1. Introduction
Recently, with the gradual maturity of DNA recombination technology, an increasing number of useful substances are produced in the form of recombinants through bioprocesses, which overcomes the limitation of extraction from natural sources. The monitoring and control of this process are very important for obtaining high-quality products. For this reason, people have developed a variety of sensor systems. However, most of these sensor technologies target environmental variables, such as temperature, dissolved oxygen/carbon dioxide, and metabolites [
1]. To analyze target products, the subsequent purification and concentration steps are usually needed. This undoubtedly consumes a lot of labor and time for the screening of productive and high-quality products with many variables that need to be optimized [
2]. Therefore, a sensor that can directly measure the target product in bioprocesses is still in demand for analysis of the expected substance in the screening process and high-efficiency monitoring in large-scale production.
Among the recombinant proteins produced by bioprocesses, most of them have a His tag [
3]. This is a fusion tag designed to purify the desired protein by an immobilized metal-affinity column (IMAC), so that people can obtain products with high purity. In this circumstance, people have become increasingly aware of the importance of specific detection of His-tagged recombinant protein. At present, a number of detection methods for His-tagged proteins have been developed. Based on traditional immunoassay methods, such as Western blotting and enzyme-linked immunosorbent assay (ELISA), which require at least 4~5 h of operating procedure, an emerging UVHis-PAGE method was developed [
4]. It simplifies the blotting step after PAGE and eliminates the expensive instruments and consumables required for detection, but this method still needs to run SDS-PAGE followed by at least 1 h of incubation and rinse steps. Another example is the lateral flow (LF)-based test strip on the market, which can detect His-tagged protein at the concentration range of 4–20 μg/mL within 15 min (“Pro-Detect Rapid assays”, Thermo Fisher Scientific Co., Waltham, MA, USA). However, this still suffers from a high detection limit and narrow detection range. Although the latest study declared that faster detection (10 min) with a densitometric detection limit of 0.5 μM has been realized, the method is still difficult for carrying out accurate quantification [
5]. Also, by combining DNA aptamer and gold nanoparticle technologies, quick and qualitative determination of His-tagged protein was reported. However, their working range is narrow [
6,
7]. This is not conducive to knowing when to harvest the target recombinant products in large-scale production, which may result in a decreased yield due to premature recovery or protein degradation. On the other hand, a kind of fluorescent probe for His-tagged proteins has been developed [
8]. The assay based on this probe is fast (90 s) and has the detection limit of 0.8 μM for His
6-proteins, but it requires an additional reagent, a peptide that acts as the donor for phosphorescence detection. This means that multiple components are necessary to perform competitive measurement, which is not ideal as a biosensor. Therefore, to cater to the rapidly changing bioprocess, especially the cases in which a microorganism is used as an expression host, a more simplified and rapid His-tagged protein monitoring method is still expected.
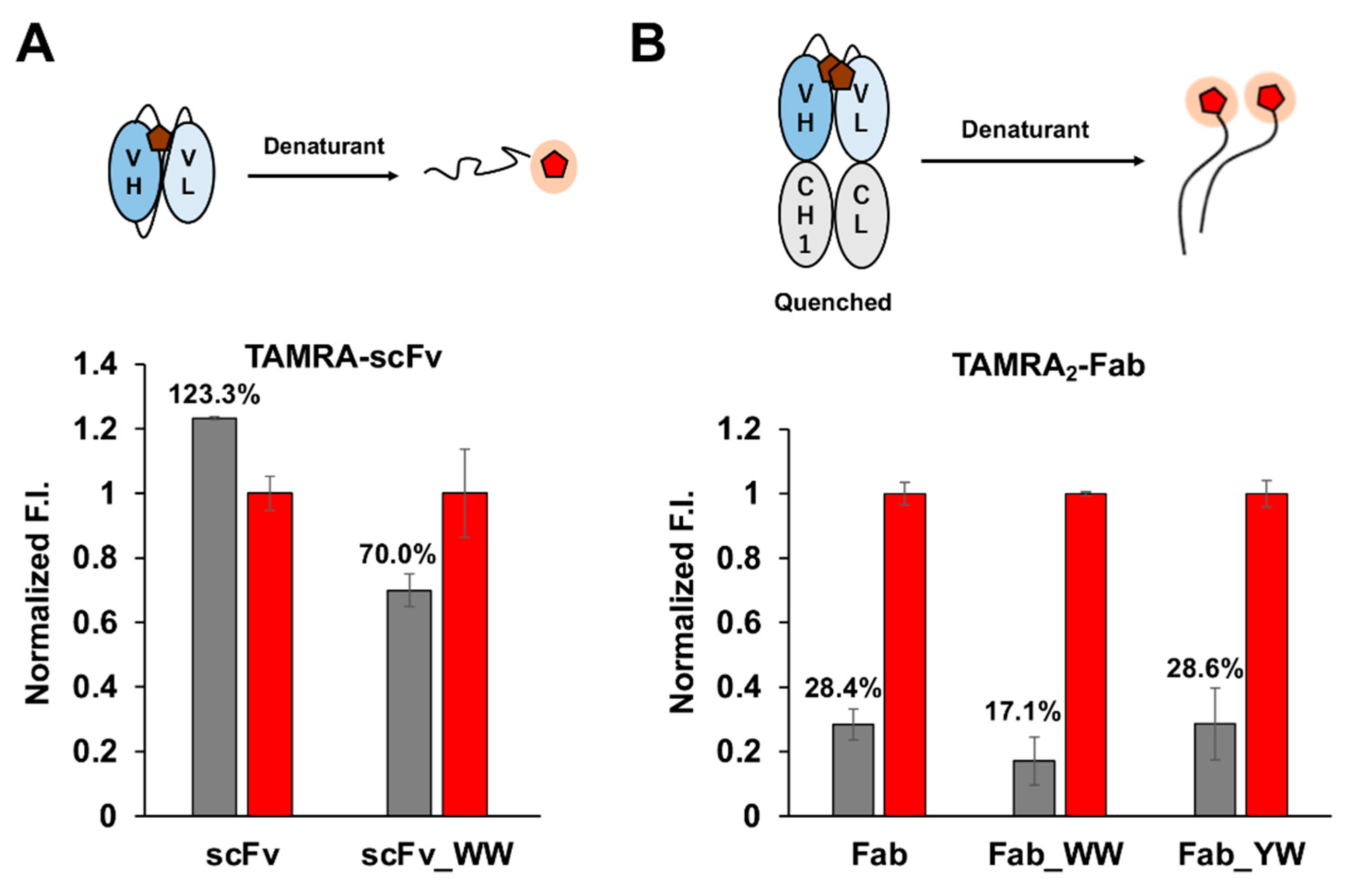
Previously, we developed a fluorescent antibody probe Quenchbody (Q-body) [
9,
10]. This is generated by certain fluorescent dyes such as tetramethylrhodamine (TAMRA) specifically labeled near the antigen-binding site of the antibody. Under the effect of tryptophan (Trp) residues inside the antibody, the fluorescent dye can be quenched by a photo-induced electron transfer (PeT) mechanism. Upon the binding of antigen to the antibody, this quenching effect is disturbed, so that the dye regains its fluorescence signal. The Q-body method can specifically detect various antigen molecules rapidly, and the operation is very simple. Just by adding Q-body into the sample containing the analyte and simply mixing without extra reagents, the fluorescence signal can be detected immediately. Because of these advantages, Q-body made of many formats of antibodies has been used as a convenient and fast biosensor targeting many useful substances, such as osteocalcin (BGP) and methotrexate (MTX) [
11,
12].
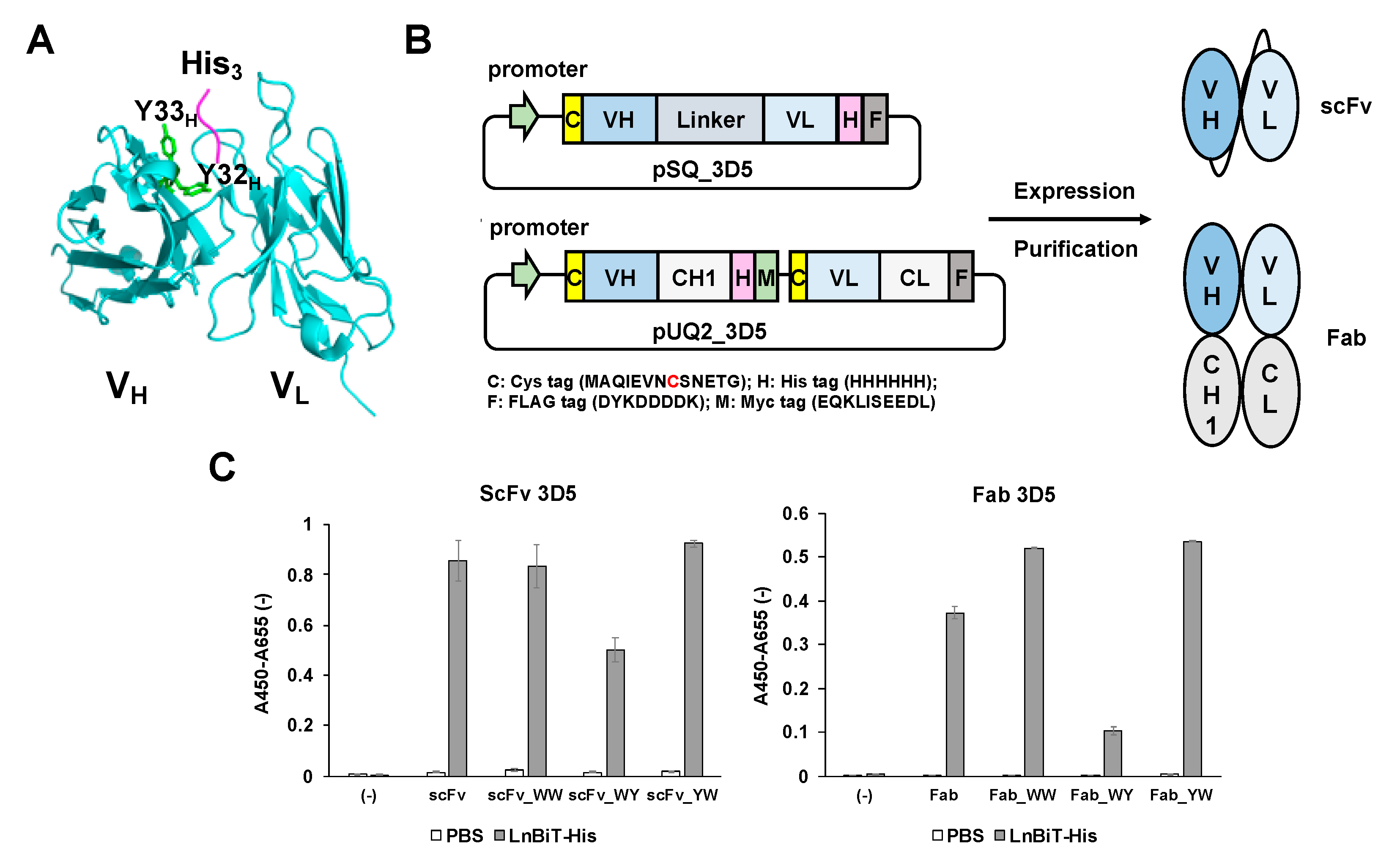
This time, with the aim of easier monitoring of secreted His-tagged recombinant proteins during bioprocesses, we tried to develop a Q-body that can quickly and specifically recognize recombinant proteins with a C-terminal His tag in the culture environment. In this study, we used anti-C-terminal His tag antibody 3D5 (PDB 1KTR) [
13] labeled with maleimide fluorescent dyes at the N-terminal region of its heavy and light chains to construct a Q-body that shows sufficient fluorescence quenching and antigen-dependent de-quenching. In addition, we investigated the effect of adding Trp residues at the antigen-binding site and successfully improved not only the fluorescence response but also the antigen-binding affinity and detection sensitivity of the Q-body. Finally, the constructed Q-body was applied to monitor the amount of the His-tagged nanobody protein secreted into the
Brevibacillus culture media.
Brevibacillus is a Gram-positive bacterium known for its ability in secretive expression of exogeneous proteins, and does not secrete proteases [
14] that can cause degradation of the secreted proteins in the culture media. The production and use of the V
HH nanobody, which is derived from a Camelid single chain antibody, are becoming increasingly popular due to its small size and superior stability [
15,
16,
17]. We tried to monitor the expression of anti-SARS-CoV-2 spike V
HH nanobody from the cultured
Brevibacillus cells.
2. Materials and Methods
2.1. Materials
Restriction endonuclease was from New England Biolabs Japan (Tokyo, Japan). The In-Fusion HD cloning kit, pGro7 chaperonine plasmid, Talon metal affinity resin, Talon disposable gravity column, and a
Brevibacillus In Vivo Cloning (BIC) system were from Takara Bio (Otsu, Japan). The KOD Plus mutagenesis kit was from Toyobo Biochemicals (Osaka, Japan). The PureYield Plasmid Miniprep System and Wizard SV Gel and PCR Clean-Up System were from Promega (Tokyo, Japan). MicroSpin G-25 Columns were from GE Healthcare (Amersham, UK). The Luria-Bertani medium was from Beckton-Dickinson (Tokyo, Japan). Immunoblock was from KAC (Hyogo, Japan). 5-TAMRA C6 maleimide was from AAT Bioquest (Sunnyvale, CA, USA). Oligonucleotides and the gene for 3D5 scFv were synthesized by Eurofins Genomics (Tokyo, Japan). The sequence of primers used is summarized in
Table S1. Ultrapure water was prepared using a Milli-Q machine (Millipore Japan, Tokyo, Japan). Biotinylated His
6 peptide was synthesized using a PSSM-8 peptide synthesizer (Shimadzu, Kyoto, Japan). Other chemicals and reagents, unless otherwise indicated, were from Sigma-Aldrich (St. Louis, MO, USA) or Fujifilm-Wako pure chemicals (Osaka, Japan).
2.2. Construction of scFv and Fab Expression Plasmids
A single-chain Fv (scFv) expression vector pSQ (KTM219) [
18] encoding a Cys-tag containing a cysteine (Cys) residue, anti-osteocalcin scFv, a His
6 (HHHHHH)- and a FLAG (DYKDDDDK)- tag at its C- terminal was digested by
AgeI-HF and
XhoI-HF to remove the original scFv fragment and ligated with
AgeI-HF- and
SalI-HF-digested V
H-V
L type 3D5 scFv fragment in pEX-K4J2-1KTR, which was amplified by primers 3D5_Age_back and 3D5_Sal_for, and KOD-plus Neo DNA polymerase.
An antigen-binding fragment (Fab) expression vector pUQ2(KTM219) encoding two Cys-tags, each containing a Cys residue at the N-terminus of both VH and VL was appended with a His6 -myc tag at the C- terminus of the heavy chain and a FLAG tag at the C- terminus of the light chain. This plasmid was firstly digested by AgeI-HF and XhoI-HF to remove the original VH fragment and in-fused with a 3D5 VH fragment, which was amplified using the primers Age_back and In-fusion_3D5_VH_for and pEX-K4J2-1KTR encoding (GGGS)4-VH-VL 3D5 scFv as a template, using KOD-plus neo DNA polymerase. After that, it was digested by SpeI-HF and HindIII-HF to remove the original VL fragment and in-fused with the 3D5 VL fragment, which was amplified from the same template by primer In-fusion3D5VLback and In-fusion3D5VLfor. The vector thus made was digested with AgeI-HF to remove the (GGGS)4 linker between Cys-tag and VH to make a linker-less pUQ2 (3D5).
To introduce Y32W and/or Y33W mutations in the VH region of scFv and Fab, a short DNA fragment was amplified by a primer pair of 3D5_Age_back and 3D5VH31for, and a longer DNA fragment was amplified by another primer pair of either 3D5VH_WY_back, 3D5VH_WW_back or 3D5VH_YW_back and 3D5_Sal_for. After running agarose electrophoresis (1.5% agarose), the bands of 164 bp and 701 bp were recovered. Overlap extension PCR was performed on these DNA fragments, and the product was amplified using VHH_Age_back and 3D5_Sal_for. After being digested by AgeI-HF and XhoI-HF, the 3D5_WY/WW/YW_VH insert was ligated with pSQ (3D5) or pUQ2 (3D5) digested with the same enzymes using ligation high ver2 enzyme, to yield WY/WW/YW-mutated pSQ (3D5) and pUQ2 (3D5), respectively.
2.3. Protein Expression and Purification
SHuffle T7 Express E. coli cells were co-transformed with pGro7 chaperone plasmid and one of pSQ/pUQ2 (3D5) plasmid and cultured at 30 °C overnight on LBAC (Luria-Bertani medium containing 50 μg/mL ampicillin and 20 μg/mL chloramphenicol) plate containing 1.5% agar. A colony was picked and cultured at 30 °C, 200 rpm in 4 mL LBAC medium for one night. The next day, transferred 4 mL bacterial culture into 400 mL LBAC medium and cultured at 30 °C, 200 rpm until OD600 reached 0.4~0.6. After 0.4 mM isopropyl β-d-1-thiogalactopyranoside induction, the bacterial solution was incubated at 16 °C, 200 rpm for 16 h, and centrifuged at 4 °C, 5000 g for 15 min. The E. coli cells were resuspended with 9 mL of extraction buffer (20 mM Na2HPO4, 20 mM NaH2PO4, 500 mM NaCl, pH 7.4) and disrupted by a cell disruptor One Shot Model (Constant Systems, Daventry, UK). After being centrifuged at 4 °C, 8000× g for 10 min, the supernatant was incubated with 200 μL Talon IMAC resin on a rotating wheel at 4 °C for 1 h. After centrifugation at 4 °C, 700× g for 1 min, the resin was transferred to a 2 mL disposable gravity column. The resin was washed with 500 μL of binding buffer (20 mM Na2HPO4, 20 mM NaH2PO4, 500 mM NaCl, 20 mM imidazole, pH 7.4) 3 times, and the target protein was eluted with 400 μL elution buffer (10 mM Na2HPO4, 10 mM NaH2PO4, 250 mM NaCl, 500 mM imidazole, pH 7.4). The yield and purity of the target protein were confirmed by SDS-PAGE, which was loaded with the same volume of 2 × SDS loading buffer (125 mM Tris-HCl, 4% SDS, 20% glycerol, 0.02% bromophenol blue, 0.2 M dithiothreitol, pH 6.8), and boiled at 95 °C for 5 min. The protein concentration was determined by using a densitometer WSE-6100 and a CS Analyzer 4 software (ATTO, Tokyo, Japan) by comparing it with several concentrations of bovine serum albumin (BSA) as a standard.
2.4. Enzyme-Linked Immunosorbent Assay
Each well of a Costar 3590 96-well microplate (Corning, Tokyo, Japan) was coated with or without 100 μL of thioredoxin (Trx)-fused LnBiT-His protein (2 μg/mL, His
6 tag at the C-terminus) [
19] in PBS and incubated at 4 °C overnight. The plate was washed 3 times with 200 μL PBS containing 0.1% Tween20 (PBST) and blocked with 200 μL of PBST containing 20% Immunoblock (KAC, Japan) at 25 °C for 1 h. After the plate was washed three times with PBST, 100 μL of anti-His scFv/Fab 3D5 (2 μg/mL) in PBST containing 5% Immunoblock was added, and then the plate was incubated at room temperature for 2 h. After washing the plate three times, bound scFv/Fab 3D5 was probed with 100 μL of 1:2000 diluted horseradish peroxidase (HRP)-labeled anti-FLAG M2 (Sigma) in PBST containing 5% Immunoblock at 25 °C for 1 h. After it was washed three times with PBST, 100 μL of substrate solution (100 mM CH
3COONa, 0.2 mg/mL TMBZ, 0.09% H
2O
2, pH 6.0) was added, and 50 μL of stop solution (10% H
2SO
4) was added when the solution turned blue. The absorbance at 450 nm with a reference at 655 nm was monitored by a microplate reader SH-1000 (Corona Electric, Ibaraki, Japan).
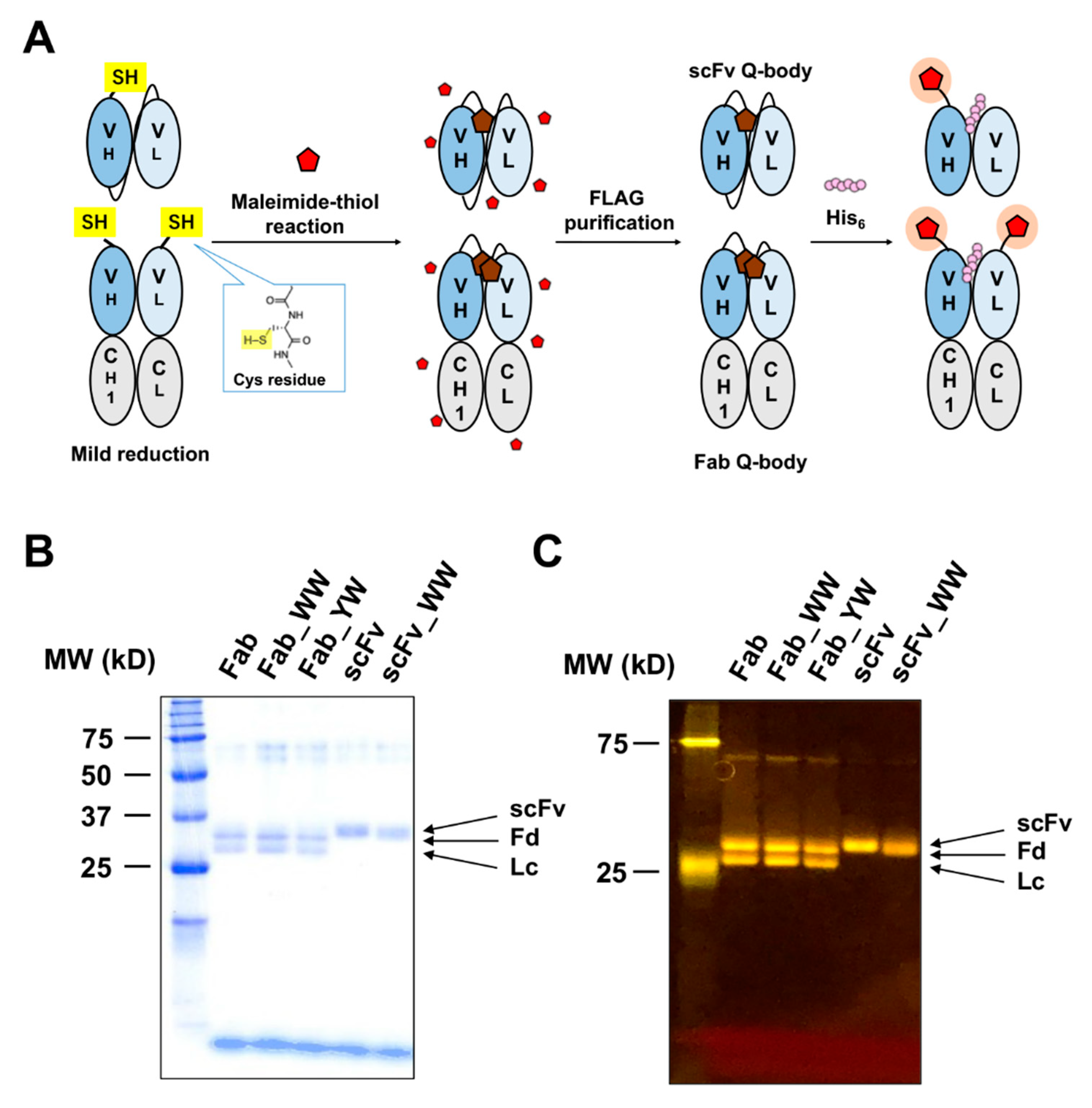
2.5. Fluorescence Labeling and Purification
The purified protein (10 μM) in 120 μL PBST was mixed with 8 mM tris (2-carboxyethyl) phosphine (TCEP)-HCl in a 1.5 mL tube for 20 min at 25 °C. To inactivate TCEP, 20 mM 4-azidobenzoic acid (ABA), pH 7.0, was added [
20], and the tube was put in a vacuum for 15 min to remove air bubbles. Afterwards, 20 × mol of 5-TAMRA C6 maleimide or ATTO520-C2 maleimide in DMSO was added and it was incubated at 4 °C for 16 h or 25 °C for 1 h. Anti-FLAG M2 magnetic beads (Sigma, 10 μL) were washed with PBST, and then the beads were added to the reaction mixture. After incubation on a rotating wheel at 25 °C for 1 h, the beads were washed 12 times with PBS containing 0.1% Brij35 and 2 times with PBST. Then 100 μL of 150 μg/mL 3×FLAG peptide in PBST was used to elute the labeled 3D5 Q-body. The 3×FLAG peptide was removed by using a MicroSpin G-25 Column (GE Healthcare, Amersham, UK), which was pre-equilibrated with PBST.
2.6. Fluorescence Measurements
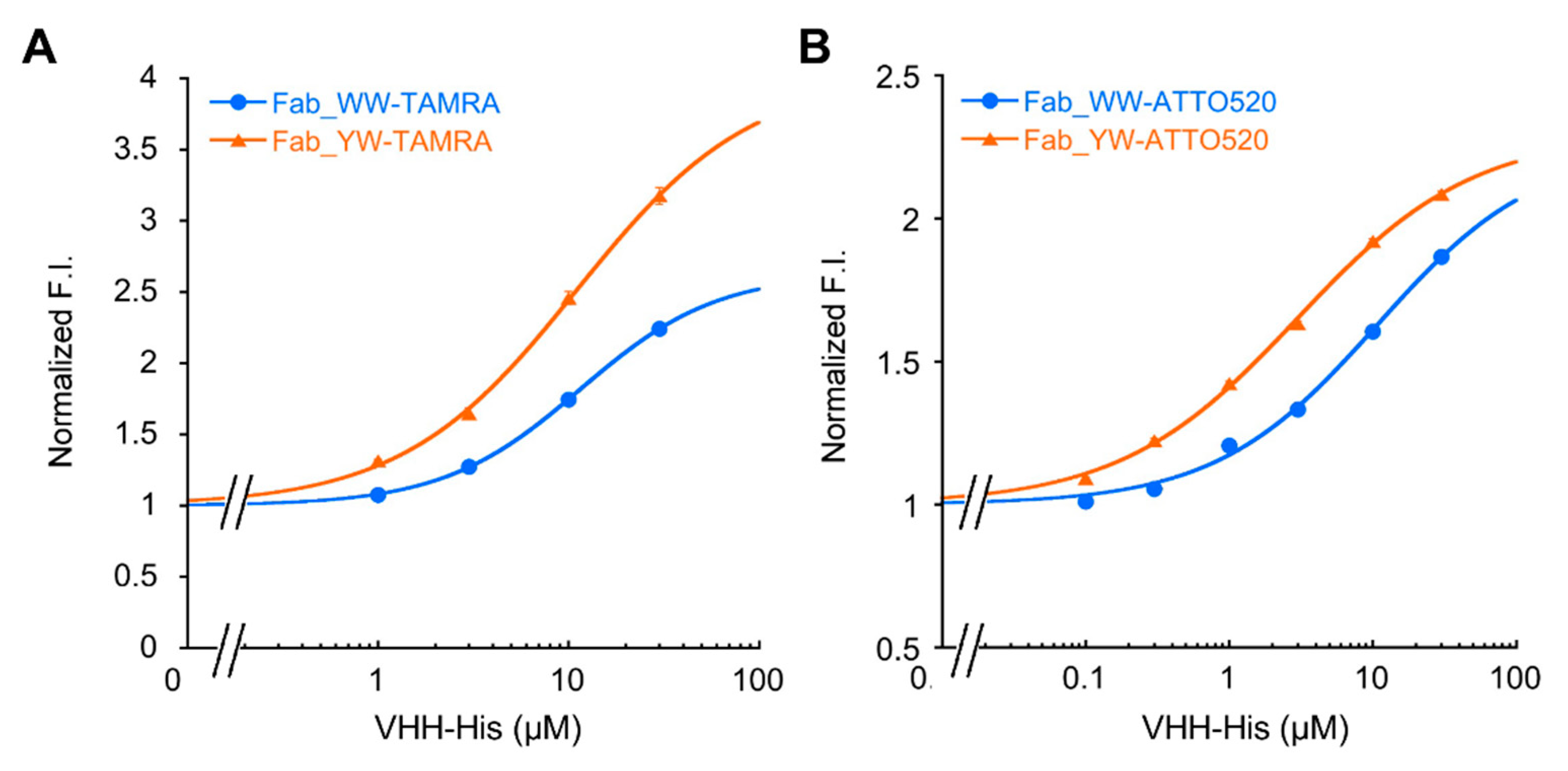
The Q-body sample solution (100 μL) (n = 3) was applied into a well of black 96-well half area microplate (Greiner Bio-One GmbH, Frickenhausen, Germany). The fluorescence measurement was carried out immediately on a CLARIOstar plate reader (BMG Labtech, Ortenberg, Germany). The wavelengths of 535/20 nm and 585/30 nm for TAMRA, and 490/20 nm and 550/40 nm for ATTO520 were used for the excitation and emission, respectively. In each measurement, the fluorescence background of the sample solution without Q-body was subtracted. To derive EC50 and limit of detection (LOD), antigen dose-response curves were fitted to a four-parameter logistic equation using Kaleidagraph 4.5 (Synergy Software, Reading, Mount Penn, PA, USA) as follows:
where a was set to 1. EC50 was taken from c, and LOD calculated as the concentration corresponding to the mean blank value plus three times the standard deviation for each assay.
2.7. Antigen-Binding Kinetics Analysis
To evaluate the antigen-binding affinity of the proteins, bio-layer interferometry (BLI) measurements were performed on an Octet K2 system (Pall FortéBio, Fremont, CA, USA). Streptavidin-conjugated (SA) biosensors were soaked in Kinetic buffer (10 mM Na
2HPO
4, 10 mM NaH
2PO
4, 150 mM NaCl, 0.002% Tween20, 0.1% BSA, pH 7.4) for 10 min. After that, 100 nM biotinylated His
6 peptide (bio-EGGGSHHHHHH-COOH, synthesized as in
Figure S1,
Supplementary materials) was loaded on an SA biosensor and equilibrated in Kinetic buffer before analysis. For each measurement, 80 μL of unlabeled protein or Q-body in Kinetic buffer in two-fold gradient concentrations were applied, which was followed by the cycle of: 1000 rpm shake speed, baseline measurement in Kinetic buffer for 1 min, association measurement in a sample for 200~400 s, and dissociation measurement in Kinetic Buffer for 200~400 s. The biosensors were regenerated in 500 mM phosphoric acid (pH 1.0) for 1 min and equilibrated with Kinetic buffer for 30 s before the next cycle. The data were imported into Data Acquisition 11.0 (FortéBio) and the kinetic constants were calculated by Data Analysis 11.0 (FortéBio) using double reference subtraction assuming a 1:1 binding model.
2.8. Secretive Expression and Analysis of His-Tagged Nanobody
The anti-SARS-CoV2 VHH gene tagged with a hinge, 3 × FLAG and C-terminal His tags (Kindly provided by Dr. Akikazu Murakami in RePhagen Inc., Okinawa, Japan) was PCR amplified with primers pBIC_pET1-4VHH_Rv and pBIC3-forwardVHH, and co-transformed to Brevibacillus choshinensis HPD31 cells with pBIC3 vector (Takara Bio) so that the plasmid encoding target gene with a C-terminal His tag was formed by homologous recombination in Brevibacillus cells. The cells were spread on an MTNm agar plate (TM medium, which is 10 g/L glucose, 10 g/L phytone peptone, 5 g/L Ehrlich bonito extract, 2 g/L yeast extract, 10 mg/L FeSO4·7H2O, 10 mg/L MnSO4·4H2O, 1 mg/L ZnSO4·7H2O, pH 7.0, containing 50 μg/mL neomycin, 20 mM MgCl2, and 1.5% agar), cultured at 37 °C for 16 h and colony PCR was performed to confirm the presence of the insert DNA.
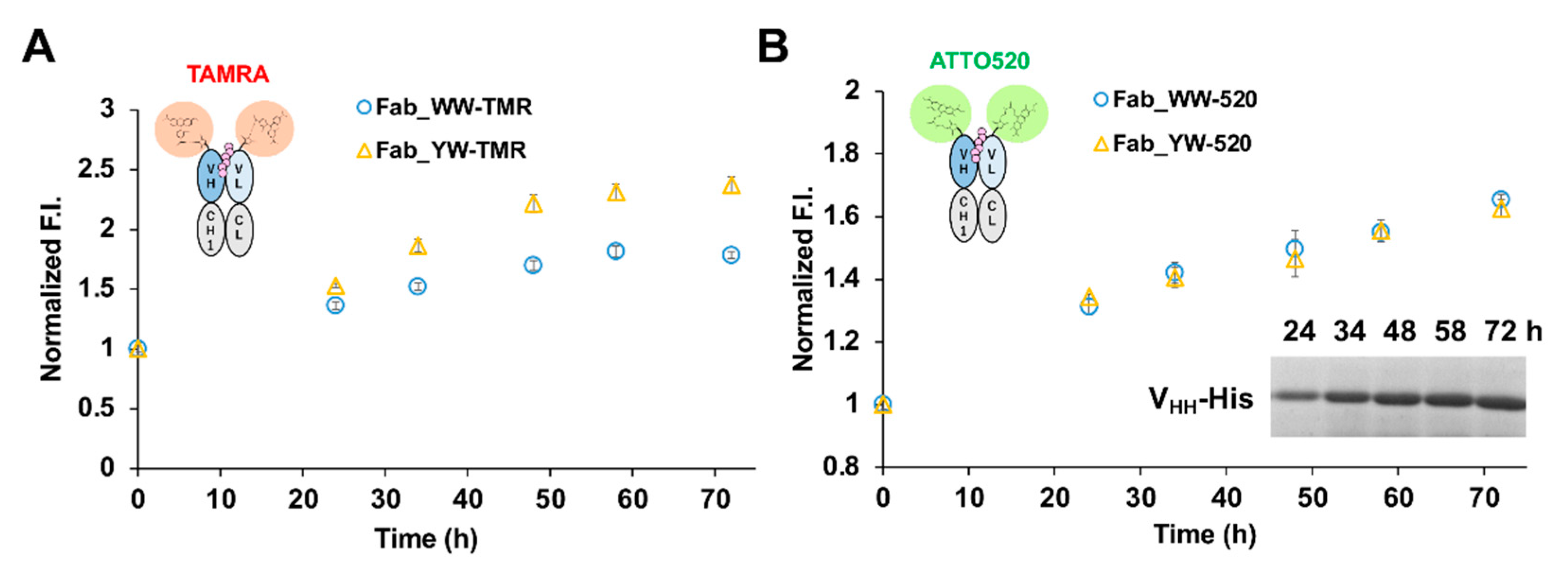
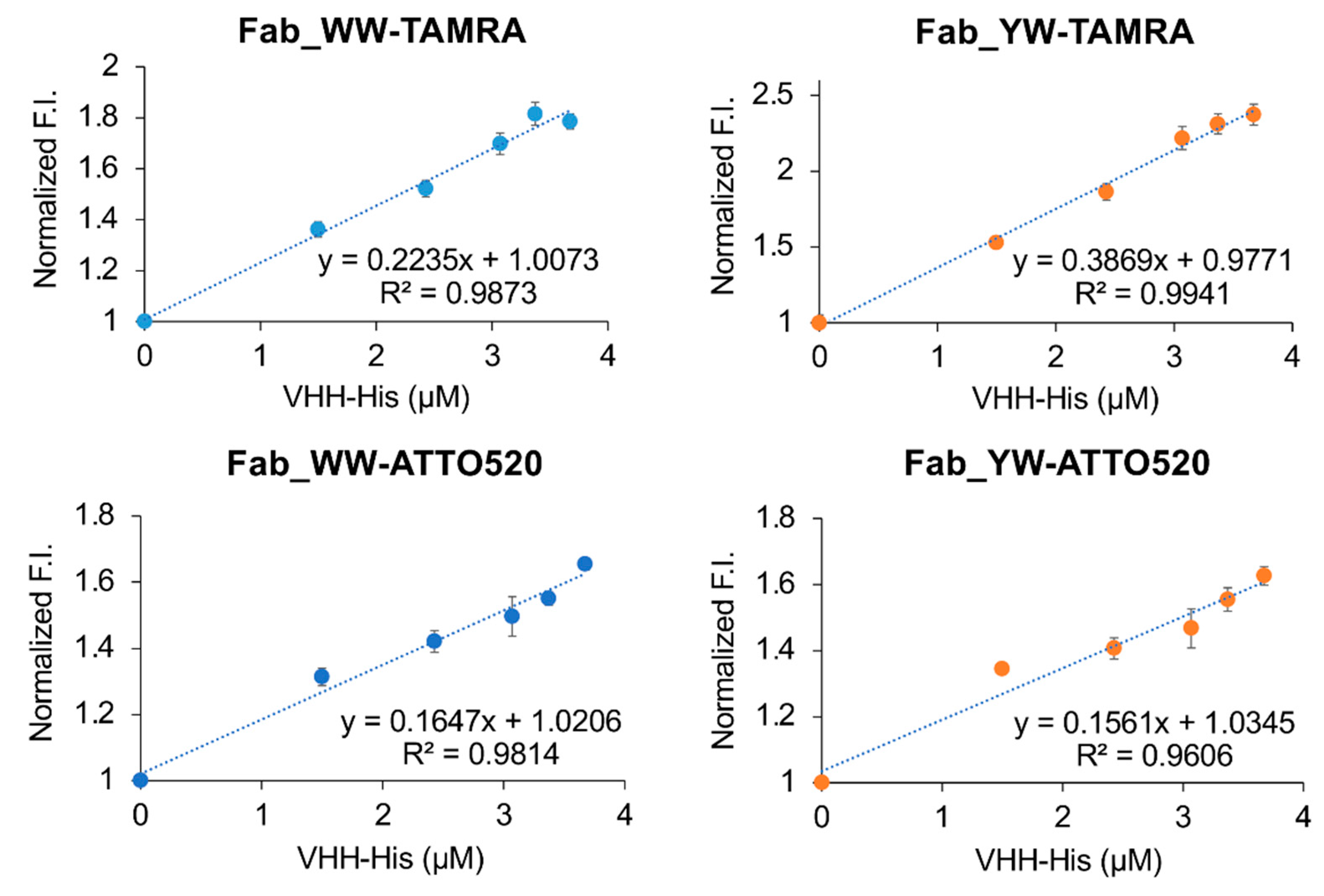
For the analysis of VHH secretion level in M9 medium (1 g/L NH4Cl, 3 g/L KH2PO4, 6 g/L Na2HPO4·7H2O, 1 mM MgSO4, 0.00005% Vitamin B1, 0.1% casamino acid, 1% glucose), a single colony was selected and pre-cultured in 4 mL TMNm (TM medium containing 50 μg/mL neomycin) medium at 30 °C, 150 rpm for 48 h. The cultured bacterial solution was centrifuged at 22 °C, 5000 rpm for 5 min. After it was washed by M9Nm (M9 medium containing 50 μg/mL neomycin) medium, the pellets were cultured in 4 mL M9Nm medium 30 °C, 150 rpm. For the analysis of VHH production by the Q-body assay, a single colony was inoculated in a 4 mL TMNm medium and cultured at 30 °C, 150 rpm for one night. The next day, 2 mL of the culture was inoculated into 100 mL TMNm medium to continue the culture at 30 °C, 150 rpm, until OD600 = 2. After the bacterial culture was centrifuged at 22 °C, 5000 rpm for 5 min, the pellets were washed with M9Nm medium and cultured in 100 mL M9Nm medium at 30 °C, 120 rpm. The culture time mentioned in this study was all counted from the exchange of M9Nm medium. The culture media at several time points were collected and centrifuged under the same condition as above. The supernatant was diluted with the same volume of PBST, and the VHH secretion level was analyzed by the Q-body assay and SDS-PAGE and Coomassie Brilliant Blue (CBB) staining.
2.9. Docking Analysis
A docking simulation based on the PDB structure 1KTR with bound ligand (His
4) was performed using CDOCKER, a grid-based molecular dynamics docking algorithm [
21], in Discovery Studio v18.1.0.1 (DS, Dassault Systèmes BIOVIA, Vélizy-Villacoublay, France). First, the “Prepare Protein” command in DS was used to automatically correct structural problems such as missing linker sequences and missing amino acid residues in the scFv. Then, the energy of the complex structure was minimized in 200 steps using the smart minimize algorithm. After that, a site sphere with a radius of 10.3 Å was built based on the ligand of the complex structure, before the His
4 ligand was removed from the model. The docking simulation was carried out using the remaining ligand. CHARMm was selected as the force field, the heating steps were set to 2000 and the heating target temperature was set to 700, the cooling steps were set to 5000 and the cooling target temperature was set to 300. The 10 generated docking poses were then ranked based on their -CDOCKER Energy and -CDOCKER Interaction Energy. -CDOCKER Energy and -CDOCKER Interaction Energy were used as indicators for the quality of molecular docking. The high positive value of these indicators presented a good interaction between the ligand and the receptor. The poses with the highest -CDOCKER Energy and -CDOCKER Interaction Energy were selected as the best poses for further analysis. 1KTR with a modified structure was mutated to minimize the energy for each mutant, and docking simulation was performed using the same procedure as for the wild-type.
4. Conclusions
In this study, we successfully constructed a fluorescent immunosensor Q-body for His-tag detection. Because of its ability to display an antigen-dependent fluorescence signal, it can also be used for the simple quantitative determination of the target antigen. The Q-body assay is simple to operate and can obtain results quickly (the fluorescence signal can be detected immediately after mixing with the antigen), therefore, it can be expected to contribute to the real-time monitoring of target recombinant products both in academic research and in practical industrial bioprocessing.
To demonstrate the merit of our method more clearly, a table that compares the His-tag detection methods reported to date was compiled (
Table 5). The comparison therein clearly shows the comparative detection range and the merits of the Q-body assay, especially in the time and the number of reagents required.
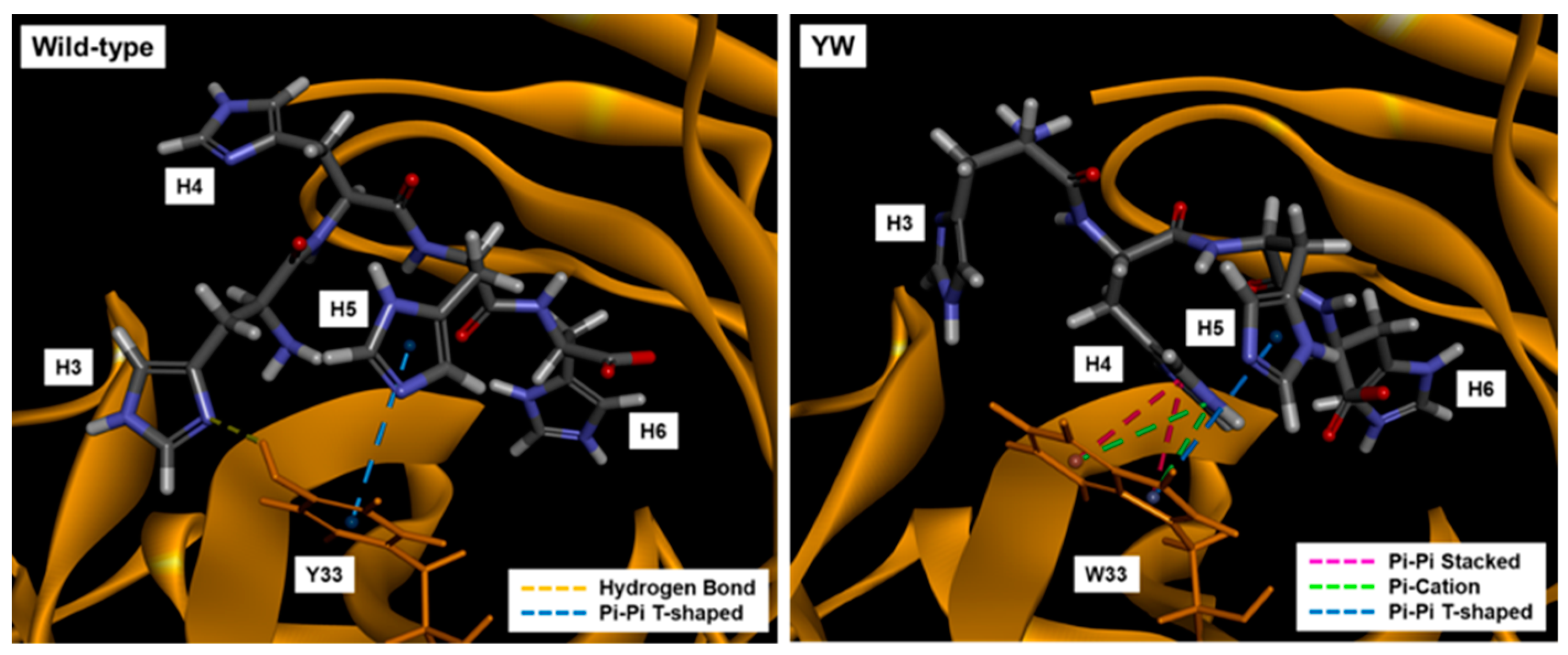
In addition, by introducing a mutation into Tyr33 in the 3D5 antibody’s VH region to increase the Trp contents, not only was improvement of the quenching effect obtained, but also higher affinity to the His6 ligand, resulting in the increased antigen detection sensitivity of the Fab-type Q-body. This approach could provide many useful ideas for designing other effective Q-bodies in the future.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}