
Electrochemical DNA Sensor Based on Acridine Yellow Adsorbed on Glassy Carbon Electrode


Abstract
:1. Introduction
2. Materials and Methods
2.1. Reagents
2.2. Apparatus
2.3. GCE Modification and DNA Sensor Assembling
3. Results
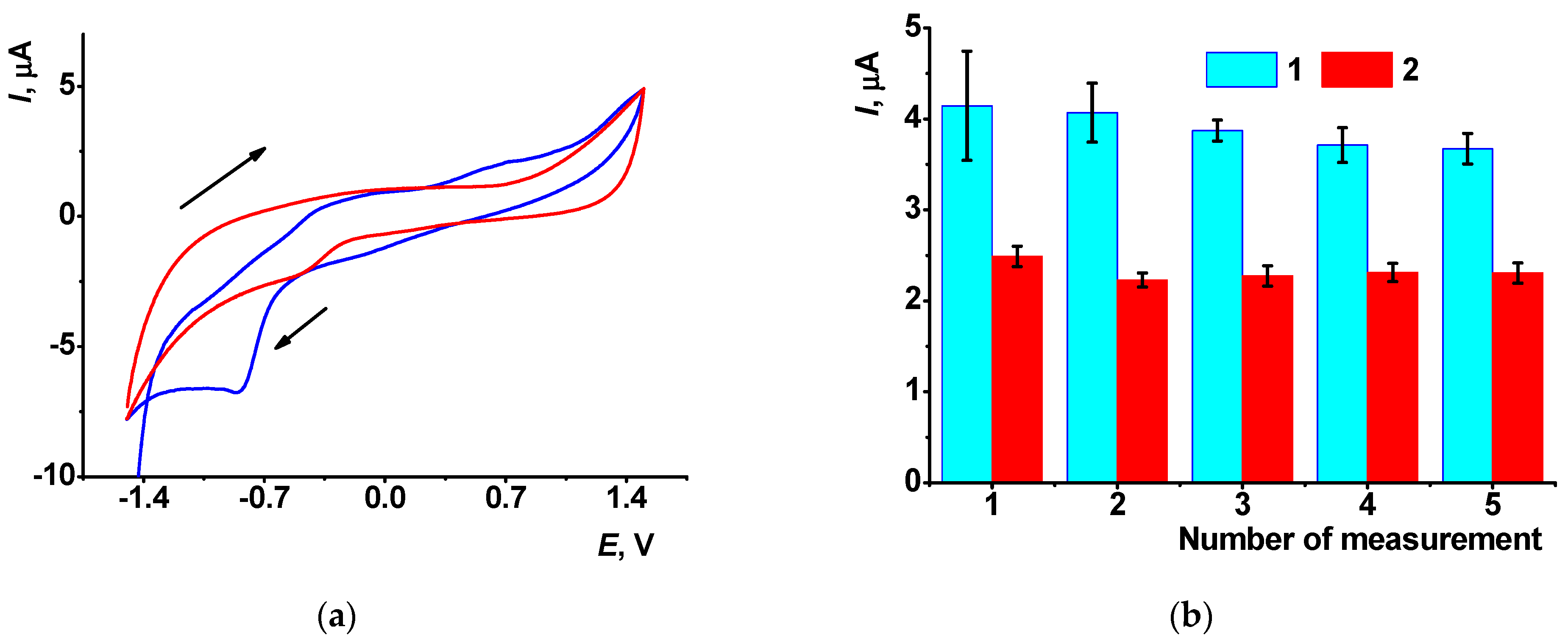
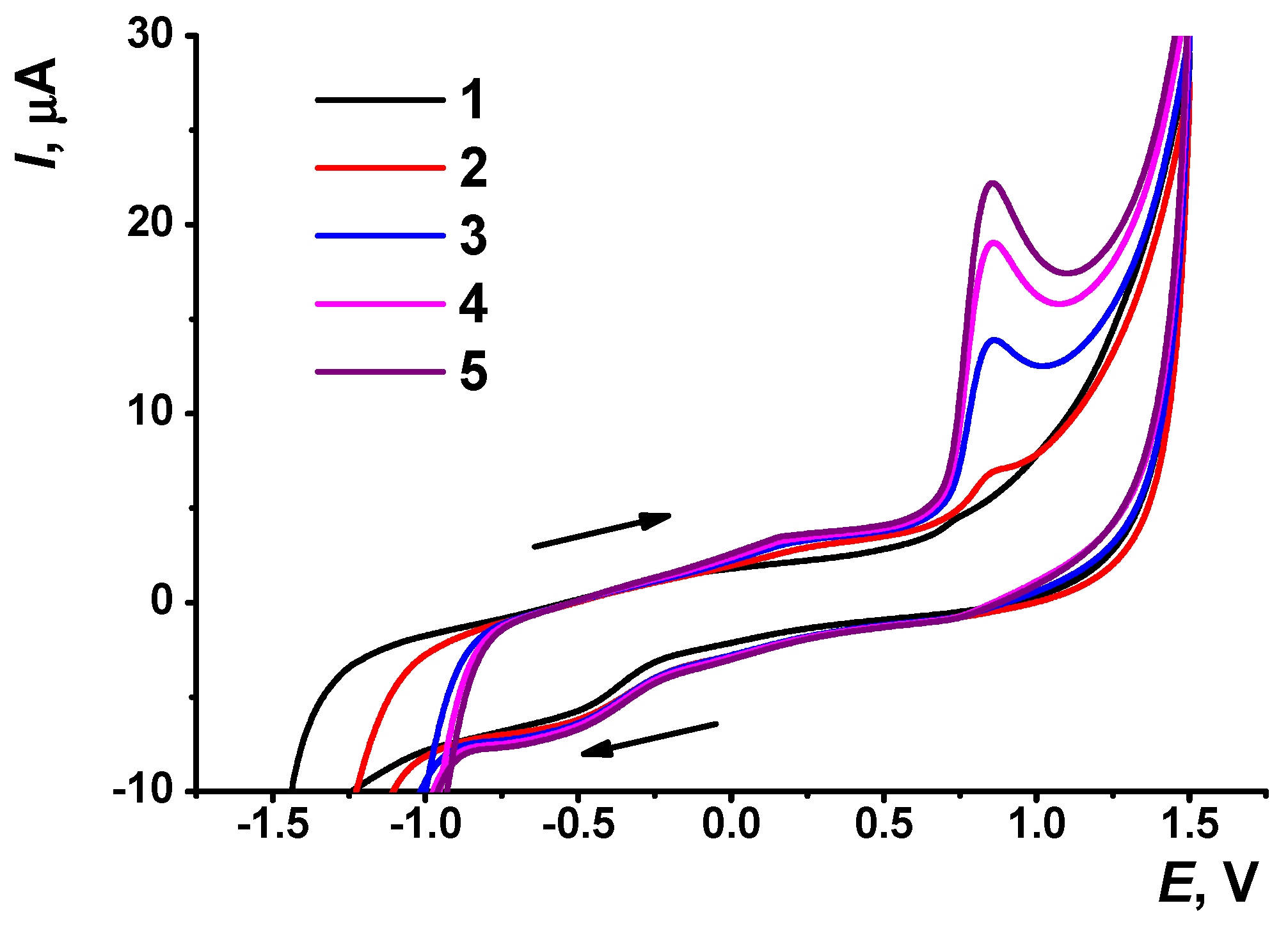
3.1. Electrochemical Properties of AY in Aqueous Solution
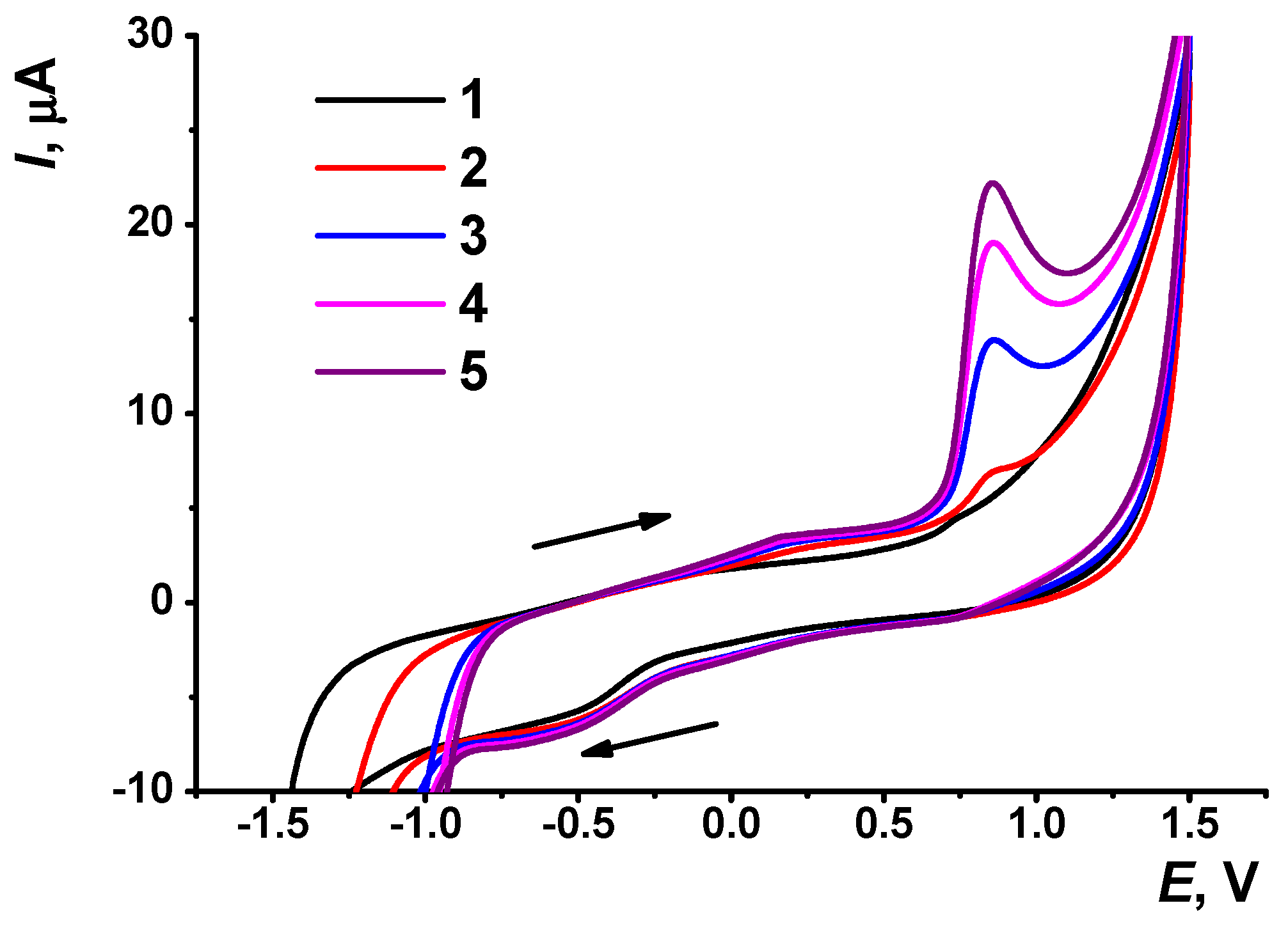
3.2. Deposition of the AY and CB on the GCE
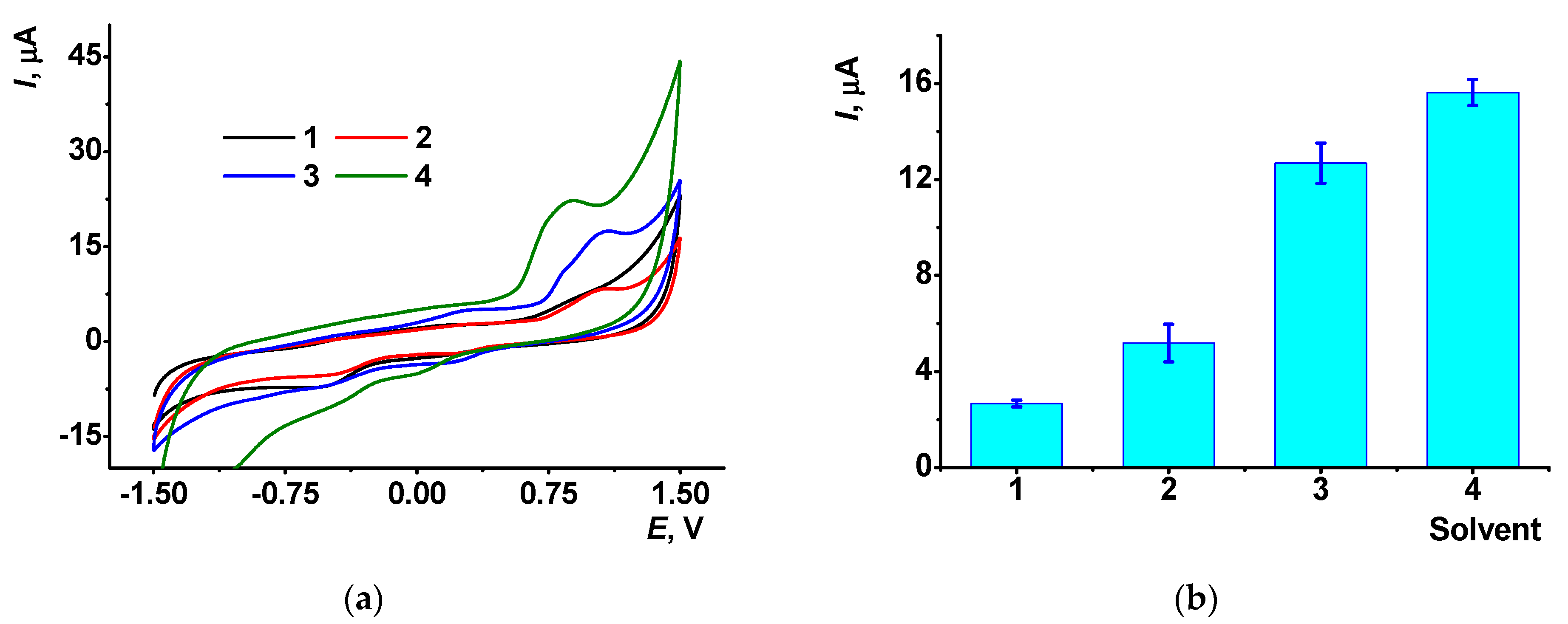
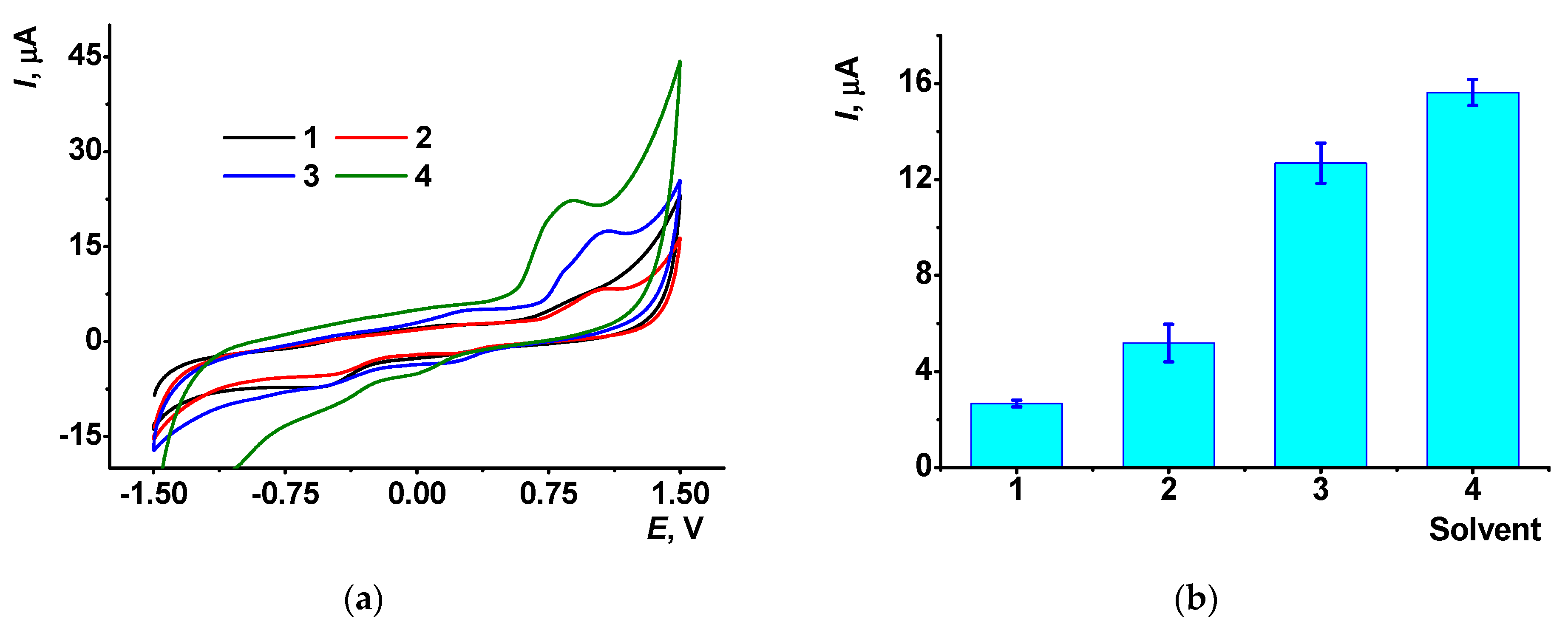
3.3. The Influence of DMF on the Electrochemical Properties of the CB/AY Modified GCE with Adsorbed DNA
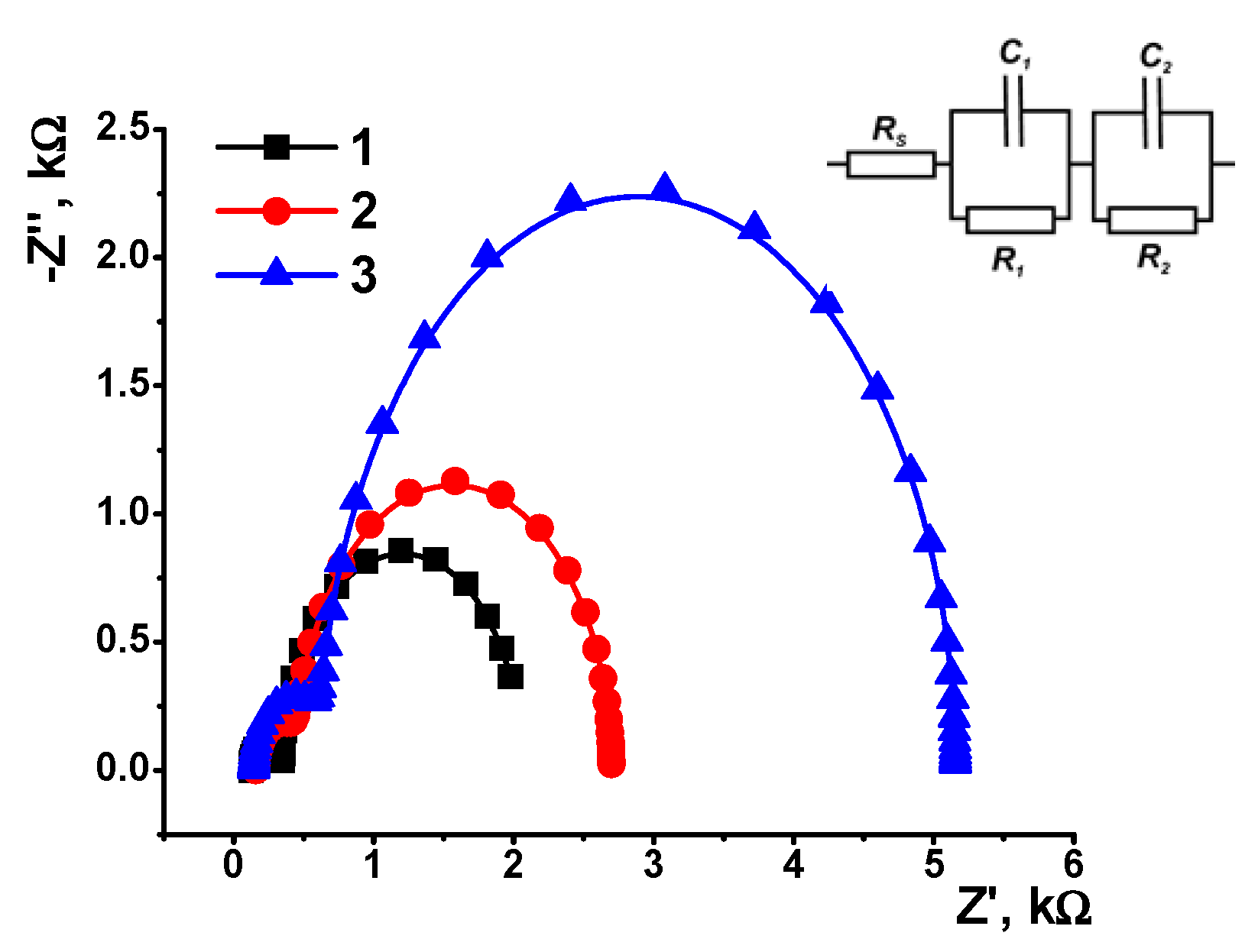
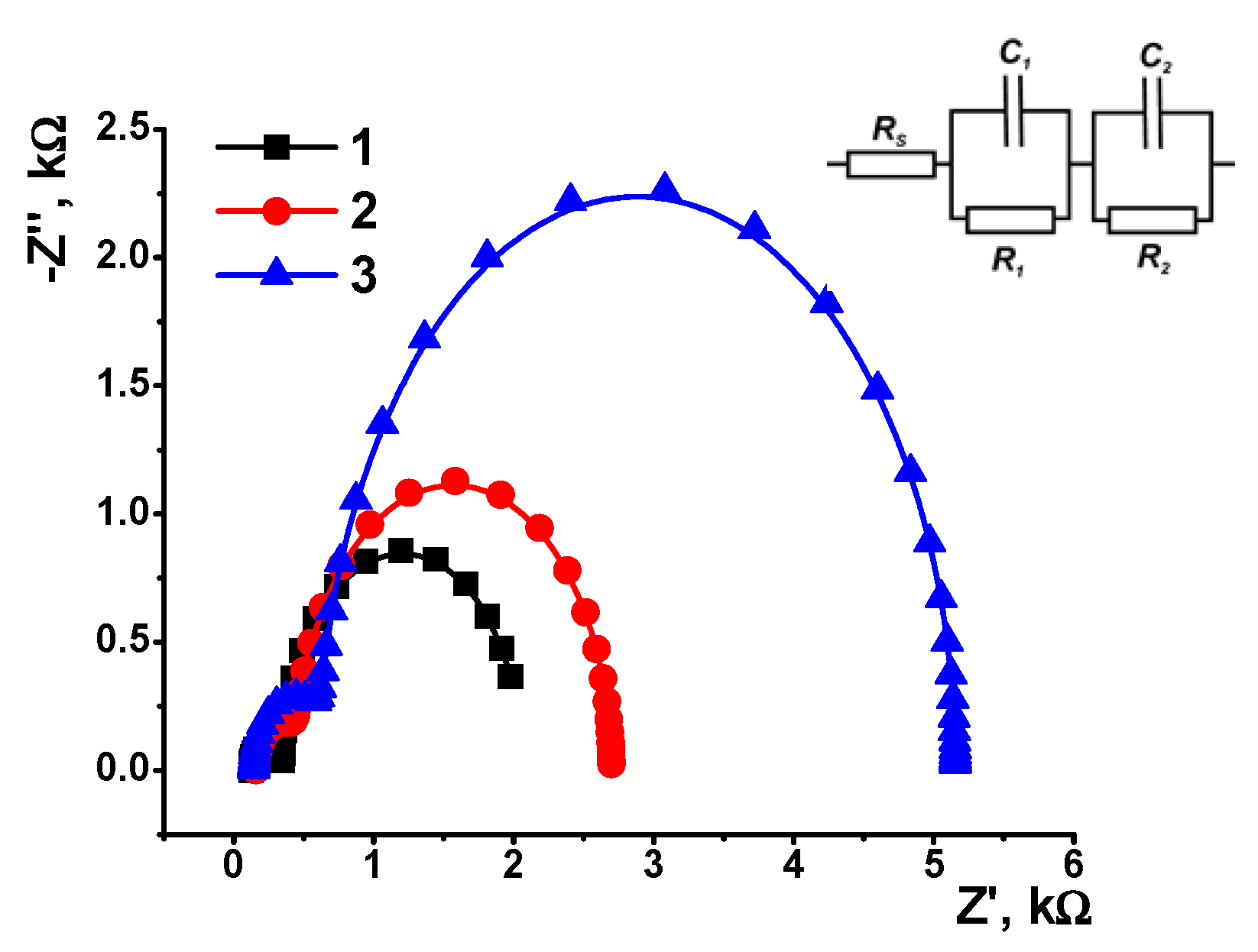
3.4. EIS and SEM Characterization
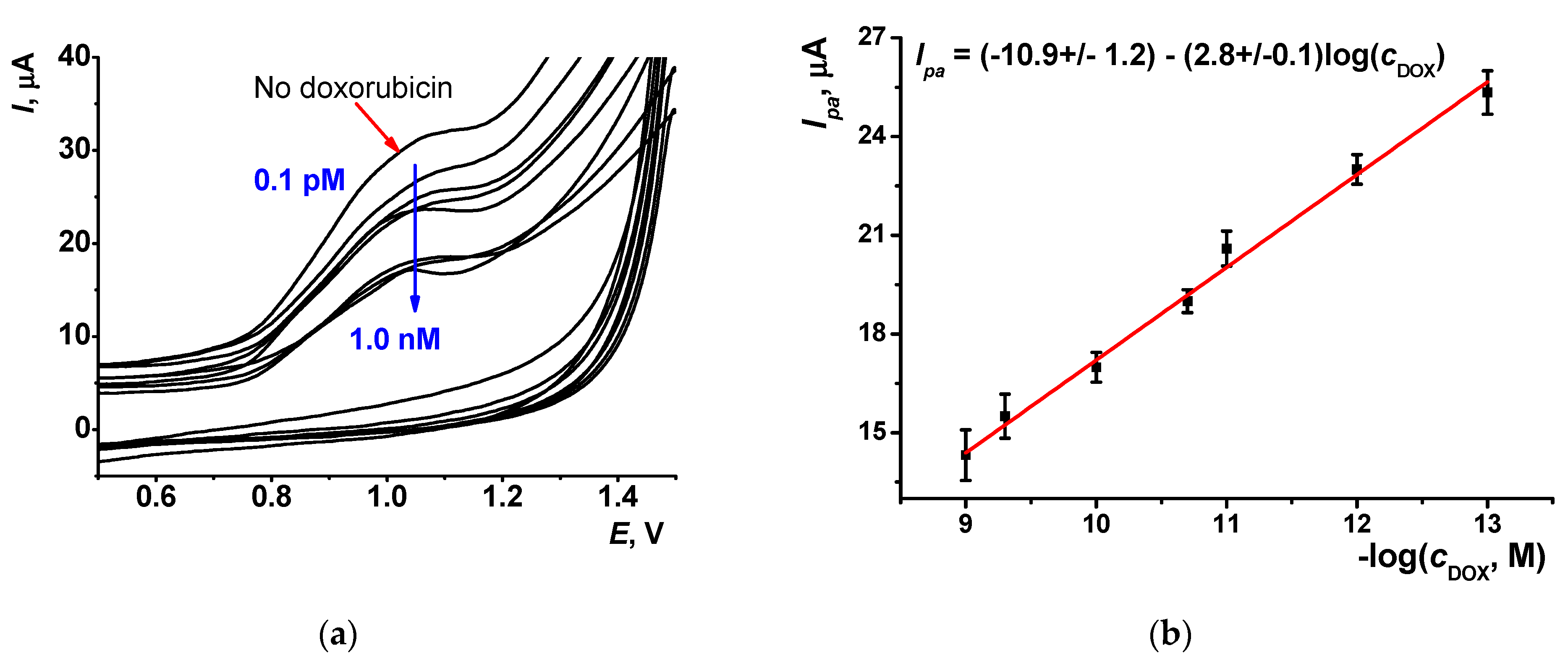
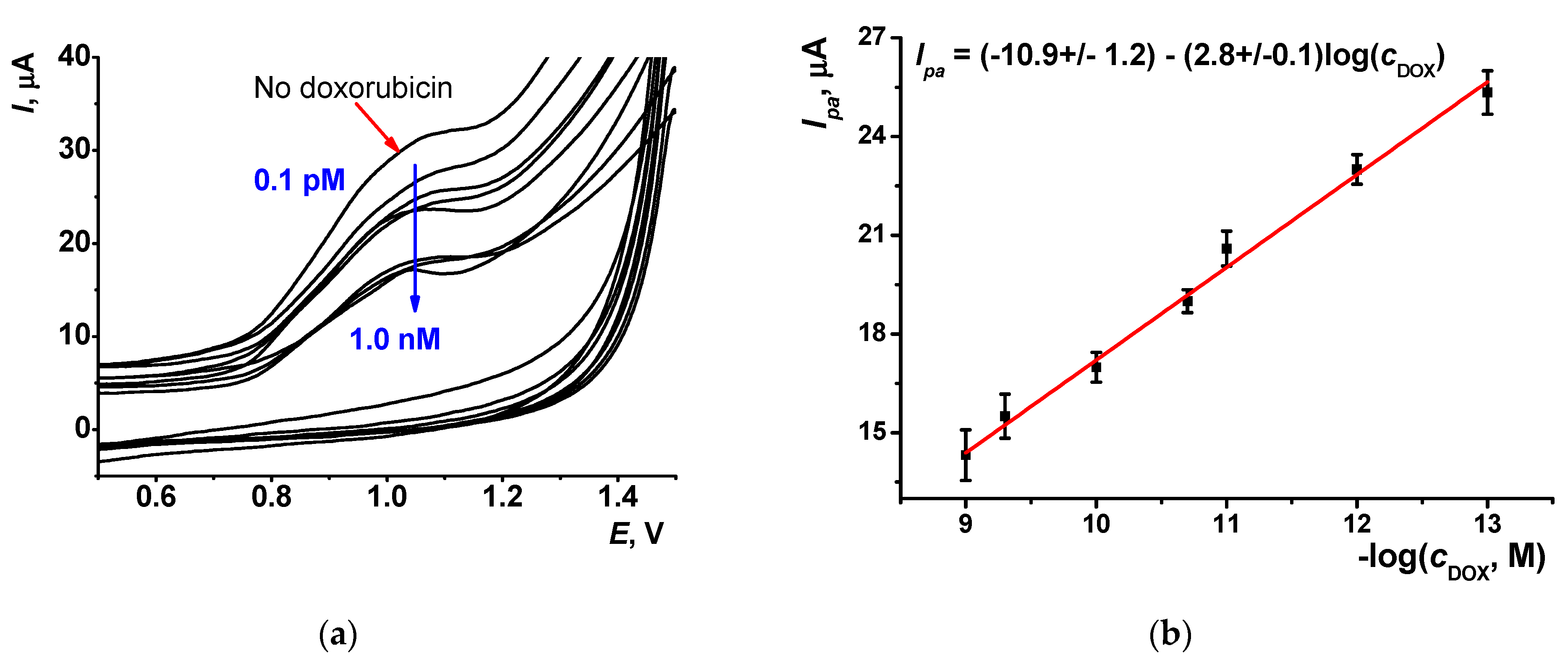
3.5. Doxorubicin Determination
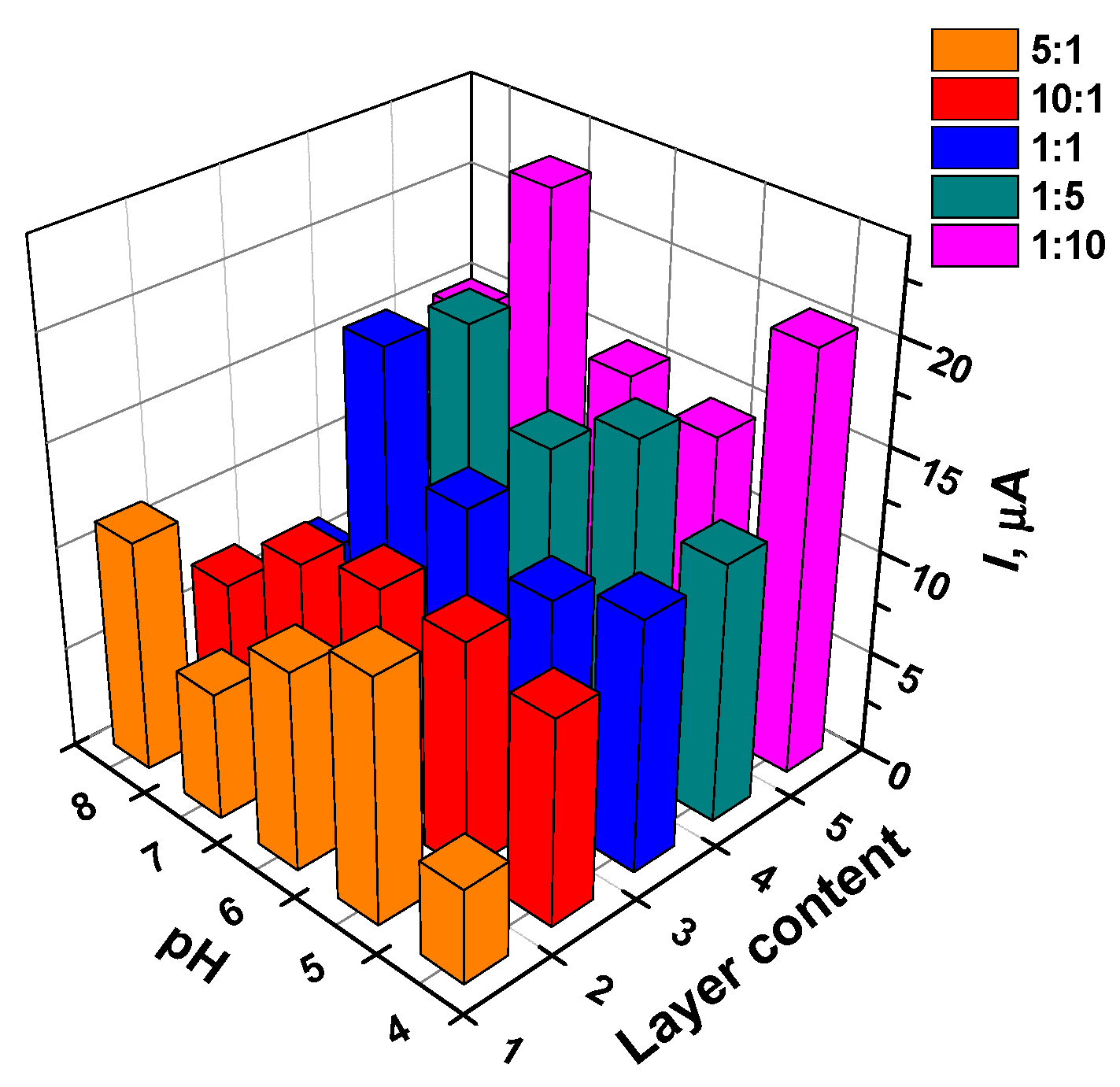
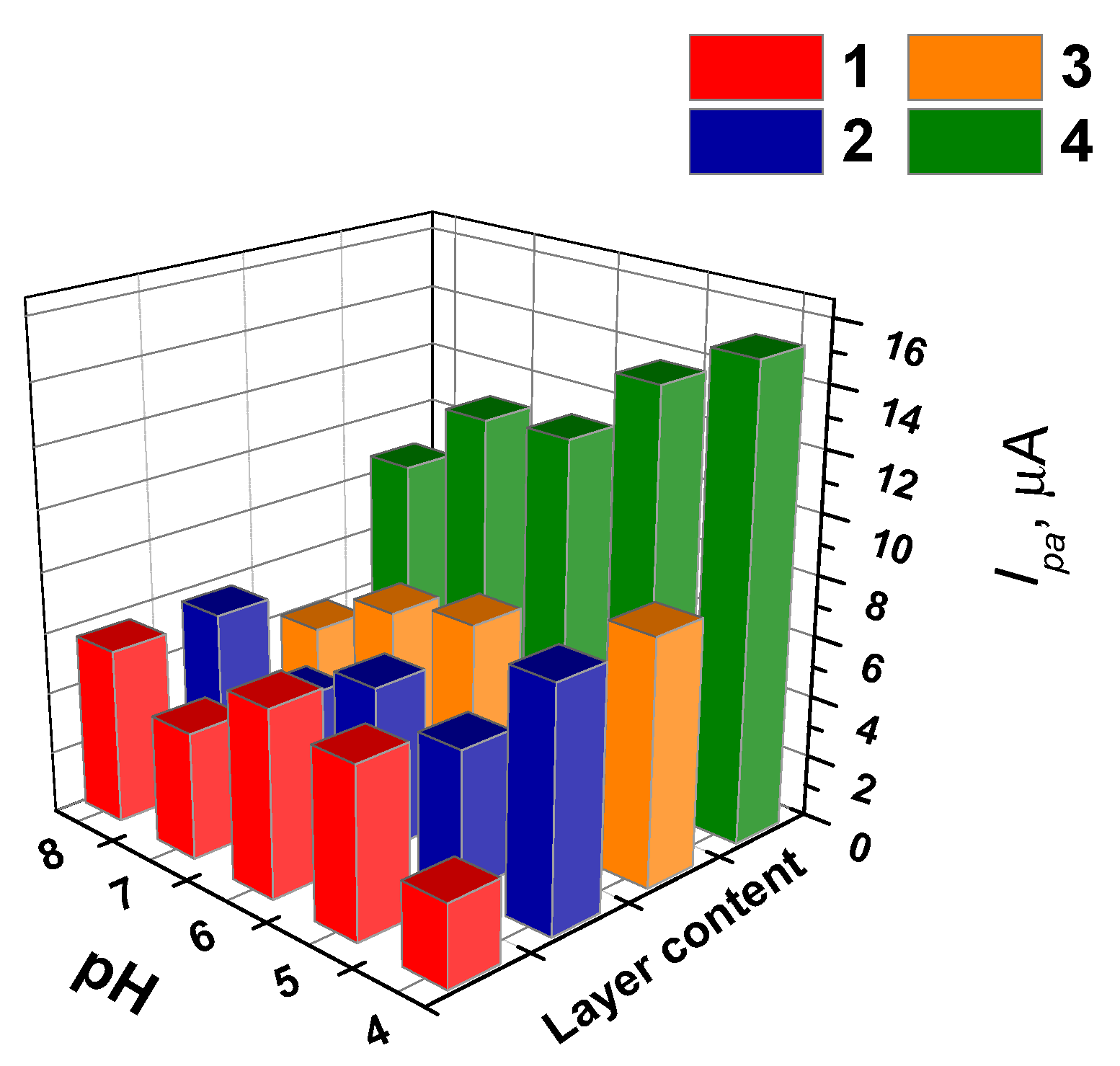
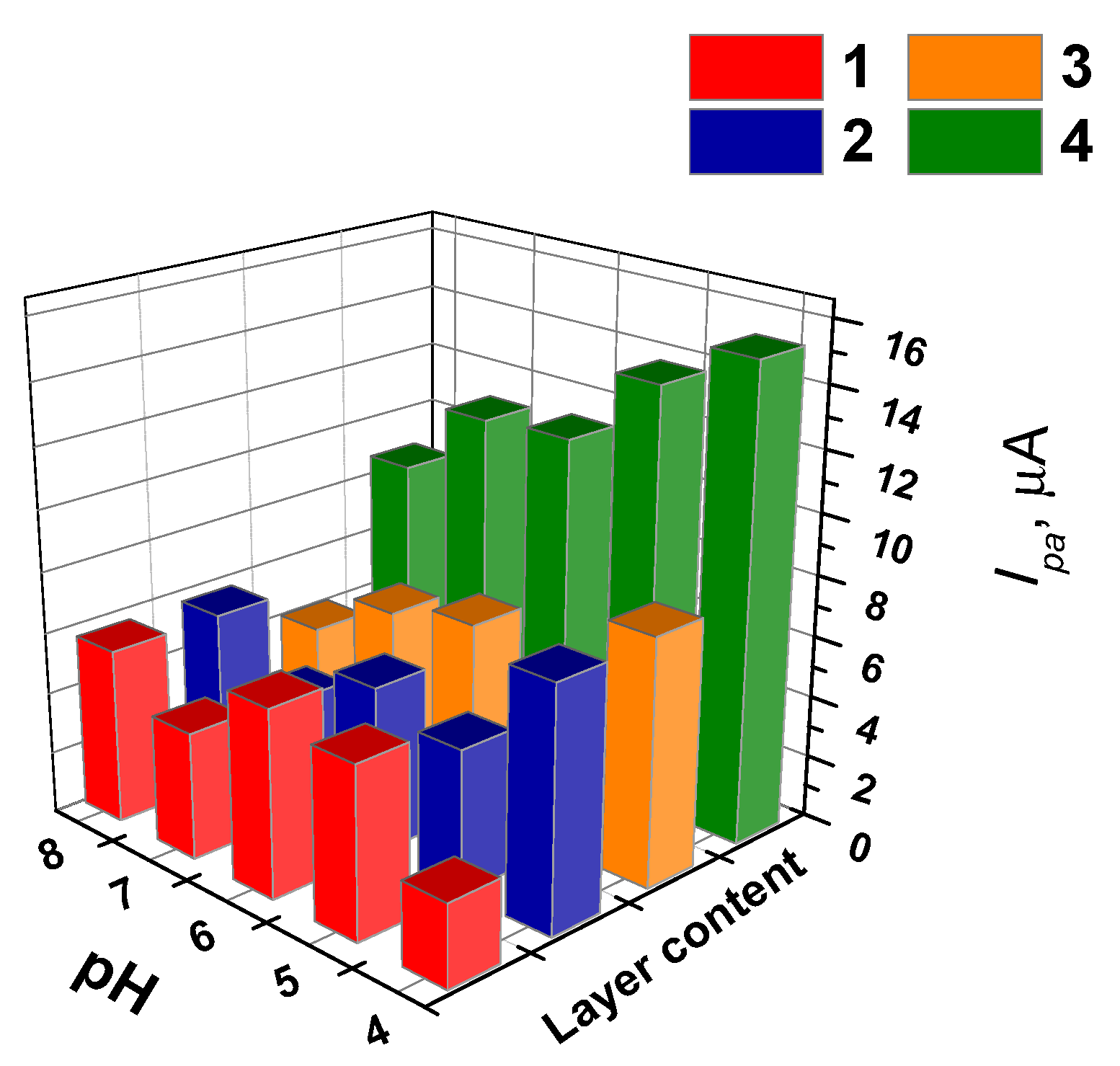
3.5.1. Selection of Working Conditions of Doxorubicin Determination
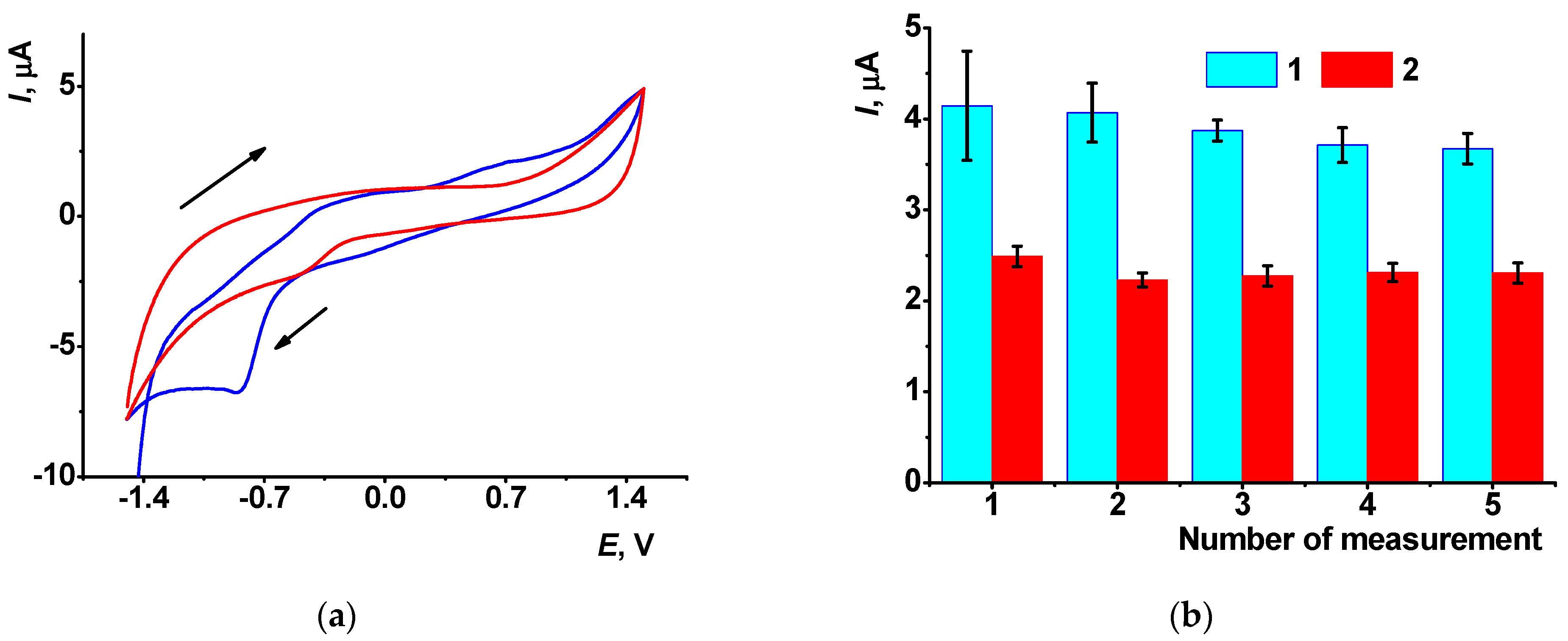
3.5.2. Measurement Precision and DNA Sensor Lifetime
3.5.3. Selectivity and Real Sample Analysis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Campuzano, S.; Yáñez-Sedeño, P.; Pingarrón, J.M. Electrochemical affinity biosensors based on selected nanostructures for food and environmental monitoring. Sensors 2020, 20, 5125. [Google Scholar] [CrossRef]
- Trotter, M.; Borst, N.; Thewes, R.; Stetten, F. Review: Electrochemical DNA sensing—Principles, commercial systems, and applications. Biosens. Bioelectron. 2020, 154, 112069. [Google Scholar] [CrossRef]
- Blair, E.O.; Corrigan, D.K. A review of microfabricated electrochemical biosensors for DNA detection. Biosens. Bioelectron. 2019, 134, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.-F.; Sun, X.-Y.; Su, M.; Wang, Y.-P.; Lv, Y.-K. Electrochemical biosensors based on antibody, nucleic acid and enzyme functionalized graphene for the detection of disease-related biomolecules. Analyst 2020, 145, 1550–1562. [Google Scholar] [CrossRef]
- Mills, D.M.; Calvo-Marzal, P.; Pinzon, J.M.; Armas, S.; Kolpashchikov, D.M.; Chumbimuni-Torres, K.Y. A single electrochemical probe used for analysis of multiple nucleic acid sequences. Electroanalysis 2017, 29, 873–879. [Google Scholar] [CrossRef] [Green Version]
- Surucu, O.; Öztürk, E.; Kuralay, F. Nucleic acid integrated technologies for electrochemical point-of-care diagnostics: A comprehensive review. Electroanalysis 2021, in press. [Google Scholar] [CrossRef]
- Tadini-Buoninsegni, F.; Palchetti, I. Label-Free bioelectrochemical methods for evaluation of anticancer drug effects at a molecular level. Sensors 2020, 20, 1812. [Google Scholar] [CrossRef] [Green Version]
- De la Cruz Morales, K.; Alarcón-Angeles, G.; Merkoçi, A. Nanomaterial-Based sensors for the study of DNA interaction with drugs. Electroanalysis 2019, 31, 1845–1867. [Google Scholar] [CrossRef]
- Kurbanoglu, S.; Dogan-Topal, B.; Rodriguez, E.P.; Bozal-Palabiyik, B.; Ozkan, S.A.; Uslu, B. Advances in electrochemical DNA biosensors and their interaction mechanism with pharmaceuticals. J. Electroanal. Chem. 2016, 775, 8–26. [Google Scholar] [CrossRef]
- Fojta, M.; Daňhel, A.; Havran, L.; Vyskočil, V. Recent progress in electrochemical sensors and assays for DNA damage and repair. TrAC—Trends Anal. Chem. 2016, 79, 160–167. [Google Scholar] [CrossRef]
- Diculescu, V.C.; Paquim, A.-M.C.; Oliveira-Brett, A.M. Electrochemical DNA sensors for detection of DNA damage. Sensors 2005, 5, 377–393. [Google Scholar] [CrossRef]
- Palchetti, I.; Mascini, M. Nucleic acid biosensors for environmental pollution monitoring. Analyst 2008, 133, 846–854. [Google Scholar] [CrossRef]
- Nishizawa, S.; Sato, Y.; Teramae, Y. Recent progress in abasic site-binding small molecules for detecting single-base mutations in DNA. Anal. Sci. 2014, 30, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Chiorcea-Paquim, A.-M.; Oliveira-Brett, A.M. Nanostructured material–based electrochemical sensing of oxidative DNA damage biomarkers 8-oxoguanine and 8-oxodeoxyguanosine: A comprehensive review. Microchim. Acta 2021, 188, 58. [Google Scholar] [CrossRef]
- Barroso, M.F.; de-los-Santos-Álvarez, N.; Delerue-Matosa, C.; Oliveira, M.B.P.P. Towards a reliable technology for antioxidant capacity and oxidative damage evaluation: Electrochemical (bio)sensors. Biosens. Bioelectron. 2011, 30, 1–12. [Google Scholar] [CrossRef] [Green Version]
- De-los-Santos-Álvarez, P.; Lobo-Castañón, M.J.; Miranda-Ordieres, A.J.; Tuñón-Blanco, P. Current strategies for electrochemical detection of DNA with solid electrodes. Anal. Bioanal. Chem. 2004, 378, 104–118. [Google Scholar] [CrossRef]
- Takahashi, S.; Anzai, J. Recent progress in ferrocene-modified thin films and nanoparticles for biosensors. Materials 2013, 6, 5742–5762. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Ali, S.; White, R.J. Electrocatalytic mechanism for improving sensitivity and specificity of electrochemical nucleic acid-based sensors with covalent redox tags—Part I. ACS Sens. 2020, 5, 3833–3841. [Google Scholar] [CrossRef]
- Lin, X.; Ni, Y.; Kokot, S. An electrochemical DNA-sensor developed with the use of methylene blue as a redox indicator for the detection of DNA damage induced by endocrine-disrupting compounds. Anal. Chim. Acta 2015, 867, 29–37. [Google Scholar] [CrossRef]
- Ziółkowski, R.; Jarczewska, M.; Górski, L.; Malinowska, E. Oligonucleotide-Based electrochemical biosensor for Hg2+ using methylene blue as a redox indicator. J. Electrochem. Soc. 2013, 160, B152–B155. [Google Scholar] [CrossRef]
- Wei, M.-Y.; Guo, L.-H.; Famouri, P. DNA biosensors based on metallo-intercalator probes and electrocatalytic amplification. Microchim. Acta 2011, 172, 247–260. [Google Scholar] [CrossRef]
- Evtugyn, G.; Hianik, T. Electrochemical DNA sensors and aptasensors based on electropolymerized materials and polyelectrolyte complexes. TrAC—Trends Anal. Chem. 2016, 79, 168–178. [Google Scholar] [CrossRef]
- Park, S.J.; Kwon, O.S.; Lee, J.E.; Jang, J.; Yoon, H. Conducting polymer-based nanohybrid transducers: A potential route to high sensitivity and selectivity sensors. Sensors 2014, 14, 3604–3630. [Google Scholar] [CrossRef] [PubMed]
- Shoaie, N.; Daneshpour, M.; Azimzadeh, M.; Mahshid, S.; Khoshfetrat, S.M.; Jahanpeyma, F.; Gholaminejad, A.; Omidfar, K.; Foruzandeh, M. Electrochemical sensors and biosensors based on the use of polyaniline and its nanocomposites: A review on recent advances. Microchim. Acta 2019, 186, 465. [Google Scholar] [CrossRef]
- Dhand, C.; Das, M.; Datt, M.; Malhotra, B.D. Recent advances in polyaniline based biosensors. Biosens. Bioelectron. 2011, 26, 2811–2821. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Jadon, N.; Pawaiya, A. Polypyrrole based next generation electrochemical sensors and biosensors: A review. TrAC—Trends Anal. Chem. 2017, 97, 363–373. [Google Scholar] [CrossRef]
- Cosnier, S. Recent advances in biological sensors based on electrogenerated polymers: A review. Anal. Lett. 2007, 40, 1260–1279. [Google Scholar] [CrossRef]
- Wang, D.; Wang, J. A sensitive and label-free electrochemical microRNA biosensor based on Polyamidoamine Dendrimer functionalized Polypyrrole nanowires hybrid. Microchim. Acta 2021, 188, 173. [Google Scholar] [CrossRef]
- Olowu, R.A.; Arotiba, O.; Mailu, S.N.; Waryo, T.T.; Baker, P.; Iwuoha, E. Electrochemical aptasensor for endocrine disrupting 17β-estradiol based on a poly(3,4-ethylenedioxylthiopene)-gold nanocomposite platform. Sensors 2010, 10, 9872–9890. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Esokkiya, A.; Sudalaimani, S.; Kumar, K.S.; Sampathkumar, P.; Suresh, C.; Giribabu, K. Poly(methylene blue)-based electrochemical platform for label-free sensing of acrylamide. ACS Omega 2021, 6, 9528–9536. [Google Scholar] [CrossRef]
- Da Silva, W.; Queiroz, A.C.; Brett, C.M.A. Nanostructured poly(phenazine)/Fe2O3 nanoparticle film modified electrodes formed by electropolymerization in ethaline—Deep eutectic solvent. Microscopic and electrochemical characterization. Electrochim. Acta 2020, 347, 136284. [Google Scholar] [CrossRef]
- Li, X.; Kan, X. A boronic acid carbon nanodots/poly(thionine) sensing platform for the accurate and reliable detection of NADH. Bioelectrochemistry 2019, 130, 107344. [Google Scholar] [CrossRef]
- Al-Jawadi, E.; Pöller, S.; Haddad, R.; Schuhmann, W. NADH oxidation using modified electrodes based on lactate and glucose dehydrogenase entrapped between an electrocatalyst film and redox catalyst-modified polymers. Microchim. Acta 2012, 177, 405–410. [Google Scholar] [CrossRef]
- Naik, T.S.S.K.; Swam, B.E.K. Modification of carbon paste electrode by electrochemical polymerization of neutral red and its catalytic capability towards the simultaneous determination of catechol and hydroquinone: A voltammetric study. J. Electroanal. Chem. 2017, 804, 7886. [Google Scholar] [CrossRef]
- Stepanova, V.B.; Shurpik, D.N.; Evtugyn, V.G.; Stoikov, I.I.; Evtugyn, G.A.; Osin, Y.N.; Hianik, T. Label-Free electrochemical aptasensor for cytochrome c detection using pillar[5]arene bearing neutral red. Sens. Actuators B 2016, 225, 57–65. [Google Scholar] [CrossRef]
- Porfireva, A.; Vorobev, V.; Babkina, S.; Evtugyn, G. Electrochemical sensor based on poly(Azure B)-DNA composite for doxorubicin determination. Sensors 2019, 19, 2085. [Google Scholar] [CrossRef] [Green Version]
- Porfireva, A.; Evtugyn, G. Electrochemical DNA sensor based on the copolymer of proflavine and azure B for doxorubicin determination. Nanomaterials 2020, 10, 924. [Google Scholar] [CrossRef]
- Porfireva, A.; Plastinina, K.; Evtugyn, V.; Kuzin, Y.; Evtugyn, G. Electrochemical DNA sensor based on poly(azure A) obtained from the buffer saturated with chloroform. Sensors 2021, 21, 2949. [Google Scholar] [CrossRef] [PubMed]
- Wemmer, D.E.; Dervan, P.B. Targeting the minor groove of DNA. Curr. Opin. Struct. Biol. 1997, 7, 355–361. [Google Scholar] [CrossRef]
- Prasher, P.; Sharma, M. Medicinal chemistry of acridine and its analogues. MedChemComm 2018, 9, 1589–1618. [Google Scholar] [CrossRef] [PubMed]
- Vaníčková, M.; Labuda, J.; Bučková, M.; Surugiu, I.; Mecklenburg, M.; Danielsson, B. Investigation of catechin and acridine derivatives using voltammetric and fluorimetric DNA-based sensors. Collect. Czech. Chem. Commun. 2000, 65, 1055–1066. [Google Scholar] [CrossRef]
- Svitkova, V.; Hanzelyová, M.; Macková, H.; Blaškovičová, J.; Vyskočil, V.; Farkašová, D.; Labuda, J. Behaviour and detection of acridine-type DNA intercalators in urine using an electrochemical DNA-based biosensor with the protective polyvinylalcohol membrane. J. Electroanal. Chem. 2018, 821, 87–91. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods. Fundamentals and Applications, 2nd ed.; John Wiley & Sons: Hoboken, NJ, USA, 2001. [Google Scholar]
- Hortobágyi, G.N. Anthracyclines in the treatment of cancer. An overview. Drugs 1997, 54, 1–7. [Google Scholar] [CrossRef]
- Li, M.; Russo, M.; Pirozzi, F.; Tocchetti, C.G.; Ghigo, A. Autophagy and cancer therapy cardiotoxicity: From molecular mechanisms to therapeutic opportunities. Biochim. Biophys. Acta 2020, 1867, 118493. [Google Scholar] [CrossRef] [PubMed]
- Rudnicki, K.; Sipa, K.; Brycht, M.; Borgul, P.; Skrzypek, S.; Poltorak, L. Electrochemical sensing of fluoroquinolone antibiotics. TrAC—Trends Anal. Chem. 2020, 128, 115907. [Google Scholar] [CrossRef]
- Wang, S.; Yan, X.; Yang, Y.; Qi, X.; Zhao, Y.; Li, L.; Ma, R.; Wang, L.; Dong, Y.; Sun, J.; et al. Advances and perspectives of aptasensors for the detection of tetracyclines: A class of model compounds of food. Food Chem. 2021, 364, 130361. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Wang, X.-M. Highly sensitive detection of daunorubicin based on carbon nanotubes-drug supramolecular interaction. Electrochem. Commun. 2009, 11, 126–129. [Google Scholar] [CrossRef]
- Alavi-Tabari, S.A.R.; Khalilzadeh, M.A.; Karimi-Maleh, H. Simultaneous determination of doxorubicin and dasatinib as two breast anticancer drugs uses an amplified sensor with ionic liquid and ZnO nanoparticle. J. Electroanal. Chem. 2018, 811, 84–88. [Google Scholar] [CrossRef]
- Liu, J.; Bo, X.; Zhou, M.; Guo, L. A nanocomposite prepared from metal-free mesoporous carbon nanospheres and graphene oxide for voltammetric determination of doxorubicin. Microchim. Acta 2019, 186, 639. [Google Scholar] [CrossRef]
- Vacek, J.; Havran, L.; Fojta, M. Ex situ voltammetry and chronopotentiometry of doxorubicin at a pyrolytic graphite electrode: Redox and catalytic properties and analytical applications. Electroanalysis 2009, 21, 21399–22144. [Google Scholar] [CrossRef]
- Skalová, Š.; Langmaier, J.; Barek, J.; Vyskočil, V.; Navrátils, T. Doxorubicin determination using two novel voltammetric approaches: A comparative study. Electrochim. Acta 2020, 330, 135180. [Google Scholar] [CrossRef]
- Kulikova, T.; Porfireva, A.; Evtugyn, G.; Hianik, T. Electrochemical DNA sensors with layered polyaniline-DNA coating for detection of specific DNA interactions. Sensors 2019, 19, 469. [Google Scholar] [CrossRef] [Green Version]
- Peng, A.; Xu, H.; Luo, C.; Ding, H. Application of a disposable doxorubicin sensor for direct determination of clinical drug concentration in patient blood. Int. J. Electrochem. Sci. 2016, 11, 6266–6278. [Google Scholar] [CrossRef]
- Evtugyn, A.; Porfireva, A.; Stepanova, V.; Budnikov, H. Electrochemical biosensors based on native DNA and nanosized mediator for the detection of anthracycline preparations. Electroanalysis 2015, 27, 629–637. [Google Scholar] [CrossRef]
- Moghadam, F.H.; Taher, M.A.; Karimi-Maleh, H. Doxorubicin anticancer drug monitoring by ds-DNA-based electrochemical biosensor in clinical samples. Micromachines 2021, 12, 808. [Google Scholar] [CrossRef] [PubMed]
- Porfir’eva, A.V.; Shibaeva, K.S.; Evtyugin, V.G.; Yakimova, L.S.; Stoikov, I.I.; Evtyugin, G.A. An electrochemical DNA sensor for doxorubicin based on a polyelectrolyte complex and aminated thiacalixF4]arene. J. Anal. Chem. 2019, 74, 707–714. [Google Scholar] [CrossRef]
- Hongpaisan, J.; Roomans, G.M. Retaining ionic concentrations during in vitro storage of tissue for microanalytical studies. J. Microsc. 1999, 193, 257–267. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Layer Content | Electrode–Film Interface | Film–Solution Interface | ||||
|---|---|---|---|---|---|---|
| R1, kΩ | C1, μF | n1 | R2, kΩ | C2, μF | n2 | |
| CB | 0.224 ± 0.010 | 3.60 ± 0.20 | 0.990 | 1.71 ± 0.11 | 52.3 ± 1.42 | 0.993 |
| CB/AY | 0.290 ± 0.015 | 2.30 ± 0.22 | 0.998 | 2.25 ± 0.15 | 19.3 ± 0.55 | 0.997 |
| CB/AY, DNA adsorption | 0.480 ± 0.028 | 0.42 ± 0.18 | 0.998 | 4.53 ± 0.26 | 4.86 ± 1.12 | 0.997 |
| Electrode Modifier/Electrode | Concentration Range, μM | Limit of Detection, nM | Ref. |
|---|---|---|---|
| Electrochemical sensors | |||
| Multiwalled carbon nanotubes/GC | 0.02–0.5 | - | [48] |
| ZnO dispersed in graphite paste | 0.07–5000 | 9.0 | [49] |
| Composite of mesoporous carbon nanospheres and reduced graphene oxide/GCE | 0.01–10 | 1.5 | [50] |
| Pyrographite | 0.01–1.0 | 10 | [51] |
| Silver solid amalgam | 0.6–10 | 440 | [52] |
| Electrochemical DNA sensors | |||
| Electropolymerized Azure B | 0.0001–0.1 | 0.07 | [36] |
| Electropolymerized aniline | 1 × 10−6–1000 | 0.0006 | [53] |
| Carbon nanotubes/poly(L-lysine) composite | 0.0025–0.25 | 1.0 | [54] |
| Electropolymerized Neutral red | 0.01–100 | 0.1 | [55] |
| Single-walled carbon nanotubes | 0.001–20 | 0.6 | [56] |
| Polyelectrolyte complexes with poly(styrene sulfonate) and aminated thiacalix[4]arene | 0.1–100 | 0.1 | [57] |
| AY and DNA adsorbed on CB/GCE | 1 × 10−5–0.001 | 0.0007 | This work |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kulikova, T.; Porfireva, A.; Rogov, A.; Evtugyn, G. Electrochemical DNA Sensor Based on Acridine Yellow Adsorbed on Glassy Carbon Electrode. Sensors 2021, 21, 7763. https://doi.org/10.3390/s21227763
Kulikova T, Porfireva A, Rogov A, Evtugyn G. Electrochemical DNA Sensor Based on Acridine Yellow Adsorbed on Glassy Carbon Electrode. Sensors. 2021; 21(22):7763. https://doi.org/10.3390/s21227763
Chicago/Turabian StyleKulikova, Tatjana, Anna Porfireva, Alexey Rogov, and Gennady Evtugyn. 2021. "Electrochemical DNA Sensor Based on Acridine Yellow Adsorbed on Glassy Carbon Electrode" Sensors 21, no. 22: 7763. https://doi.org/10.3390/s21227763
APA StyleKulikova, T., Porfireva, A., Rogov, A., & Evtugyn, G. (2021). Electrochemical DNA Sensor Based on Acridine Yellow Adsorbed on Glassy Carbon Electrode. Sensors, 21(22), 7763. https://doi.org/10.3390/s21227763