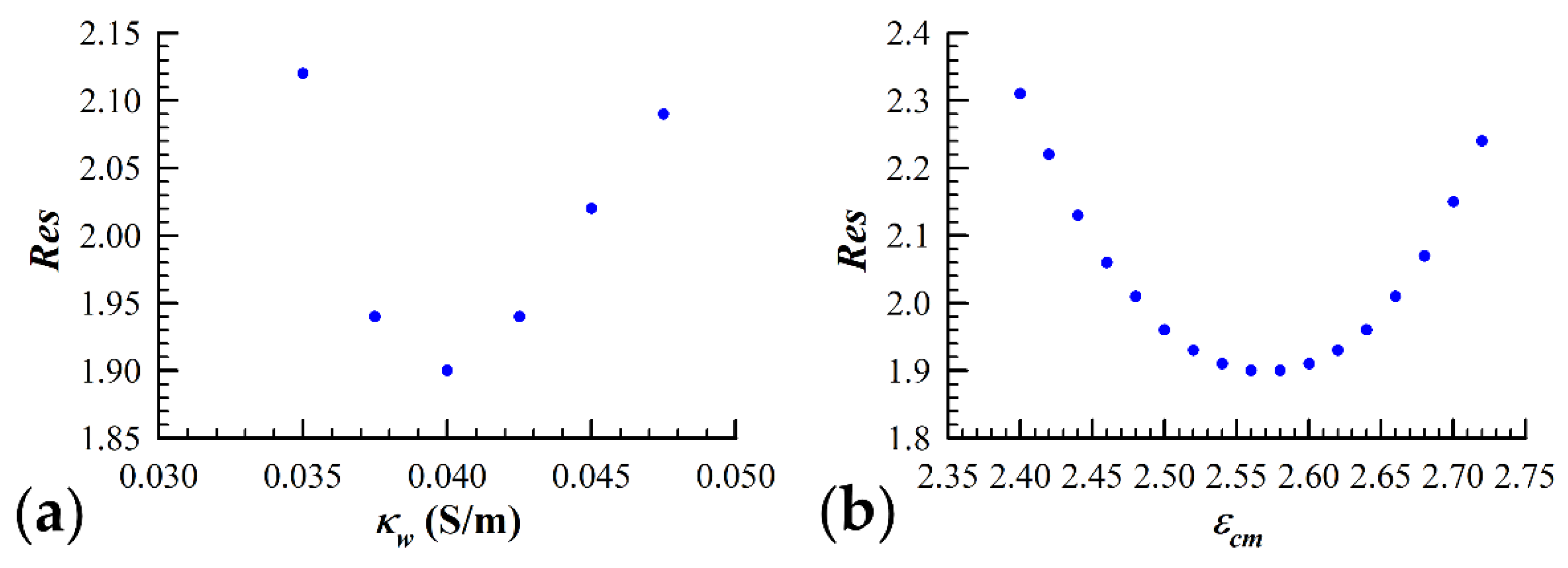
2.3.3. Dielectric Data Fitting Algorithms and Procedures
3. Results and Discussions
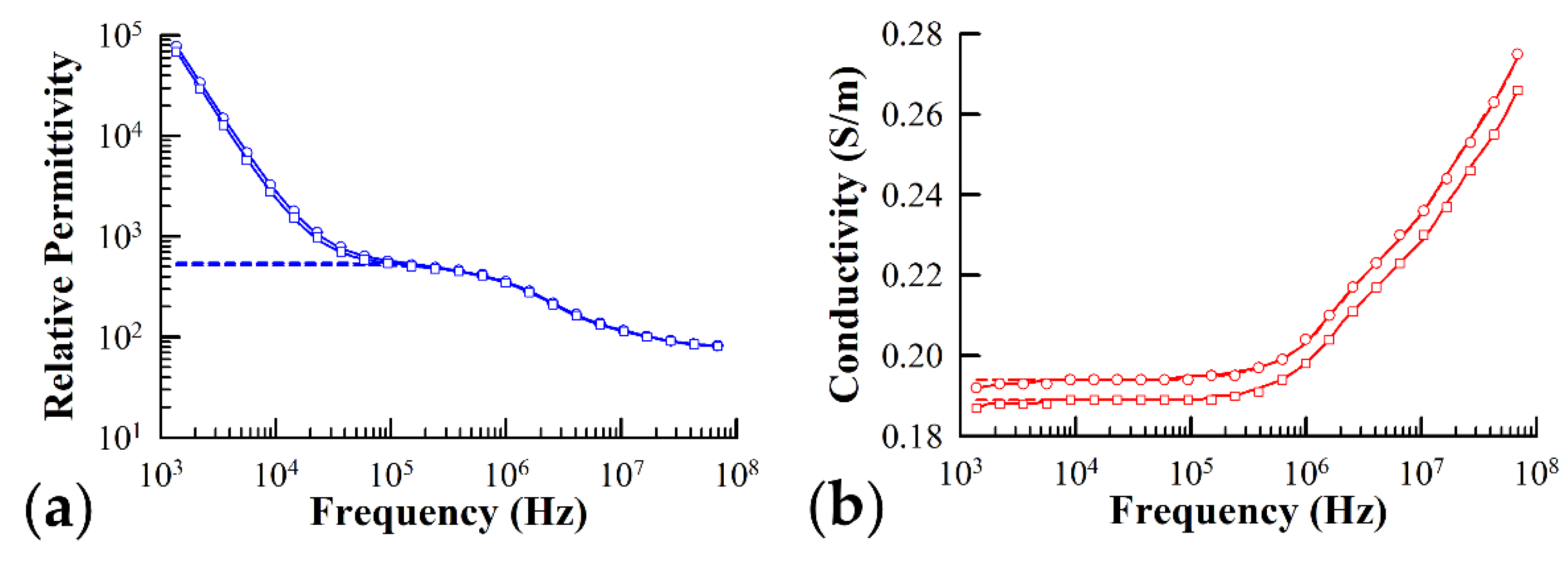
3.1. Dielectric Measurements of Ste2-Expressing Yeast Cells Exposed to α-Factor
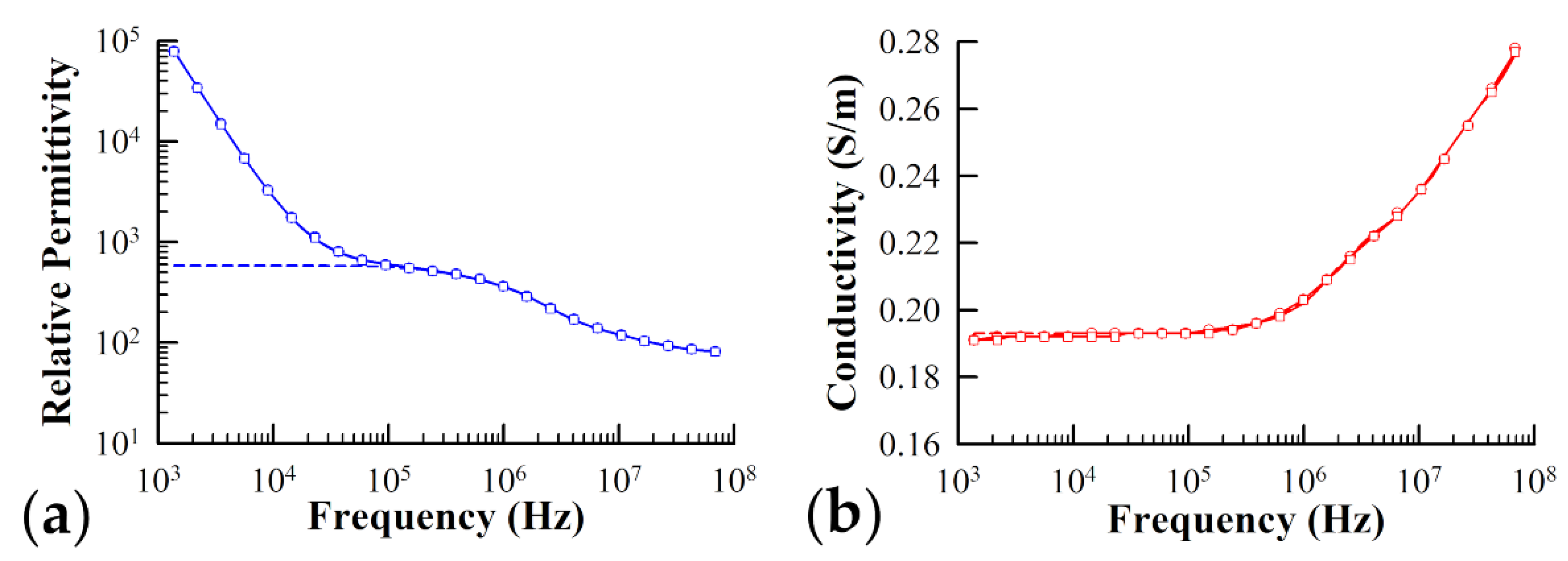
3.2. Dielectric Measurements of Yeast Cells Lacking Ste2 Receptors
3.3. Testing the Effect of Osmotic Pressure Variation on the Cytoplasm Dielectric Properties
3.4. Determining the Extent of Nonspecific Internalization Using the Fluorescent Molecule D-Luciferin
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fredriksson, R.; Schioth, H.B. The repertoire of G-protein-coupled receptors in fully sequenced genomes. Mol. Pharm. 2005, 67, 1414–1425. [Google Scholar] [CrossRef] [PubMed]
- Pierce, K.L.; Premont, R.T.; Lefkowitz, R.J. Seven-transmembrane receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Takeda, S.; Kadowaki, S.; Haga, T.; Takaesu, H.; Mitaku, S. Identification of G protein-coupled receptor genes from the human genome sequence. FEBS Lett. 2002, 520, 97–101. [Google Scholar] [CrossRef]
- Zhang, R.; Xie, X. Tools for GPCR drug discovery. Acta Pharmacol. Sin. 2012, 33, 372–384. [Google Scholar] [CrossRef] [PubMed]
- Eglen, R.M. Functional G protein-coupled receptor assays for primary and secondary screening. Comb. Chem. High. T Scr. 2005, 8, 311–318. [Google Scholar] [CrossRef]
- Ward, R.J.; Milligan, G. Structural and biophysical characterisation of G protein-coupled receptor ligand binding using resonance energy transfer and fluorescent labelling techniques. Bba-Biomembranes 2014, 1838, 3–14. [Google Scholar] [CrossRef]
- Sridharan, R.; Zuber, J.; Connelly, S.M.; Mathew, E.; Dumont, M.E. Fluorescent approaches for understanding interactions of ligands with G protein coupled receptors. Bba-Biomembranes 2014, 1838, 15–33. [Google Scholar] [CrossRef]
- Stoddart, L.A.; White, C.W.; Nguyen, K.; Hill, S.J.; Pfleger, K.D.G. Fluorescence- and bioluminescence-based approaches to study GPCR ligand binding. Brit. J. Pharmacol. 2016, 173, 3028–3037. [Google Scholar] [CrossRef]
- Middleton, R.J.; Kellam, B. Fluorophore-tagged GPCR ligands. Curr. Opin. Chem. Biol. 2005, 9, 517–525. [Google Scholar] [CrossRef] [PubMed]
- Kuder, K.; Kiec-Kononowicz, K. Fluorescent GPCR Ligands as New Tools in Pharmacology. Curr. Med. Chem. 2008, 15, 2132–2143. [Google Scholar] [CrossRef]
- Kuder, K.J.; Kiec-Kononowicz, K. Fluorescent GPCR Ligands as New Tools in Pharmacology-Update, Years 2008-Early 2014. Curr. Med. Chem. 2014, 21, 3962–3975. [Google Scholar] [CrossRef] [PubMed]
- Ilien, B.; Franchet, C.; Bernard, P.; Morisset, S.; Weill, C.O.; Bourguignon, J.J.; Hibert, M.; Galzi, J.L. Fluorescence resonance energy transfer to probe human M1 muscarinic receptor structure and drug binding properties. J. Neurochem. 2003, 85, 768–778. [Google Scholar] [CrossRef]
- Ding, F.X.; Lee, B.K.; Hauser, M.; Patri, R.; Arshava, B.; Becker, J.M.; Naider, F. Study of the binding environment of alpha-factor in its G protein-coupled receptor using fluorescence spectroscopy. J. Pept. Res. 2002, 60, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Raicu, V.; Popescu, A. Integrated Molecular and Cellular Biophysics; Springer: London, UK, 2008. [Google Scholar]
- Selvin, P.R. The renaissance of fluorescence resonance energy transfer. Nat. Struct. Mol. Biol. 2000, 7, 730–734. [Google Scholar] [CrossRef]
- Piston, D.W.; Kremers, G.J. Fluorescent protein FRET: The good, the bad and the ugly. Trends Biochem. Sci. 2007, 32, 407–414. [Google Scholar] [CrossRef] [PubMed]
- Angers, S.; Salahpour, A.; Joly, E.; Hilairet, S.; Chelsky, D.; Dennis, M.; Bouvier, M. Detection of beta 2-adrenergic receptor dimerization in living cells using bioluminescence resonance energy transfer (BRET). Proc. Natl. Acad. Sci. USA 2000, 97, 3684–3689. [Google Scholar] [CrossRef] [PubMed]
- Cornea, A.; Janovick, J.A.; Maya-Nunez, G.; Conn, P.M. Gonadotropin-releasing hormone receptor microaggregation. Rate monitored by fluorescence resonance energy transfer. J. Biol. Chem. 2001, 276, 2153–2158. [Google Scholar] [CrossRef]
- Kroeger, K.M.; Hanyaloglu, A.C.; Seeber, R.M.; Miles, L.E.; Eidne, K.A. Constitutive and agonist-dependent homo-oligomerization of the thyrotropin-releasing hormone receptor. Detection in living cells using bioluminescence resonance energy transfer. J. Biol. Chem. 2001, 276, 12736–12743. [Google Scholar] [CrossRef] [PubMed]
- Paprocki, J.D.; Biener, G.; Stoneman, M.R.; Raicu, V. In-Cell Detection of Conformational Substates of a G Protein-Coupled Receptor Quaternary Structure: Modulation of Substate Probability by Cognate Ligand Binding. J. Phys. Chem. B 2020, 124, 10062–10076. [Google Scholar] [CrossRef]
- Granier, S.; Kim, S.; Shafer, A.M.; Ratnala, V.R.P.; Fung, J.J.; Zare, R.N.; Kobilka, B. Structure and conformational changes in the C-terminal domain of the beta(2)-adrenoceptor—Insights from fluorescence resonance energy transfer studies. J. Biol. Chem. 2007, 282, 13895–13905. [Google Scholar] [CrossRef] [PubMed]
- Matsushita, S.; Nakata, H.; Kubo, Y.; Tateyama, M. Ligand-induced Rearrangements of the GABA(B) Receptor Revealed by Fluorescence Resonance Energy Transfer. J. Biol. Chem. 2010, 285, 10291–10299. [Google Scholar] [CrossRef] [PubMed]
- Overton, M.C.; Blumer, K.J. G-protein-coupled receptors function as oligomers in vivo. Curr. Biol. 2000, 10, 341–344. [Google Scholar] [CrossRef]
- Milligan, G. Applications of bioluminescence- and fluorescence resonance energy transfer to drug discovery at G protein-coupled receptors. Eur. J. Pharm. Sci. 2004, 21, 397–405. [Google Scholar] [CrossRef] [PubMed]
- Milligan, G. G protein-coupled receptor dimerization: Function and ligand pharmacology. Mol. Pharmacol. 2004, 66, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Mercier, J.F.; Salahpour, A.; Angers, S.; Breit, A.; Bouvier, M. Quantitative assessment of beta 1- and beta 2-adrenergic receptor homo- and heterodimerization by bioluminescence resonance energy transfer. J. Biol. Chem. 2002, 277, 44925–44931. [Google Scholar] [CrossRef] [PubMed]
- McVey, M.; Ramsay, D.; Kellett, E.; Rees, S.; Wilson, S.; Pope, A.J.; Milligan, G. Monitoring receptor oligomerization using time-resolved fluorescence resonance energy transfer and bioluminescence resonance energy transfer. The human delta -opioid receptor displays constitutive oligomerization at the cell surface, which is not regulated by receptor occupancy. J. Biol. Chem. 2001, 276, 14092–14099. [Google Scholar] [PubMed]
- Ayoub, M.A.; Couturier, C.; Lucas-Meunier, E.; Angers, S.; Fossier, P.; Bouvier, M.; Jockers, R. Monitoring of ligand-independent dimerization and ligand-induced conformational changes of melatonin receptors in living cells by bioluminescence resonance energy transfer. J. Biol. Chem. 2002, 277, 21522–21528. [Google Scholar] [CrossRef]
- Foster, K.R.; Schwan, H.P. Dielectric Properties of Tissues. In Handbook of Biological Effects of Electromagnetic Fields, 2nd ed.; Polk, C., Postow, E., Eds.; CRC Press: Boca Raton, FL, USA, 1996; pp. 25–102. [Google Scholar]
- Raicu, V.; Feldman, Y. Dielectric Relaxation in Biological Systems Physical Principles, Methods, and Applications Preface; Oxford University Press: Oxford, UK, 2015. [Google Scholar] [CrossRef]
- Asami, K. Characterization of biological cells by dielectric spectroscopy. J. Non-Cryst. Solids 2002, 305, 268–277. [Google Scholar] [CrossRef]
- Gabriel, C. Dielectric properties of biological materials. In Bioengineering and Biophysical Aspects of Electromagnetic Fields, 3rd ed.; Barnes, F.S., Greenbaum, B., Eds.; Handbook of Biological Effects of Electromagnetic Fields; CRC Press: Boca Raton, FL, USA, 2007; pp. 51–100. [Google Scholar]
- Schwan, H.P. Electrical properties of tissue and cell suspensions. In Advances in Biological and Medical Physics; Lawrence, J.H., Tobias, C.A., Eds.; Academic Press: New York, NY, USA, 1957; Volume 5, pp. 147–209. [Google Scholar]
- Sabuncu, A.C.; Zhuang, J.; Kolb, J.F.; Beskok, A. Microfluidic impedance spectroscopy as a tool for quantitative biology and biotechnology. Biomicrofluidics 2012, 6. [Google Scholar] [CrossRef]
- Asami, K.; Hanai, T. Dielectric Monitoring of Biological Cell Sedimentation. Colloid Polym. Sci. 1992, 270, 78–84. [Google Scholar] [CrossRef]
- Irimajiri, A.; Ando, M.; Matsuoka, R.; Ichinowatari, T.; Takeuchi, S. Dielectric monitoring of rouleaux formation in human whole blood: A feasibility study. Biochim. Biophys. Acta 1996, 1290, 207–209. [Google Scholar] [CrossRef]
- Asami, K.; Gheorghiu, E.; Yonezawa, T. Real-time monitoring of yeast cell division by dielectric spectroscopy. Biophys. J. 1999, 76, 3345–3348. [Google Scholar] [CrossRef]
- Ron, A.; Shur, I.; Daniel, R.; Singh, R.R.; Fishelson, N.; Croitoru, N.; Benayahu, D.; Shacham-Diamand, Y. Dielectric screening of early differentiation patterns in mesenchymal stem cells induced by steroid hormones. Bioelectrochemistry 2010, 78, 161–172. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, K.; Raicu, V. Investigation of dielectric relaxation in systems with hierarchical organization: From time to frequency domain and back again. Phys. Lett. A 2017, 381, 1981–1988. [Google Scholar] [CrossRef]
- Badu, D.N.; Yokoi, K.; Raicu, V. Relaxation in systems with hierarchical organization: Analytical derivation of the relaxation and dispersion functions. Phys. Lett. A 2019, 383, 1063–1070. [Google Scholar] [CrossRef]
- Javed, A.; Arif, A.; Zubair, M.; Mehmood, M.Q.; Riaz, K. A Low-Cost Multiple Complementary Split-Ring Resonator-Based Microwave Sensor for Contactless Dielectric Characterization of Liquids. IEEE Sens. J. 2020, 20, 11326–11334. [Google Scholar] [CrossRef]
- Chuma, E.L.; Iano, Y.; Fontgalland, G.; Roger, L.L.B.; Loschi, H. PCB-integrated non-destructive microwave sensor for liquid dielectric spectroscopy based on planar metamaterial resonator. Sens. Actuat A-Phys. 2020, 312. [Google Scholar] [CrossRef]
- Maxwell, J.C. A Treatise on Electricity and Magnetism; Clarendon Press: Oxford, UK, 1891; Volume 1. [Google Scholar]
- Wagner, K.W. Erklärung der dielektrischen nachwirkungsvorgänge auf grund maxwellscher vorstellungen. Arch. Für Elektrotechnik 1914, 2, 371–387. [Google Scholar] [CrossRef]
- Fricke, H. The Electric Capacity of Suspensions with Special Reference to Blood. J. Gen. Physiol. 1925, 9, 137–152. [Google Scholar] [CrossRef]
- Bai, W.; Zhao, K.S.; Asami, K. Effects of copper on dielectric properties of E. coli cells. Colloids Surf. B-Biointerfaces 2007, 58, 105–115. [Google Scholar] [CrossRef]
- Raicu, V.; Gusbeth, C.; Anghel, D.F.; Turcu, G. Effects of cetyltrimethylammonium bromide (CTAB) surfactant upon the dielectric properties of yeast cells. Biochim. Et Biophys. Acta-Gen. Subj. 1998, 1379, 7–15. [Google Scholar] [CrossRef]
- Gimsa, J.; Schnelle, T.; Zechel, G.; Glaser, R. Dielectric spectroscopy of human erythrocytes: Investigations under the influence of nystatin. Biophys. J. 1994, 66, 1244–1253. [Google Scholar] [CrossRef][Green Version]
- Raicu, V.; Raicu, G.; Turcu, G. Dielectric properties of yeast cells as simulated by the two-shell model. Biochim. Et Biophys. Acta-Bioenerg. 1996, 1274, 143–148. [Google Scholar] [CrossRef]
- Asami, K.; Yonezawa, T. Dielectric behavior of wild-type yeast and vacuole-deficient mutant over a frequency range of 10 kHz to 10 GHz. Biophys. J. 1996, 71, 2192–2200. [Google Scholar] [CrossRef]
- Van Laarhoven, P.J.M.; Arts, E.H.L. Simulated Annealing: Theory and Applications; Kluwer Academic Publishers: New York, NY, USA, 1987. [Google Scholar]
- Overton, M.C.; Chinault, S.L.; Blumer, K.J. Oligomerization of G-protein-coupled receptors: Lessons from the yeast Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 1963–1970. [Google Scholar] [CrossRef]
- Stoneman, M.R.; Paprocki, J.D.; Biener, G.; Yokoi, K.; Shevade, A.; Kuchin, S.; Raicu, V. Quaternary structure of the yeast pheromone receptor Ste2 in living cells. Bba-Biomembranes 2017, 1859, 1456–1464. [Google Scholar] [CrossRef] [PubMed]
- Raicu, V.; Jansma, D.B.; Miller, R.J.; Friesen, J.D. Protein interaction quantified in vivo by spectrally resolved fluorescence resonance energy transfer. Biochem. J. 2005, 385, 265–277. [Google Scholar] [CrossRef]
- Zimmermann, T.; Rietdorf, J.; Girod, A.; Georget, V.; Pepperkok, R. Spectral imaging and linear un-mixing enables improved FRET efficiency with a novel GFP2-YFP FRET pair. FEBS Lett. 2002, 531, 245–249. [Google Scholar] [CrossRef]
- Shaner, N.C.; Steinbach, P.A.; Tsien, R.Y. A guide to choosing fluorescent proteins. Nat. Methods 2005, 2, 905–909. [Google Scholar] [CrossRef]
- Raicu, V.; Stoneman, M.R.; Fung, F.; Melnichuk, M.; Jansma, D.B.; Rath, S.; Pisterzi, L.F.; Fox, M.P.; Wells, J.W.; Saldin, D.K. Determination of supramolecular structure and spatial distribution of protein complexes in living cells. Nat. Photonics 2009, 3, 107–113. [Google Scholar] [CrossRef]
- Biener, G.; Stoneman, M.R.; Acbas, G.; Holz, J.D.; Orlova, M.; Komarova, L.; Kuchin, S.; Raicu, V. Development and Experimental Testing of an Optical Micro-Spectroscopic Technique Incorporating True Line-Scan Excitation. Int. J. Mol. Sci. 2014, 15, 261–276. [Google Scholar] [CrossRef] [PubMed]
- Pauly, H.; Schwan, H.P. On the dielectric properties of a suspension of spherical particles covered by a shell. Z. Naturforsch. 1959, 14b, 125–131. [Google Scholar] [CrossRef]
- Irimajiri, A.; Hanai, T.; Inouye, A. A dielectric theory of “multi-stratified shell” model with its application to a lymphoma cell. J. Theor. Biol. 1979, 78, 251–269. [Google Scholar] [CrossRef]
- Bruggeman, D.A.G. Berechnug vershiedener physikalischen Konstanten von heterogenen Substanzen. Ann. Phys. 1935, 416, 636–664. [Google Scholar] [CrossRef]
- Hanai, T. Theory of the dielectric disperison due to the interfacial polarization and its application to emulsion. Kolloid Z. 1960, 171, 23–31. [Google Scholar] [CrossRef]
- Hanai, T. Electrical properties of emulsions. In Emulsion Science; Sherman, P., Ed.; Academic Press: New York, NY, USA, 1968; pp. 353–478. [Google Scholar]
- Raicu, V.; Saibara, T.; Enzan, H.; Irimajiri, A. Dielectric properties of rat liver in vivo: Analysis by modeling hepatocytes in the tissue architecture. Bioelectrochem. Bioenerg. 1998, 47, 333–342. [Google Scholar] [CrossRef]
- Schwan, H.P. Determination of biological impedances. In Physical Techniques in Biological Research; Nastuk, W.L., Ed.; Academic Press: New York, NY, USA, 1963; Volume 6, pp. 323–407. [Google Scholar]
- Feldman, Y.; Nigmatullin, R.; Polygalov, E.; Texter, J. Fractal-polarization correction in time domain dielectric spectroscopy. Phys. Rev. E 1998, 58, 7561–7565. [Google Scholar] [CrossRef]
- Bordi, F.; Cametti, C.; Gili, T. Reduction of the contribution of electrode polarization effects in the radiowave dielectric measurements of highly conductive biological cell suspensions. Bioelectrochemistry 2001, 54, 53–61. [Google Scholar] [CrossRef]
- Raicu, V.; Saibara, T.; Irimajiri, A. Dielectric properties of rat liver in vivo: A noninvasive approach using an open-ended coaxial probe at audio/radio frequencies. Bioelectrochem. Bioenerg. 1998, 47, 325–332. [Google Scholar] [CrossRef]
- Bao, J.Z.; Davis, C.C.; Schmukler, R.E. Frequency domain impedance measurements of erythrocytes. Constant phase angle impedance characteristics and a phase transition. Biophys J. 1992, 61, 1427–1434. [Google Scholar] [CrossRef]
- Stoneman, M.R.; Kosempa, M.; Gregory, W.D.; Gregory, C.W.; Marx, J.J.; Mikkelson, W.; Tjoe, J.; Raicu, V. Correction of electrode polarization contributions to the dielectric properties of normal and cancerous breast tissues at audio/radiofrequencies. Phys. Med. Biol. 2007, 52, 6589–6604. [Google Scholar] [CrossRef]
- Kaatze, U. Complex Permittivity of Water as a Function of Frequency and Temperature. J. Chem. Eng. Data 1989, 34, 371–374. [Google Scholar] [CrossRef]
- Kirkpatrick, S.; Gelatt, C.D.J.; Vecchi, M.P. Optimization by Simulated Annealing. Science 1983, 220, 671–680. [Google Scholar] [CrossRef] [PubMed]
- Vitols, E.; North, R.J.; Linnane, A.W. Studies on the oxidative metabolism of Saccharomyces cerevisiae: I. Observations on the fine structure of the yeast cell. J. Cell Biol. 1961, 9, 689–699. [Google Scholar] [CrossRef] [PubMed]
- Son, C.D.; Sargsyan, H.; Naider, F.; Becker, J.M. Identification of ligand binding regions of the Saccharomyces cerevisiae alpha-factor pheromone receptor by photoaffinity cross-linking. Biochemistry 2004, 43, 13193–13203. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.J.I.; Harmon, S.K.; O’Malley, K.L. Intracellular GPCRs Play Key Roles in Synaptic Plasticity. Acs Chem. Neurosci. 2018, 9, 2162–2172. [Google Scholar] [CrossRef] [PubMed]
- Vilardaga, J.P.; Jean-Alphonse, F.G.; Gardella, T.J. Endosomal generation of cAMP in GPCR signaling. Nat. Chem. Biol. 2014, 10, 700–706. [Google Scholar] [CrossRef] [PubMed]
- O’Malley, K.L.; Jong, Y.J.I.; Gonchar, Y.; Burkhalter, A.; Romano, C. Activation of metabotropic glutamate receptor mGlu5 on nuclear membranes mediates intranuclear Ca2+ changes in heterologous cell types and neurons. J. Biol. Chem. 2003, 278, 28210–28219. [Google Scholar] [CrossRef] [PubMed]
- Jong, Y.J.I.; Harmon, S.K.; O’Malley, K.L. GPCR signalling from within the cell. Brit. J. Pharmacol. 2018, 175, 4026–4035. [Google Scholar] [CrossRef]
- Khelashvili, G.; Mondal, S.; Andersen, O.S.; Weinstein, H. Cholesterol Modulates the Membrane Effects and Spatial Organization of Membrane-Penetrating Ligands for G-Protein Coupled Receptors. J. Phys. Chem. B 2010, 114, 12046–12057. [Google Scholar] [CrossRef] [PubMed]
- Rohrer, J.; Benedetti, H.; Zanolari, B.; Riezman, H. Identification of a novel sequence mediating regulated endocytosis of the G protein-coupled alpha-pheromone receptor in yeast. Mol. Biol. Cell 1993, 4, 511–521. [Google Scholar] [CrossRef] [PubMed]
- Yesilaltay, A.; Jenness, D.D. Homo-oligomeric complexes of the yeast alpha-factor pheromone receptor are functional units of endocytosis. Mol. Biol. Cell 2000, 11, 2873–2884. [Google Scholar] [CrossRef] [PubMed]
- Schandel, K.A.; Jenness, D.D. Direct evidence for ligand-induced internalization of the yeast alpha-factor pheromone receptor. Mol. Cell. Biol. 1994, 14, 7245–7255. [Google Scholar] [CrossRef] [PubMed]
- Patowary, S.; Pisterzi, L.F.; Biener, G.; Holz, J.D.; Oliver, J.A.; Wells, J.W.; Raicu, V. Experimental verification of the kinetic theory of FRET using optical microspectroscopy and obligate oligomers. Biophys J. 2015, 108, 1613–1622. [Google Scholar] [CrossRef]
- Nakamura, M.; Yasuda, D.; Hirota, N.; Shimizu, T. Specific Ligands as Pharmacological Chaperones: The Transport of Misfolded G-protein Coupled Receptors to the Cell Surface. Iubmb Life 2010, 62, 453–459. [Google Scholar] [CrossRef]
- Tao, Y.X.; Conn, P.M. Chaperoning G Protein-Coupled Receptors: From Cell Biology to Therapeutics. Endocr. Rev. 2014, 35, 602–647. [Google Scholar] [CrossRef]
- Jean-Alphonse, F.; Perkovska, S.; Frantz, M.C.; Durroux, T.; Mejean, C.; Morin, D.; Loison, S.; Bonnet, D.; Hibert, M.; Mouillac, B.; et al. Biased Agonist Pharmacochaperones of the AVP V2 Receptor May Treat Congenital Nephrogenic Diabetes Insipidus. J. Am. Soc. Nephrol. 2009, 20, 2190–2203. [Google Scholar] [CrossRef]
- Petaja-Repo, U.E.; Hogue, M.; Bhalla, S.; Laperriere, A.; Morello, J.P.; Bouvier, M. Ligands act as pharmacological chaperones and increase the efficiency of delta opioid receptor maturation. EMBO J. 2002, 21, 1628–1637. [Google Scholar] [CrossRef]
- Bossak, K.; Mital, M.; Poznanski, J.; Bonna, A.; Drew, S.; Bal, W. Interactions of alpha-Factor-1, a Yeast Pheromone, and Its Analogue with Copper(II) Ions and Low-Molecular-Weight Ligands Yield Very Stable Complexes. Inorg. Chem. 2016, 55, 7829–7831. [Google Scholar] [CrossRef]
- Stoneman, M.; Chaturvedi, A.; Jansma, D.B.; Kosempa, M.; Zeng, C.; Raicu, V. Protein influence on the plasma membrane dielectric properties: In vivo study utilizing dielectric spectroscopy and fluorescence microscopy. Bioelectrochemistry 2007, 70, 542–550. [Google Scholar] [CrossRef]
- Ashcroft, R.G.; Coster, H.G.; Smith, J.R. The molecular organisation of bimolecular lipid membranes. The dielectric structure of the hydrophilic/hydrophobic interface. Biochim. Biophys. Acta 1981, 643, 191–204. [Google Scholar] [CrossRef]
- Mondal, S.; Khelashvili, G.; Shan, J.F.; Andersen, O.S.; Weinstein, H. Quantitative Modeling of Membrane Deformations by Multihelical Membrane Proteins: Application to G-Protein Coupled Receptors. Biophys. J. 2011, 101, 2092–2101. [Google Scholar] [CrossRef] [PubMed]
- Shan, J.F.; Khelashvili, G.; Mondal, S.; Mehler, E.L.; Weinstein, H. Ligand-Dependent Conformations and Dynamics of the Serotonin 5-HT2A Receptor Determine Its Activation and Membrane-Driven Oligomerization Properties. PLoS Comput. Biol. 2012, 8, e1002473. [Google Scholar] [CrossRef]
- Fallahi-Sichani, M.; Linderman, J.J. Lipid raft-mediated regulation of G-protein coupled receptor signaling by ligands which influence receptor dimerization: A computational study. PLoS ONE 2009, 4, e6604. [Google Scholar] [CrossRef] [PubMed]
- Spencer, D.; Morgan, H. High-Speed Single-Cell Dielectric Spectroscopy. ACS Sens. 2020, 5, 423–430. [Google Scholar] [CrossRef]
- Luongo, K.; Holton, A.; Kaushik, A.; Spence, P.; Ng, B.; Deschenes, R.; Sundaram, S.; Bhansali, S. Microfluidic device for trapping and monitoring three dimensional multicell spheroids using electrical impedance spectroscopy. Biomicrofluidics 2013, 7, 034108. [Google Scholar] [CrossRef]
- Sabuncu, A.C.; Stacey, M.; Craviso, G.L.; Semenova, N.; Vernier, P.T.; Leblanc, N.; Chatterjee, I.; Zaklit, J. Dielectric properties of isolated adrenal chromaffin cells determined by microfluidic impedance spectroscopy. Bioelectrochemistry 2018, 119, 84–91. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| −0.13 ± 2.4 | 0.0 ± 0.0 | 0.06 ± 0.05 | −11.4 ± 9.2 | −0.035 ± 0.027 | |
| p | 0.90 | 1.0 | 0.026 | 0.029 | 0.025 |
| −2.4 ± 3.5 | 0.0 ± 0.0 | 0.00 ± 0.04 | −3.5 ± 2.1 | −0.015 ± 0.006 | |
| p | 0.21 | 1.0 | 0.84 | 0.020 | 0.007 |
| 0.1 ± 3.8 | 1.25 ± 2.5 | 0.00 ± 0.07 | −0.2 ± 14.3 | −0.002 ± 0.04 | |
| p | 0.96 | 0.39 | 1.0 | 0.98 | 0.94 |
| Ste2-Expressing Cells | Wild Type Cells | |||
|---|---|---|---|---|
| R (µm) | Ro (µm) | R (µm) | Ro (µm) | |
| −0.01 ± 0.05 | 0.00 ± 0.08 | −0.05 ± 0.06 | −0.02 ± 0.02 | |
| p | 0.85 | 0.95 | 0.29 | 0.18 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stoneman, M.R.; Raicu, V. Dielectric Spectroscopy Based Detection of Specific and Nonspecific Cellular Mechanisms. Sensors 2021, 21, 3177. https://doi.org/10.3390/s21093177
Stoneman MR, Raicu V. Dielectric Spectroscopy Based Detection of Specific and Nonspecific Cellular Mechanisms. Sensors. 2021; 21(9):3177. https://doi.org/10.3390/s21093177
Chicago/Turabian StyleStoneman, Michael R., and Valerică Raicu. 2021. "Dielectric Spectroscopy Based Detection of Specific and Nonspecific Cellular Mechanisms" Sensors 21, no. 9: 3177. https://doi.org/10.3390/s21093177
APA StyleStoneman, M. R., & Raicu, V. (2021). Dielectric Spectroscopy Based Detection of Specific and Nonspecific Cellular Mechanisms. Sensors, 21(9), 3177. https://doi.org/10.3390/s21093177