
Electrochemical Discrimination of Salbutamol from Its Excipients in VentolinTM at Nanoporous Gold Microdisc Arrays

 and
and 
Abstract
1. Introduction
2. Materials and Methods
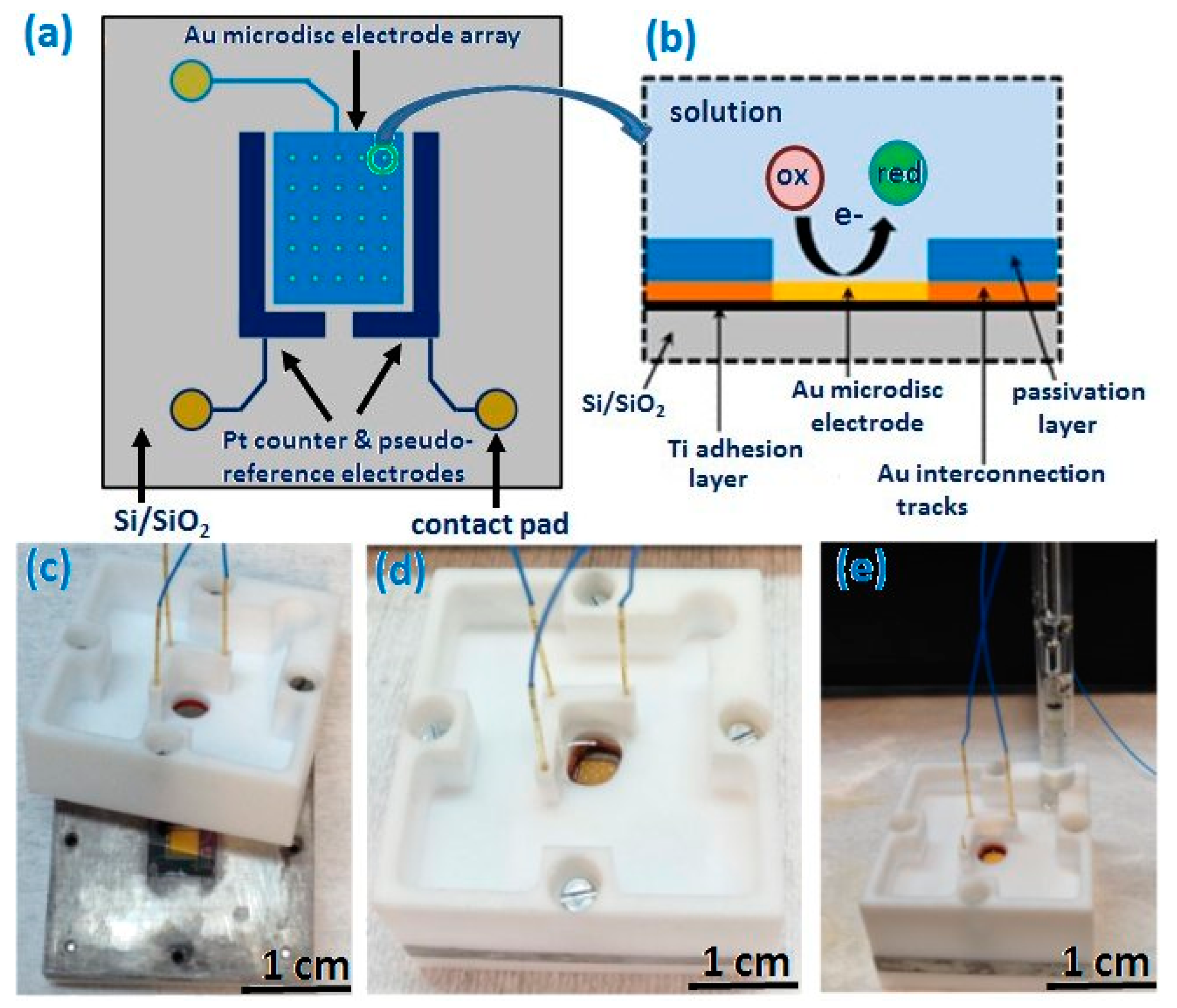
2.1. Fabrication of Recessed Gold Microdisc Electrode Arrays
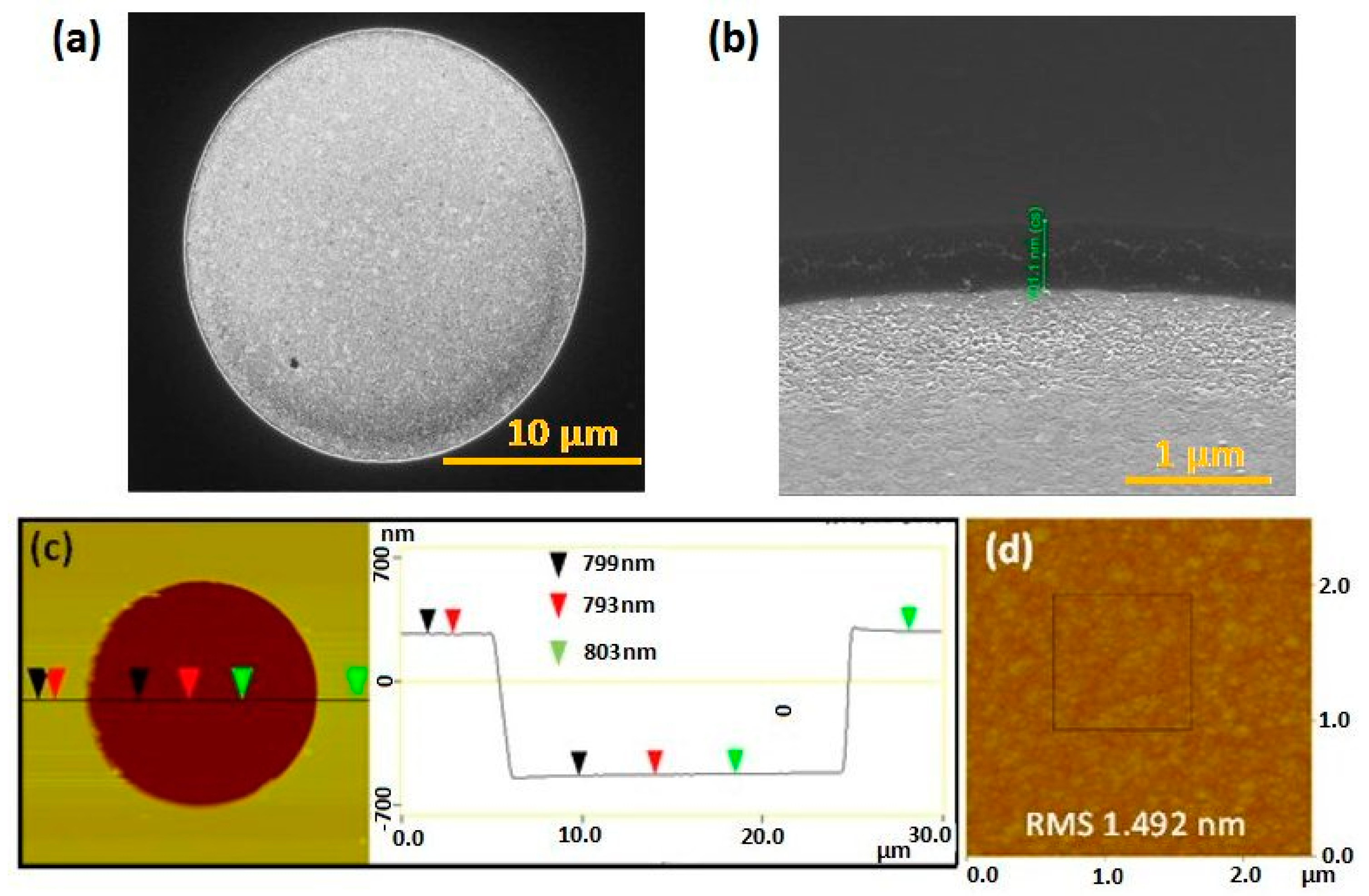
2.2. Structural and Elemental Characterisation
2.3. Three-Electrodes Electrochemical Cell
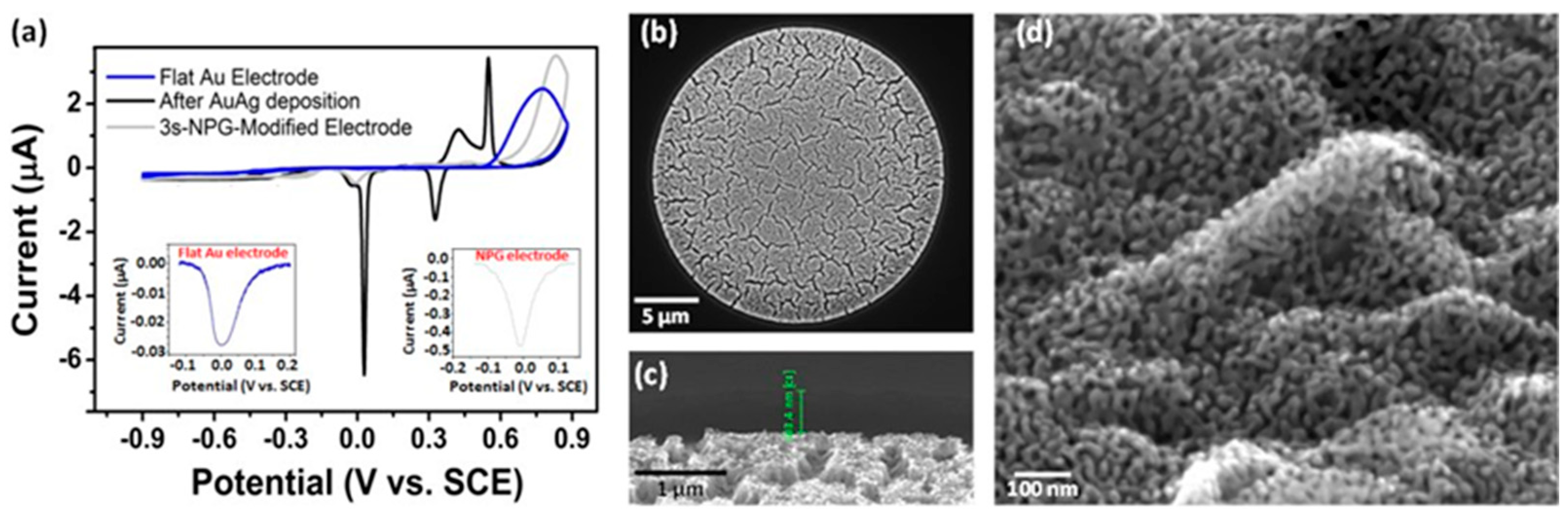
2.4. Nanoporous Gold Modification
2.5. Electrochemical Analysis
2.6. Chemicals and Glassware
3. Results and Discussion
3.1. Microscopical Characterisation of Gold Microdisc Array Electrode
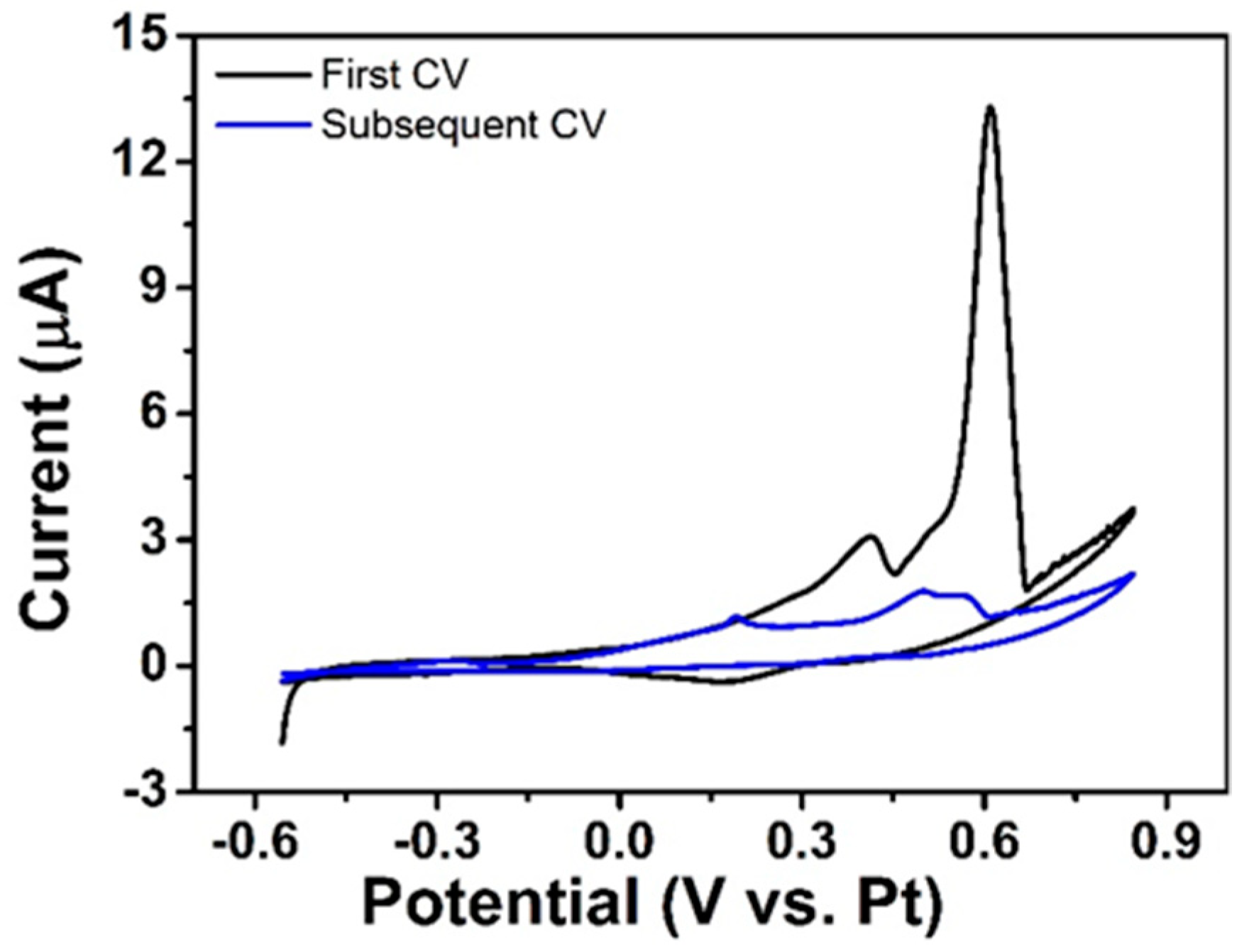
3.2. NPG Modification of Microdisc Arrays with High Reproducibility
3.3. Functionality of Microdisc Array Electrodes
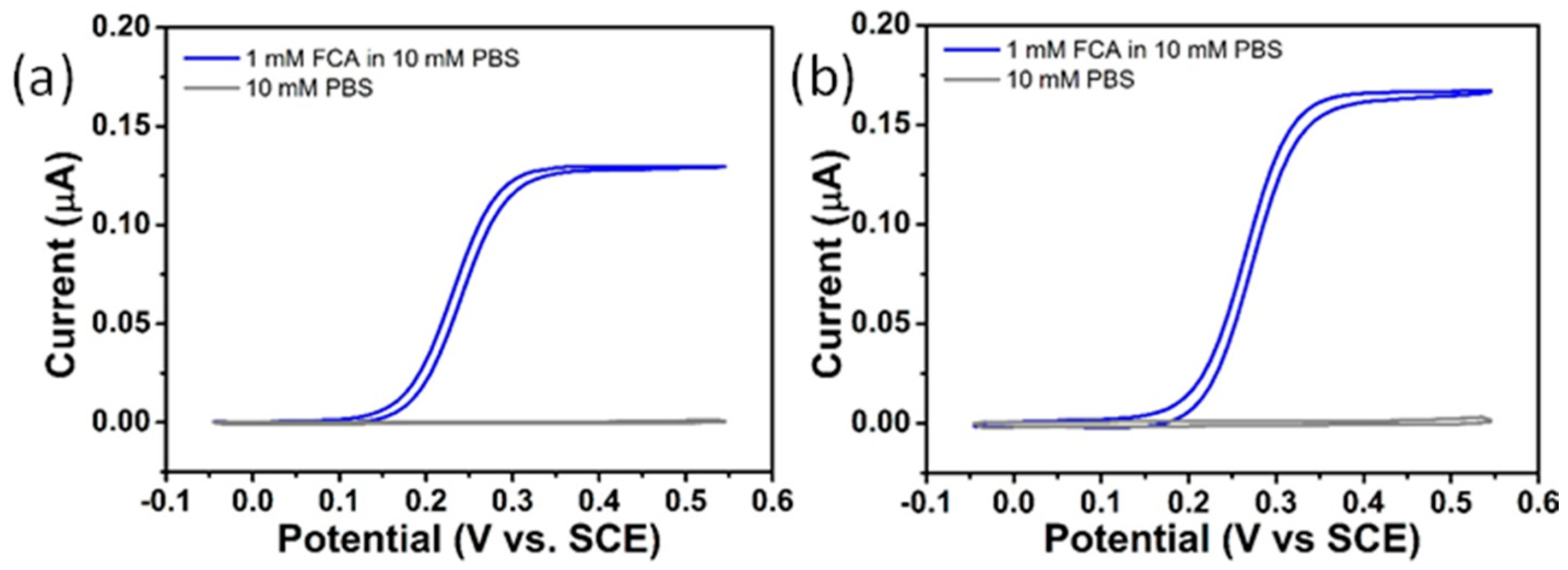
3.3.1. Bare Gold Microdisc Array Electrodes
3.3.2. NPG-Modified Gold Microdisc Array Electrodes
3.4. Detection of Salbutamol API and Chloride Excipient in VentolinTM at Bare Gold and NPG Electrodes
3.4.1. CV of VentolinTM at NPG Electrodes
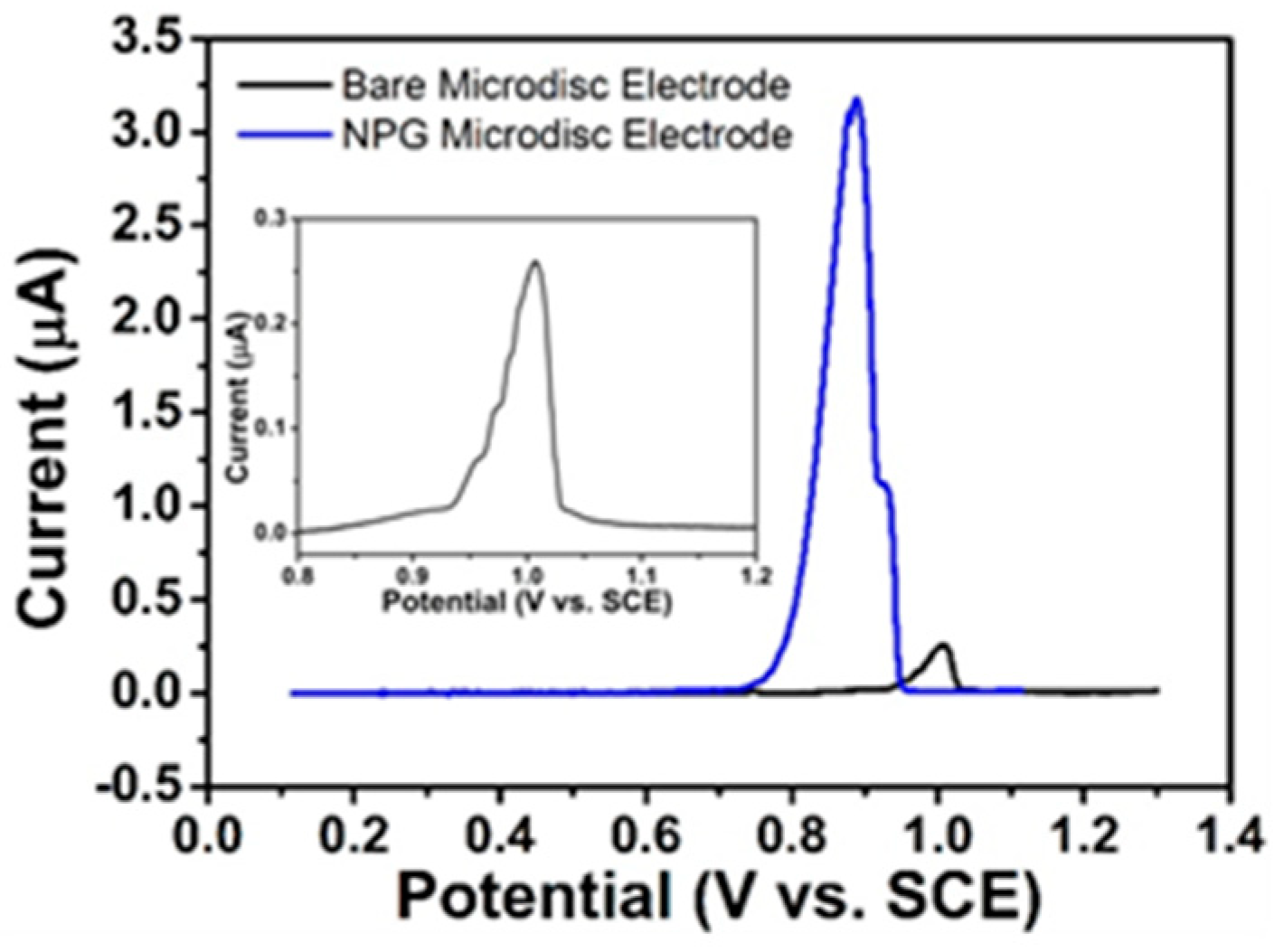
3.4.2. LSV of Excipients in VentolinTM at Bare Gold and NPG Electrodes
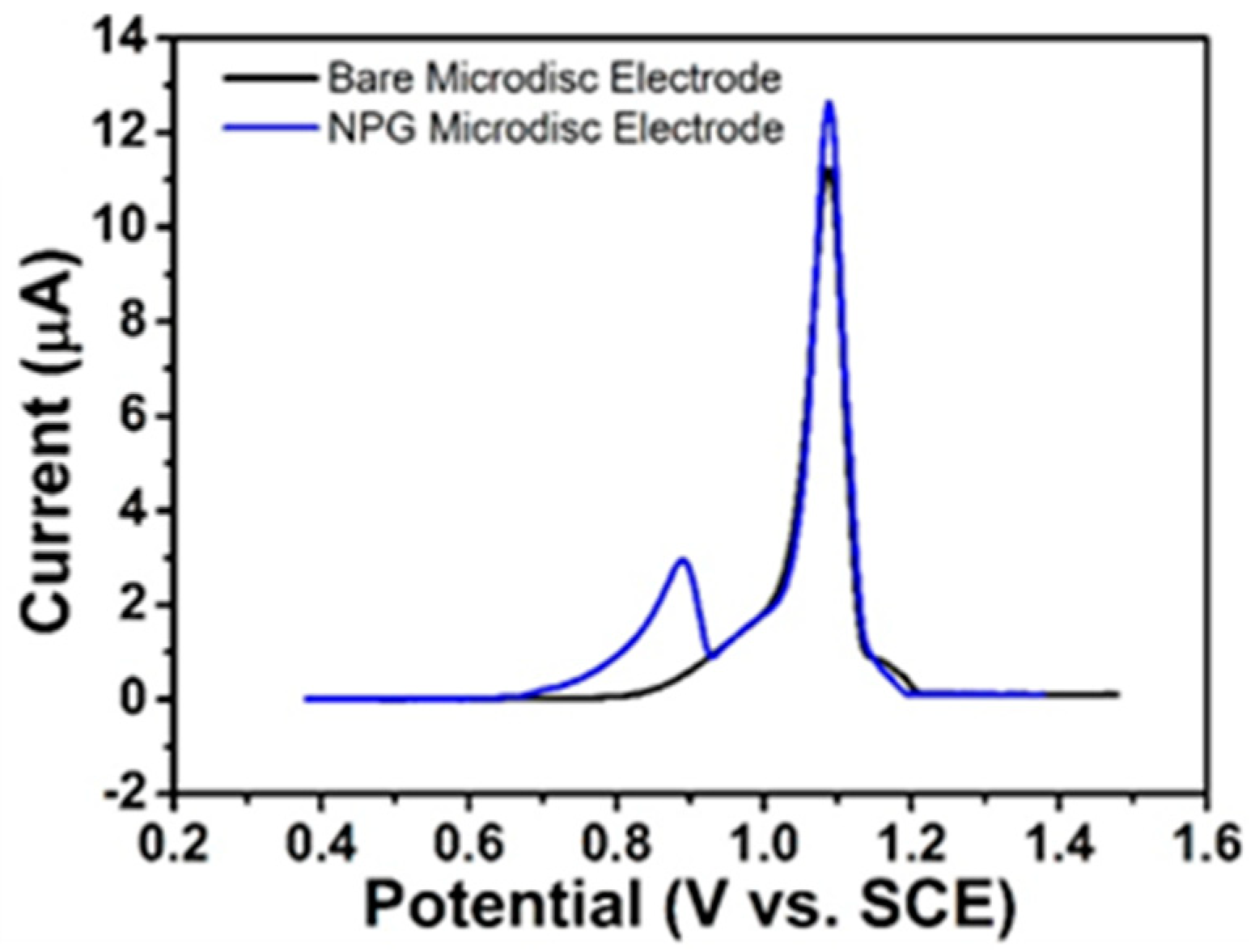
3.4.3. LSV of VentolinTM at Bare Gold and NPG Electrodes
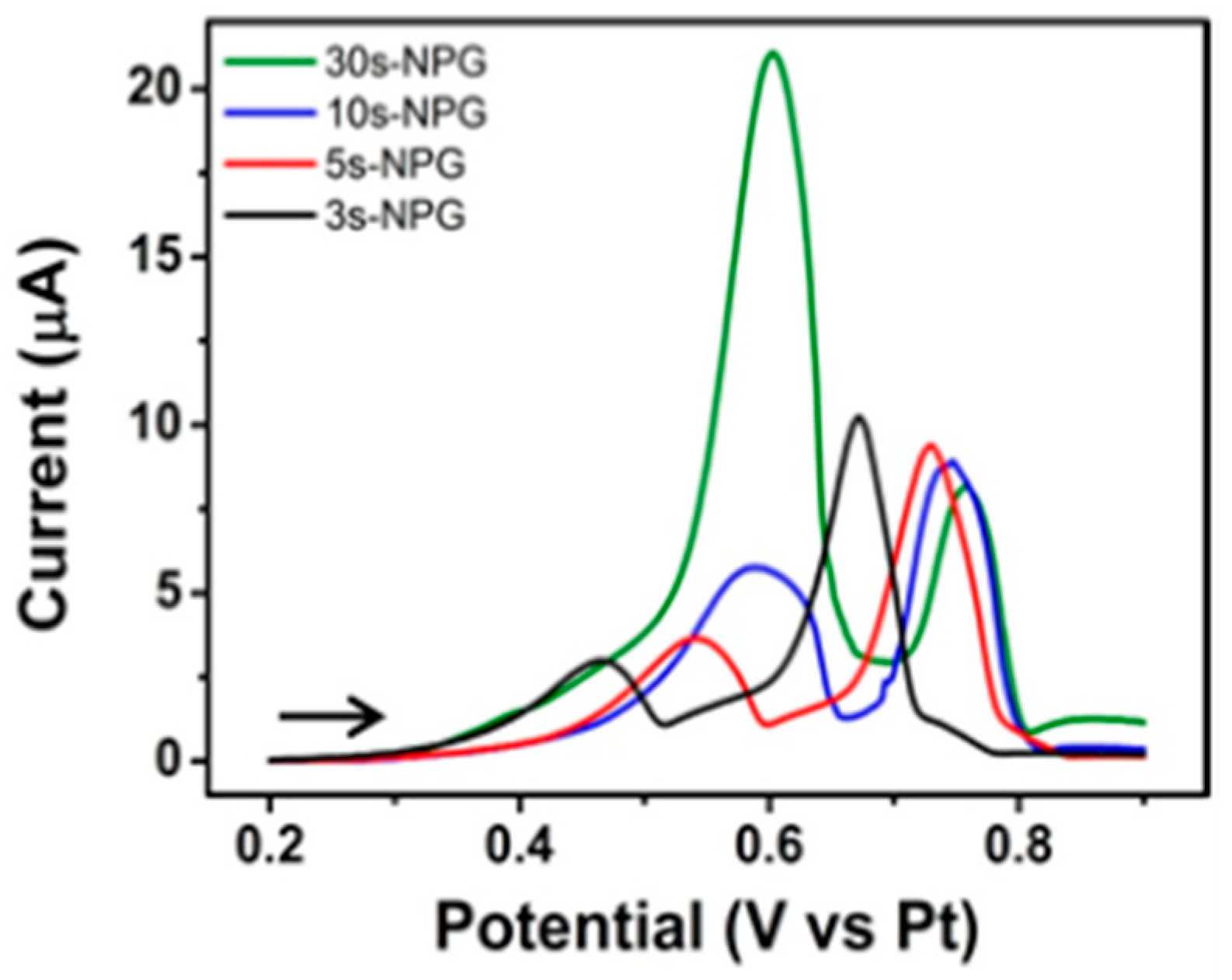
3.4.4. Selective Amplification Verified at Thicker NPG Electrodes
3.4.5. Nature of Salbutamol and Chloride Reactions at NPG Electrodes
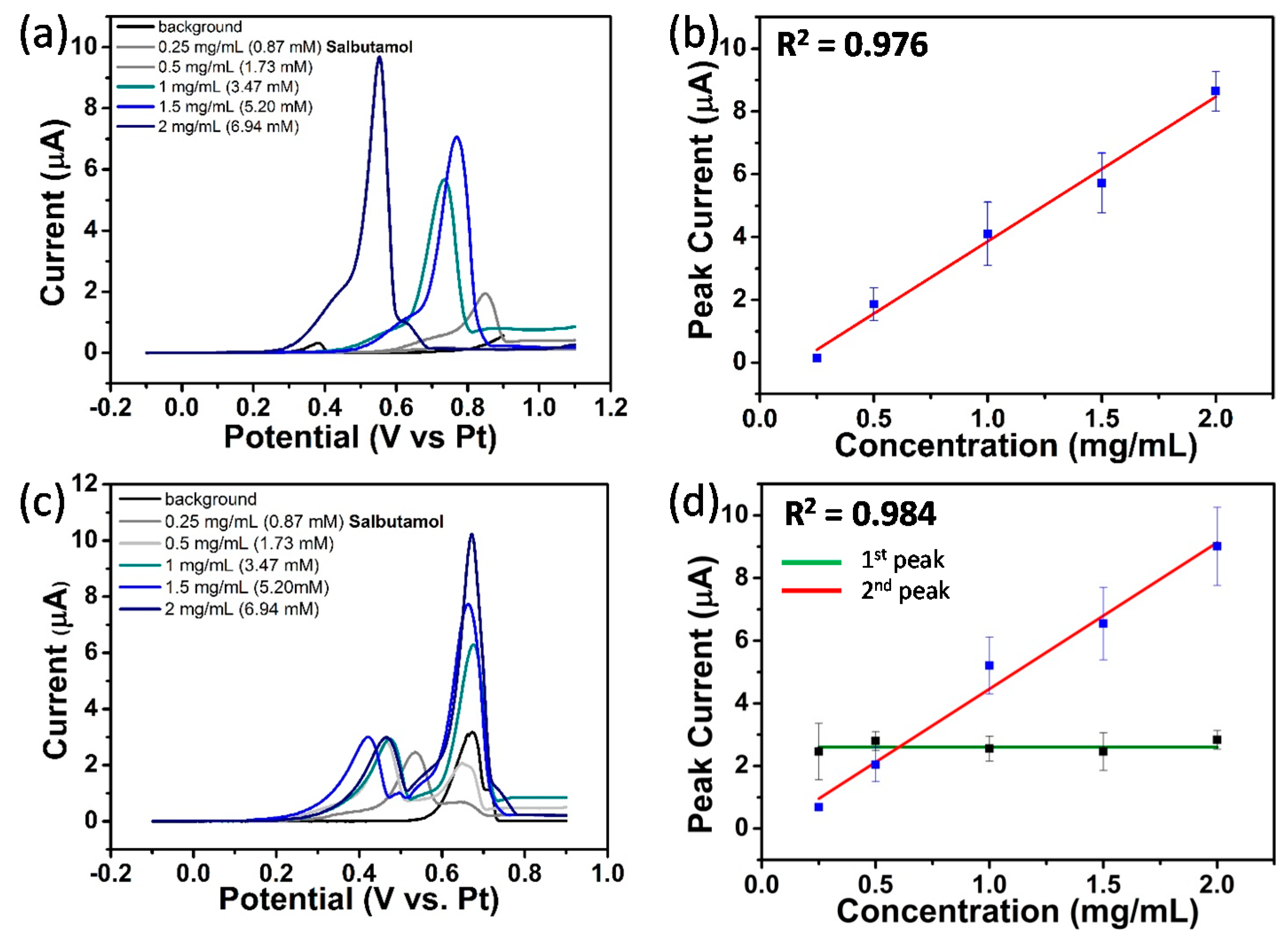
3.4.6. Quantification of Salbutamol at NPG Electrodes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Li, Y.F.; Ye, Z.; Luo, P.L.; Li, Y.; Ye, B.X. A sensitive voltammetric sensor for salbutamol based on MWNTs composite nano-Au film modified electrode. Anal. Methods 2014, 6, 61928–61935. [Google Scholar] [CrossRef]
- Caiyun, L.; Long, W. Research on determination of clenbuterol and salbutamol in pork by SPE-HPLC. Int. Conf. New Technol. Agric. 2011, 1024–1026. [Google Scholar] [CrossRef]
- Rotta, E.T.; Amantéa, S.L.; Froehlich, P.E.; Becker, A. Determination of plasma salbutamol concentrations after nebulization in a pediatric emergency department. J. Pediatr. 2007, 83, 481–484. [Google Scholar] [CrossRef]
- Fuentes, A.V.; Pineda, M.D.; Nagulapalli-Venkata, K.C. Comprehension of Top 200 Prescribed Drugs in the US as a Resource for Pharmacy Teaching, Training and Practice. Pharmacy 2018, 6, 43. [Google Scholar] [CrossRef] [PubMed]
- ISO 27427:2013. Anaesthetic and Respiratory Equipment—Nebulizing Systems and Components; ISO: Geneva, Switzerland, 2013. [Google Scholar]
- Bennett, G.; Joyce, M.; Sweeney, L.; Mac Loughlin, R. In Vitro Determination of the Main Effects in the Design of High-Flow Nasal Therapy Systems with Respect to Aerosol Performance. Pulm. Ther. 2018, 4, 73–86. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, L.; McCloskey, A.P.; Higgins, G.; Ramsey, J.M.; Cryan, S.; Mac Loughlin, R. Effective nebulization of interferon-γ using a novel vibrating mesh. Respir. Res. 2019, 20, 66–77. [Google Scholar] [CrossRef]
- Vencken, S.; Foged, C.; Ramsey, J.M.; Sweeney, L.; Cryan, S.; MacLoughlin, R.J.; Greene, C.M. Nebulised lipid–polymer hybrid nanoparticles for the delivery of a therapeutic anti-inflammatory microRNA to bronchial epithelial cells. ERJ Open Res. 2019, 5. [Google Scholar] [CrossRef] [PubMed]
- GlaxoSmithKline. Monografía de Producto: VENTOLIN® SALBUTAMOL 100mcg. 2015. Available online: https://www.3eonline.com/EeeOnlinePortal/DesktopDefault.aspx (accessed on 20 April 2021).
- Liu, Z.; Zhang, B.; Sun, J.; Yi, Y.; Li, M.; Du, D.; Zhu, F.; Luan, J. Highly efficient detection of salbutamol in environmental water samples by an enzyme immunoassay. Sci. Total Environ. 2018, 613–614, 861–865. [Google Scholar] [CrossRef]
- Palacios-Arreola, L.E.; Esquivel-Sánchez, L.P.; Balderas-Hernández, P.; Roa-Morales, G. Degradation of Salbutamol in Solution by Electrochemical Methods. ECS Trans. 2021, 101, 87–93. [Google Scholar] [CrossRef]
- Suherman, S.; Kinichi, M.; Toshikazu, K. Highly selective and sensitive detection of β-agonists using a surface plasmon resonance sensor based on an alkanethiol monolayer functionalized on a Au surface. Biosens. Bioelectron. 2015, 67, 356–363. [Google Scholar] [CrossRef]
- Dickens, G.R.; McCoy, R.A.; West, R.; Stapczynski, J.S.; Clion, G.D. Effect of Nebulized Albuterol on Serum Potassium and Cardiac Rhythm in Patients with Asthma or Chronic Obstructive Pulmonary Disease. Pharmacotherapy 1994, 14, 729–733. [Google Scholar]
- Bennett, J.A.; Smyth, E.T.; Pavord, I.D.; Wilding, P.J.; Tattersfeld, A.E. Systemic effects of salbutamol and salmeterol in patients with asthma. Thorax 1994, 49, 771–774. [Google Scholar] [CrossRef][Green Version]
- Fink, J.B.; Dailey, P.; Martin, A.R.; Kuhl, P.J.; Mac Loughlin, R.; Muellinger, B.; Corcoran, T.E.; Dhand, R. Reducing Aerosol-Related Risk of Transmission in the Era of COVID-19: An Interim Guidance Endorse by the International Society of Aerosols in Medicine. J. Aerosol Med. Pulm. Drug Deliv. 2020, 33, 1–5. [Google Scholar] [CrossRef]
- Mc Grath, J.A.; O’Sullivan, A.; Bennett, G.; O’Toole, C.; Joyce, M.; Byrne, M.A.; Mac Loughlin, R. Investigation of the Quantity of Exhaled Aerosols Released into the Environment during Nebulisation. Pharmaceutics 2019, 11, 75. [Google Scholar] [CrossRef]
- Mc Grath, J.A.; O’Toole, C.; Bennett, G.; Joyce, M.; Byrne, M.A.; Mac Loughlin, R. Investigation of Fugitive Aerosols Released into the Environment during High-Flow Therapy. Pharmaceutics 2019, 11, 254. [Google Scholar] [CrossRef]
- O’Toole, C.; Joyce, M.; Mc Grath, J.A.; O’Sullivan, A.; Byrne, M.A.; Mac Loughlin, R. Fugitive Aerosols in the intensive care unit: A narrative review. Ann. Transl. Med. 2021, 9, 592–601. [Google Scholar] [CrossRef] [PubMed]
- Joyce, M.; Mc Grath, J.A.; Mac Giolla Eain, M.; O’Sullivan, A.; Byrne, M.A.; Mac Loughlin, R. Nebuliser Type Influences Both Patient-Derived Bioaerosol Emissions and Ventilation Parameters during Mechanical Ventilation. Pharmaceutics 2021, 13, 199. [Google Scholar] [CrossRef] [PubMed]
- Frank, E.; Ishau, S.; Maier, A.; Reutman, S.; Reichard, J.F. An occupational exposure limit (OEL) approach to protect home healthcare workers exposed to common nebulized drugs. Regul. Toxicol. Pharmacol. 2019, 106, 251–261. [Google Scholar] [CrossRef] [PubMed]
- Sessink, P.J.M.; Nyulasi, T.; Haraldsson, E.L.M.; Rebic, B. Reduction of Contamination with Antibiotics on Surfaces and in Environmental Air in Three European Hospitals Following Implementation of a Closed-System Drug Transfer Device. Ann. Work Expo. Health. 2019, 63, 459–467. [Google Scholar] [CrossRef]
- Sears, M.R.J. Adverse effects of β-agonists. Allergy Clin. Immunol. 2002, 110, S322–S328. [Google Scholar] [CrossRef]
- Committee for Medicinal Products for Human Use (CHMP). Guideline on the Quality Requirements for Drug-Device 5 Combinations; European Medicines Agency: London, UK, 2019; EMA/CHMP/QWP/BWP/259165/2019. Available online: https://www.ema.europa.eu/en/documents/scientific-guideline/draft-guideline-quality-requirements-drug-device-combinations_en.pdf (accessed on 20 April 2021).
- Stanescu, O. Considerations on Regulatory Requirements for Registration of Drug-Device Combination Products in Canada and the European Union, Wissenschaftliche Prüfungsarbeit zur Erlangung des Titels. Master’s Thesis, der Mathematisch-Naturwissenschaftlchen Fakultat der Rheinischen Friedrich-Wilhelmns-Universitat Bonn, Bonn, Germany, 2020. [Google Scholar]
- Srivastava, M.; Jaiswal, P. Global Drug-Device Combination Product Market 2018. Available online: https://www.alliedmarketresearch.com/press-release/drug-device-combination-products-market.html (accessed on 20 April 2021).
- Erram, S.V.; Fanska, C.B.; Asif, M. Determination of albuterol sulfate and its related substances in albuterol sulfate inhalation solution, 0.5% by RP-HPLC. J. Pharm Biomed. Anal. 2006, 40, 864–874. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Conrado, N.; Dell’Aica, M.; León-González, M.E.D.; Pérez-Arribas, L.V.; Polo-Díez, L.M. Determination of salbutamol by direct chiral reversed-phase HPLC using teicoplanin as stationary phase and its application to natural water analysis. Biomed. Chromatogr. 2013, 27, 1413–1422. [Google Scholar] [CrossRef] [PubMed]
- Montes Nino, A.M.; Granja, R.H.M.M.; Reche, K.V.G.; Wanschel, A.C.B.A.; Salerno, A.G. Laboratory validation of an LC-MS/MS method for the detection of ractopamine, clenbuterol and salbutamol in bovine and swine muscle at sub-μg kg−1 regulatory limits. Food Addit. Contam. Part A Chem. 2017, 34, 785–792. [Google Scholar]
- Chan, S.H.; Lee, W.; Asmawi, M.Z.; Tan, S.C. Chiral liquid chromatography-mass spectrometry (LC-MS/MS) method development for the detection of salbutamol in urine samples. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2016, 1025, 83–91. [Google Scholar] [CrossRef]
- Rele, R.V. UV-spectrophotometric third order derivative method for simultaneous estimation of ambroxal hydrochloride and salbutamol sulphate by in combined pharmaceutical dosage form. Pharm. Lett. 2016, 8, 261–266. [Google Scholar]
- Rele, R.V. Simultaneous UV-spectrophotometric estimation of bromhexine hydrochloride and salbutamol sulphate by second order derivative method in combined dosage form. Res. J. Pharm Tech. 2015, 8, 702–706. [Google Scholar] [CrossRef]
- Samir, A.; Lotfy, H.M.; Salem, H.; Abdelkawy, M. Development and validation of simultaneous spectrophotometric and TLC-spectrodensitometric methods for determination of beclomethasone dipropionate and salbutamol in combined dosage form. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 128, 127–136. [Google Scholar] [CrossRef] [PubMed]
- Dong, T.; Tang, Q.; Zhao, K.; Deng, A.; Li, J. Ultrasensitive electrochemiluminescent salbutamol immunoassay with dual-signal amplification using CdSe@SiO2as label and gold nanoparticles as substrate. Microchim. Acta 2017, 184, 961–968. [Google Scholar] [CrossRef]
- Goyal, R.N.; Bishnoi, S.; Agrawal, B. Single walled carbon nanotube-modified pyrolytic graphite electrode used as a simple sensor for the determination of salbutamol in urine. Int. J. Electrochem. 2011, 2011, 373498. [Google Scholar] [CrossRef]
- Du, P. Preparation of molecular imprinting electro-biosensor for the rapid detection of salbutamol and its application in food. Mod. Food Sci. Technol. 2017, 33, 250–255. [Google Scholar]
- Guo, X.C.; Wang, H.H.; Chen, X.J.; Kang, W.Y.; Zhou, W.H. One step electrodeposition of graphene-Au nanocomposites for highly sensitive electrochemical detection of salbutamol. Int. J. Electrochem. Sci. 2017, 12, 861–875. [Google Scholar] [CrossRef]
- Attaran, A.M.; Javanbakht, M.; Fathollahi, F.; Enhessari, M. Determination of salbutamol in pharmaceutical and serum samples by adsorptive stripping voltammetry on a carbon paste electrode modified by iron titanate nanopowders. Electroanalysis 2012, 24, 2013–2020. [Google Scholar] [CrossRef]
- Li, J.; Xu, Z.; Liu, M.; Deng, P.; Tang, S.; Jiang, J.; Feng, H.; Qian, D.; He, L. Ag/N-doped reduced graphene oxide incorporated with molecularly imprinted polymer: An advanced electrochemical sensing platform for salbutamol determination. Biosens. Bioelectron. 2017, 90, 210–216. [Google Scholar] [CrossRef] [PubMed]
- Dechtrirat, D.; Sookcharoenpinyo, B.; Prajongtat, P.; Sriprachuabwong, C.; Sanguankiat, A.; Tuantranont, A.; Hannongbu, S. An electrochemical MIP sensor for selective detection of salbutamol based on a graphene/PEDOT:PSS modified screen printed carbon electrode. RSC Adv. 2018, 8, 206–212. [Google Scholar] [CrossRef]
- Talay, P.P.; Ali, H.S.; Abdullah, A.A.; Yardim, Y.; Şenturk, Z. Electroanalytical determination of salbutamol in pharmaceutical formulation.ns using cathodically pretreated boron-doped diamond electrode. Marmara Pharm. J. 2018, 22, 460–468. [Google Scholar]
- Amare, M.; Menkir, G. Differential pulse voltammetric determination of salbutamol sulfate in syrup pharmaceutical formulation using poly(4-amino-3-hydroxynaphthalene sulfonic acid) modified glassy carbon electrode. Heliyon 2017, 3, e00417. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Zhu, W.; Ma, L.; Ma, J.J.; Zhang, D.E.; Tong, Z.W.; Chen, J. Enhanced simultaneous detection of ractopamine and salbutamol Via electrochemical-facial deposition of MnO2 nanoflowers onto 3D RGO/Ni foam templates. Biosens. Bioelectron. 2016, 78, 259–266. [Google Scholar] [PubMed]
- Goyal, R.N.; Oyama, M.; Singh, S.P. Fast determination of salbutamol, abused by athletes for doping, in pharmaceuticals and human biological fluids by square wave voltammetry. J. Electroanal. Chem. 2007, 611, 140–148. [Google Scholar] [CrossRef]
- Santos, A.M.; Wong, A.; Fatibello-Filho, O. Simultaneous determination of salbutamol and propranolol in biological fluid samples using an electrochemical sensor based on functionalized graphene, ionic liquid and silver nanoparticles. J. Electroanal. Chem. 2018, 824, 1–8. [Google Scholar] [CrossRef]
- Kutluay, A.; Aslanoglu, M. Selective detection of albuterol in the presence of uric acid using a glassy carbon electrode modified with multi-walled carbon nanotubes and poly (pivalic acid). Sens. Actuators B Chem. 2013, 177, 703–709. [Google Scholar] [CrossRef]
- Mishra, R.K.; Sempionatto, J.R.; Li, Z.; Brown, C.; Galdino, N.M.; Shah, R.; Liu, S.; Hubble, L.J.; Bagot, K.; Tapert, S.; et al. Simultaneous detection of salivary Δ9-tetrahydrocannabinol and alcohol using a wearable electrochemical ring sensor. Talanta 2020, 211, 120757–120765. [Google Scholar] [CrossRef]
- Barfidokht, A.; Mishra, R.K.; Seenivasan, R.; Liu, S.; Hubble, L.J.; Wang, J.; Hall, D.A. Wearable electrochemical glove-based sensor for rapid and on-site detection of fentanyl. Sens. Actuators B Chem. 2019, 296, 126422–126428. [Google Scholar] [CrossRef] [PubMed]
- Sassa, F.; Biswas, G.C.; Suzuki, H. Microfabricated electrochemical sensing devices. Lab. A Chip 2020, 20, 1358–1389. [Google Scholar] [CrossRef]
- Wittstock, A.; Wichmann, A.; Biener, M.B.; Baumer, M. Nanoporous gold: A new gold catalyst with tunable properties. J. Faraday Discuss. 2011, 152, 87–98. [Google Scholar] [CrossRef] [PubMed]
- Bhattarai, J.K.; Neupane, D.; Nepal, B.V.; Mikhaylov, V.; Demchenko, A.V.; Stine, K.J. Structure and Applications of Gold in Nanoporous Form, Noble and Precious Metals—Properties, Nanoscale Effects and Applications. Available online: https://www.intechopen.com/books/noble-and-precious-metals-properties-nanoscale-effects-and-applications/structure-and-applications-of-gold-in-nanoporous-form (accessed on 20 December 2017).
- Pia, G.; Sogne, E.; Falqui, A. Ag surface segregation in nanoporous Au catalysts during CO oxidation. Sci. Rep. 2018, 8, 15208–15217. [Google Scholar] [CrossRef] [PubMed]
- Nur Hossain, M.; Liu, Z.G.; Wen, J.; Chen, A. Enhanced catalytic activity of nanoporous Au for the efficient electrochemical reduction of carbon dioxide. Appl. Catal. B Environ. 2018, 236, 483–489. [Google Scholar] [CrossRef]
- Wang, L.C.; Zhong, Y.; Jin, H.; Widmann, D.; Weissmüller, J.; Behm, J. Catalytic activity of nanostructured Au: Scale effects versus bimetallic/bifunctional effects in low-temperature CO oxidation on nanoporous Au. Beilstein. J. Nanotechnol. 2013, 4, 111–128. [Google Scholar] [CrossRef]
- Zhen, Q.; Biener, M.M.; Kashi, A.R.; Hunegnaw, S.; Leung, A.; Ma, S.; Huo, Z.; Kuhl, K.P.; Biener, J. Electrochemical CO2 to CO reduction at high current densities using a nanoporous gold catalyst. Mater. Res. Lett. 2021, 9, 99–104. [Google Scholar]
- Seker, E.; Reed, M.L.; Begley, M.R. Nanoporous Gold: Fabrication, Characterization and Applications. Materials 2009, 2, 2188–2215. [Google Scholar] [CrossRef]
- Biener, J.; Hodge, A.M.; Hayes, J.R.; Volkert, C.A.; Zepeda-Ruiz, L.A.; Hamza, F.F.; Abraham, A.V. Size effects on the mechanical behavior of nanoporous Au. Nano Lett. 2006, 6, 2379–2382. [Google Scholar] [CrossRef]
- Fujita, T.; Guan, P.; McKenna, K.; Lang, X.; Hirata, A.; Zhang, L.; Tokunaga, T.; Arai, S.; Yamamoto, Y.; Tanaka, N.; et al. Atomic origins of the high catalytic activity of nanoporous gold. Nat. Mater. 2012, 11, 775–780. [Google Scholar] [CrossRef]
- Lopez, N.; Janssens, T.V.W.; Clausen, B.S.; Xu, Y.; Mavrikakis, M.; Bligaard, T.J.K. Nørskov. On the origin of the catalytic activity of gold nanoparticles for low-temperature CO oxidation. J. Catal. 2004, 223, 232–235. [Google Scholar] [CrossRef]
- Welch, A.J.; Du Chene, J.S.G.; Tagliabue, A.; Davoyan, W.; Cheng, H.; Atwater, A. Nanoporous Gold as a Highly Selective and Active Carbon Dioxide Reduction Catalyst. ACS Appl. Energy Mater. 2019, 2, 164–170. [Google Scholar] [CrossRef]
- Stine, K.J.; Jefferson, K.; Shulga, O.V. Nanoporous gold for enzyme immobilization. In Enzyme Stabilization and Immobilization: Methods and Protocols; Minteer, S.D., Ed.; Springer: New York, NY, USA, 2017; pp. 37–60. ISBN 978–1-60761–895–9. [Google Scholar]
- Pandey, B.; Bhattarai, J.K.; Pornsuriyasak, P.; Fujikawa, K.; Catania, R.; Demchenko, A.; Stine, K.J. Square-wave voltammetry assays for glycoproteins on nanoporous gold. J. Electroanal. Chem. 2014, 717–718, 47–60. [Google Scholar] [CrossRef]
- van der Zalm, J. Review—Recent Advances in the Development of Nanoporous Au for Sensing Applications. J. Electrochem. Soc. 2020, 167, 037532. [Google Scholar] [CrossRef]
- Ruffino, F.; Grimaldi, M.G. Nanoporous Gold-based sensing. Coatings 2020, 10, 899. [Google Scholar] [CrossRef]
- Kumar, A.; Furtado, V.L.; Gonçalves, J.M.; Bannitz-Fernandes, R.; Eduardo, L.; Netto, S.; Araki, K.; Bertotti, M. Amperometric microsensor based on nanoporous gold for ascorbic acid detection in highly acidic biological extracts. Anal. Chim. Acta 2020, 1095, 61–70. [Google Scholar] [CrossRef] [PubMed]
- Regiart, M.; Gimenez, A.M.; Lopes, A.T.; Carreno, M.N.P.; and Bertotti, M. Ultrasensitive microfluidic electrochemical immunosensor based on electrodeposited nanoporous gold for SOX-2 determination. Anal. Chim. Acta 2020, 1127, 122–130. [Google Scholar] [CrossRef]
- Twomey, K.; Nagle, L.C.; Said, A.; Barry, F.; Ogurtsov, V.I. Characterisation of Nanoporous Gold for Use in a Dissolved Oxygen Sensing Application. BioNano Sci. 2015, 5, 55–63. [Google Scholar] [CrossRef]
- Galvin, P.; Padmanathan, N.; Razeeb, K.M.; Rohan, J.F.; Nagle, L.C.; Wahl, A.; Moore, E.; Messina, W.; Twomey, K.; Ogurtsov, V.I. Nanoenabling electrochemical sensors for life sciences applications. J. Mater. Res. 2017, 32, 2883–2904. [Google Scholar] [CrossRef]
- Ma, W.; Chang, Q.; Zhao, J.; Ye, B.C. Novel electrochemical sensing platform based on ion imprinted polymer with nanoporous gold for ultrasensitive and selective determination of As3+. Microchim. Acta 2020, 187, 571–580. [Google Scholar] [CrossRef] [PubMed]
- Gilberto, J.; Junior, S.; Selva, J.S.G.; Sukeri, A.; Gonçalves, J.M.; Regiart, M.; Bertotti, M. Fabrication of dendritic nanoporous gold via a two-step amperometric approach: Application for electrochemical detection of methyl parathion in river water samples. Talanta 2021, 226, 122130–122137. [Google Scholar]
- Xingbo, G.; Wang, L.; Liu, Z.; Ding, Y. Nanoporous Gold Leaf for Amperometric Determination of Nitrite. Electroanalysis 2011, 23, 381–386. [Google Scholar]
- Fang, C.; Bandaru, N.M.; Ellis, A.V.; Voelcker, N.H. Electrochemical fabrication of nanoporous gold. Mater. Chem. 2012, 22, 2952–2957. [Google Scholar] [CrossRef]
- Manikandan, V.S.; Liu, Z.; Chen, A. Simultaneous detection of hydrazine, sulfite, and nitrite based on a nanoporous gold microelectrode. J. Electroanal. Chem. 2018, 819, 524–532. [Google Scholar] [CrossRef]
- Mie, Y.; Takayama, H.; Hirano, Y. Facile Control of Surface Crystallographic Orientation of Anodized Nanoporous Gold Catalyst and its Application for Highly Efficient Hydrogen Evolution Reaction. J. Catal. 2020, 389, 476–482. [Google Scholar] [CrossRef]
- Wang, K.; Ding, Y. Carbon-free nanoporous gold based membrane electrocatalysts for fuel cells. Prog. Nat. Sci. Mater. Int. 2020, 30, 775–786. [Google Scholar] [CrossRef]
- Nagle, L.C.; Rohan, J.F. Rohan. Nanoporous gold anode catalyst for direct borohydride fuel cell. Int. J. Hydrogen Energy 2011, 36, 10319–10326. [Google Scholar] [CrossRef]
- Nagle, L.C.; Rohan, J.F. Nanoporous Gold Catalyst for Direct Ammonia Borane Fuel Cells. J. Electrochem. Soc. 2011, 158, B772–B778. [Google Scholar] [CrossRef]
- Zeis, R.; Mathur, A.G.; Fritz, J.; Lee, J.; Erlebacher, J. Platinum-plated nanoporous gold: An efficient, low Pt loading electrocatalyst for PEM fuel cells. Power Sources 2007, 165, 65–72. [Google Scholar] [CrossRef]
- Guo, H.; Yin, H.; Yan, X.; Shi, S.; Yu, Q.; Cao, Z.; Li, J. Pt-Bi decorated nanoporous gold for high performance direct glucose fuel cell. Sci. Rep. 2016, 6, 39162–39171. [Google Scholar] [CrossRef]
- Searson, P.; Ji, C. Synthesis and characterization of nanoporous gold nanowires. J. Phys. Chem. B 2003, 107, 4494–4503. [Google Scholar]
- Liu, Z.; Searson, P.C. Single nanoporous gold nanowire sensors. J. Phys. Chem. B 2006, 110, 4318–4322. [Google Scholar] [CrossRef] [PubMed]
- Scanlon, M.D.; Salaj-Kosla, U.; Belochapkine, S.; MacAodha, D.; Leech, D.; Ding, Y.; Magner, E. Characterization of nanoporous gold electrodes for bioelectrochemical applications. Langmuir 2012, 28, 2251–2261. [Google Scholar] [CrossRef]
- Wopschall, R.H.; Shain, I. Effects of adsorption of electroactive species in stationary electrode polarography. Anal. Chem. 1967, 39, 1514–1528. [Google Scholar]
- Fang, Z.J.; Bin, W.H.; Nakashima, T.; Rao, T.N.; Fujishima, A. Electrochemical Incineration of Organic Pollutants on Boron-Doped Diamond Electrode. J. Phys. Chem. B 2003, 107, 13389–13395. [Google Scholar]
- Shayani-Jam, H. Electrochemical study of adsorption and electrooxidation of 4,4′-biphenol on the glassy carbon electrode: Determination of the orientation of adsorbed molecules. Evidence for Direct Electrochemical Oxidation Pathway. Mon. Für Chem. Chem. Mon. 2019, 150, 183–192. [Google Scholar] [CrossRef]
- Karuwan, C.; Mantim, T.; Chaisuwan, P.; Wilairat, P.; Grudpan, K.; Jittangprasert, P.; Einaga, Y.; Chailapakul, O.; Suntornsuk, L.; Anurukvorakun, O.; et al. Pulsed Amperometry for Anti-fouling of Boron-doped Diamond in Electroanalysis of β-Agonists: Application to Flow Injection for Pharmaceutical Analysis. Sensors 2006, 6, 1837–1850. [Google Scholar] [CrossRef]
- Shi, Z.; Lipkowski, J. Chloride adsorption at the Au(111) electrode surface. J. Electroanal. Chem. 1996, 403, 225–239. [Google Scholar] [CrossRef]
- Baker, T.A.; Friend, C.M.; Kaxiras, E. Nature of Cl Bonding on the Au(111) Surface: Evidence of a Mainly Covalent Interaction. J. Am. Chem. Soc. 2008, 130, 3720–3721. [Google Scholar] [CrossRef]
- Baker, T.A.; Friend, C.M.; Kaxiras, E. Chlorine interaction with defects on the Au(111) surface: A first-principles theoretical investigation. J. Chem. Phys. 2008, 129, 104702–104707. [Google Scholar] [CrossRef]
- Kumar, A.; Selva, J.S.G.; Gonçalves, J.M.; Araki, K.; Bertotti, M. Nanoporous gold-based dopamine sensor with sensitivity boosted by interferant ascorbic acid. Electrochim. Acta 2019, 322, 134772–134782. [Google Scholar] [CrossRef]
- Mie, Y.; Katagai, S.; Ikegami, M. Electrochemical Oxidation of Monosaccharides at Nanoporous Gold with Controlled Atomic Surface Orientation and Non-Enzymatic Galactose Sensing. Sensors 2020, 20, 5632. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.N.; Zhang, H.C.; Hou, S.F.; Ma, H.Y. Highly sensitive and selective electrochemical detection of L-cysteine using nanoporous gold. Microchim. Acta 2012, 177, 427–433. [Google Scholar] [CrossRef]
- Meng, F.; Yan, X.; Liu, J.; Gu, J.; Zou, Z. Nanoporous gold as non-enzymatic sensor for hydrogen peroxide. Electrochim. Acta 2011, 56, 4657–4662. [Google Scholar]
- Han, J.-H.; Lee, E.; Park, S.; Chang, R.; Chung, T.D. Effect of Nanoporous Structure on Enhanced Electrochemical Reaction. J. Phys. Chem. C 2010, 114, 9546–9553. [Google Scholar] [CrossRef]
- Park, S.; Kim, H.C.; Dong Chung, T. Electrochemical analysis based on nanoporous structures. Analyst 2012, 137, 3891–3904. [Google Scholar] [CrossRef]
- Jeong, H.; Kim, J. Electrochemical oxidation of glucose at nanoporous black gold surfaces in the presence of high concentration of chloride ions and application to amperometric detection. Electrochim. Acta 2012, 80, 383–389. [Google Scholar] [CrossRef]
- Hsiao, M.W.; Adzic, R.R.; Yeager, E.B. Electrochemical Oxidation of Glucose on Single Crystal and Polycrystalline Gold Surfaces in Phosphate Buffer. J. Electrochem. Soc. 1996, 143, 759–767. [Google Scholar] [CrossRef]
- Xia, Y.; Huang, W.; Zheng, J.F.; Niu, Z.J.; Li, Z.L. Nonenzymatic amperometric response of glucose on a nanoporous gold film electrode fabricated by a rapid and simple electrochemical method. Biosens. Bioelectron. 2011, 26, 3555–3561. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Song, Y.Y.; Yang, C.; Xia, X.H. Hydrogen bubble dynamic template synthesis of porous gold for nonenzymatic electrochemical detection of glucose. Electrochem. Commun. 2007, 9, 981–988. [Google Scholar]
- Cho, S.; Shin, H.; Kang, C. Catalytic glucose oxidation on a polycrystalline gold electrode with an amalgamation treatment (TM 05092). Electrochim. Acta 2006, 51, 3781–3786. [Google Scholar] [CrossRef]
- Seo, B.; Choi, S.; Kim, J. Simple Electrochemical Deposition of Au Nanoplates from Au(I) Cyanide Complexes and Their Electrocatalytic Activities B. ACS Appl. Mater. Interfaces 2011, 3, 441–446. [Google Scholar] [CrossRef] [PubMed]
- Collinson, M.M. Nanoporous Gold Electrodes and Their Applications in Analytical Chemistry. ISRN Anal. Chem. 2013, 2013, 692484. [Google Scholar] [CrossRef]
- Guntupalli, B.; Liang, P.; Lee, J.-H.; Yang, Y.; Yu, H.; Canoura, J.; He, J.; Li, W.Z.; Weizmann, Y.; Xiao, Y. Ambient Filtration Method to Rapidly Prepare Highly Conductive, Paper-Based Porous Gold Films for Electrochemical Biosensing. ACS Appl. Mater. Interfaces 2015, 7, 27049–27058. [Google Scholar] [CrossRef] [PubMed]
- Aleksić, M.M.; Pantić, J.; Kapetanović, V.P. Evaluation of kinetic parameters and redox mechanism of Quinoxaline at glassy carbon electrode. Facta Univ. Ser. Phys. Chem. Technol. 2014, 12, 55–63. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Electrode | Linear Range/µM | Detection Limit | References |
|---|---|---|---|
| Single-walled carbon nanotube-modified pyrolytic graphite | 0.15–7.5 | 0.129 µM | [34] |
| Gold nanoparticle–polythionine-modified electrode | 0.2–200 | 0.6 pM | [35] |
| Graphene–Au nanocomposite | 0.05–200 | Not specified | [36] |
| Iron titanate-modified carbon paste electrode | 0.0002–0.025 | 90 pM | [37] |
| Ag/N-doped reduced graphene oxide incorporated with molecularly imprinted polymer | 0.03–20 | 7 nM | [38] |
| Graphene/PEDOT:PSS-modified screen printed carbon electrode | 0.01–1.20 | 100 pM | [39] |
| Cathodically pretreated boron-doped diamond | 17.3–347 | 5 µM | [40] |
| Poly(4-amino-3-hydroxynaphthalene sulfonic acid)-modified glassy carbon electrode | 0.2–8.0 | 0.6 pM | [41] |
| MnO2 nanoflowers-modified 3D RGO/Ni foam | 0.042–1.463 | 23 nM | [42] |
| Gold nanoparticle-modified indium tin oxide electrode | 0.15–6.0 | 0.225 µM | [43] |
| Graphene–ionic liquid silver nanoparticle composite | 0.079–2.9 | 13 nM | [44] |
| Multiwalled carbon nanotubes and poly(pivalic acid)-modified glassy carbon electrode | 0.05–70 | 12 nM | [45] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagle, L.C.; Wahl, A.; Ogourstov, V.; Seymour, I.; Barry, F.; Rohan, J.F.; Mac Loughlin, R. Electrochemical Discrimination of Salbutamol from Its Excipients in VentolinTM at Nanoporous Gold Microdisc Arrays. Sensors 2021, 21, 3975. https://doi.org/10.3390/s21123975
Nagle LC, Wahl A, Ogourstov V, Seymour I, Barry F, Rohan JF, Mac Loughlin R. Electrochemical Discrimination of Salbutamol from Its Excipients in VentolinTM at Nanoporous Gold Microdisc Arrays. Sensors. 2021; 21(12):3975. https://doi.org/10.3390/s21123975
Chicago/Turabian StyleNagle, Lorraine C., Amelie Wahl, Vladimir Ogourstov, Ian Seymour, Fiona Barry, James F. Rohan, and Ronan Mac Loughlin. 2021. "Electrochemical Discrimination of Salbutamol from Its Excipients in VentolinTM at Nanoporous Gold Microdisc Arrays" Sensors 21, no. 12: 3975. https://doi.org/10.3390/s21123975
APA StyleNagle, L. C., Wahl, A., Ogourstov, V., Seymour, I., Barry, F., Rohan, J. F., & Mac Loughlin, R. (2021). Electrochemical Discrimination of Salbutamol from Its Excipients in VentolinTM at Nanoporous Gold Microdisc Arrays. Sensors, 21(12), 3975. https://doi.org/10.3390/s21123975