Reversible Oxygen Sensing Based on Multi-Emission Fluorescence Quenching

 ,
,  , , ,
, , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Synthesis of the Fluorophores
2.3. Apparatus and Methods
2.4. Preparation of Polyvinyl Alcohol (PVA)/U-PPD16 Film
2.5. Oxygen Response Measurement of Polyvinyl Alcohol (PVA)/U-PPD16 Film
3. Results
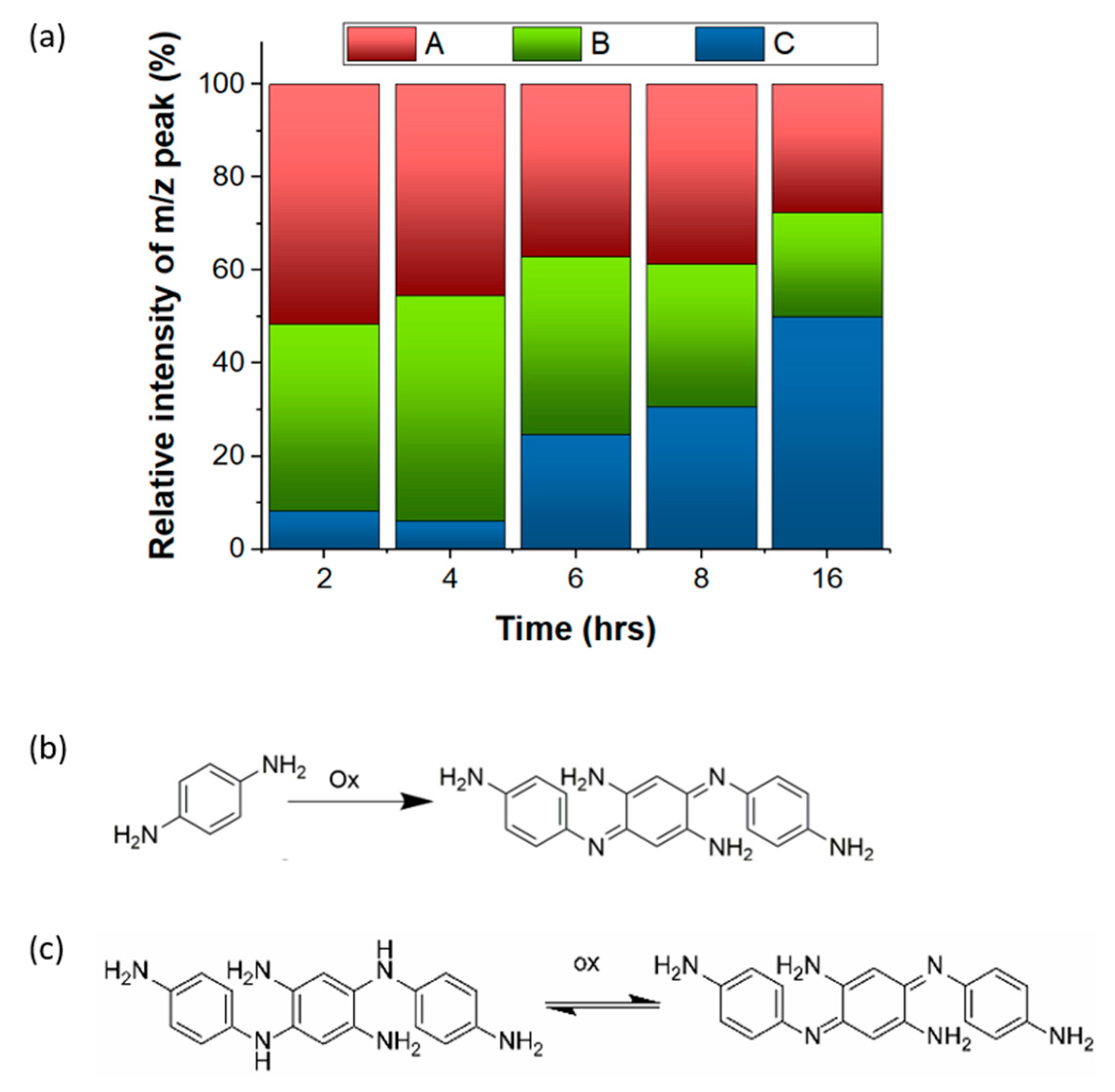
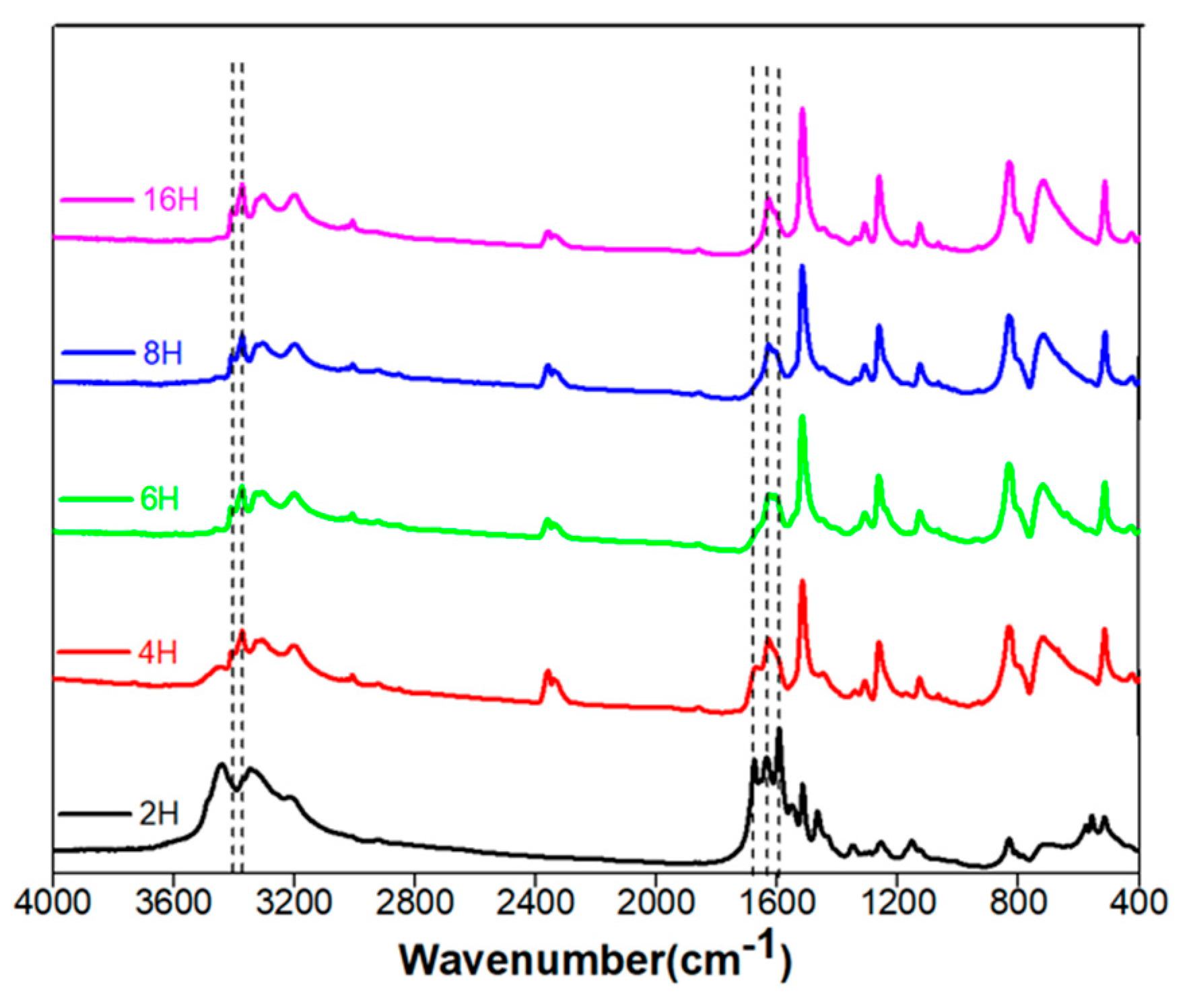
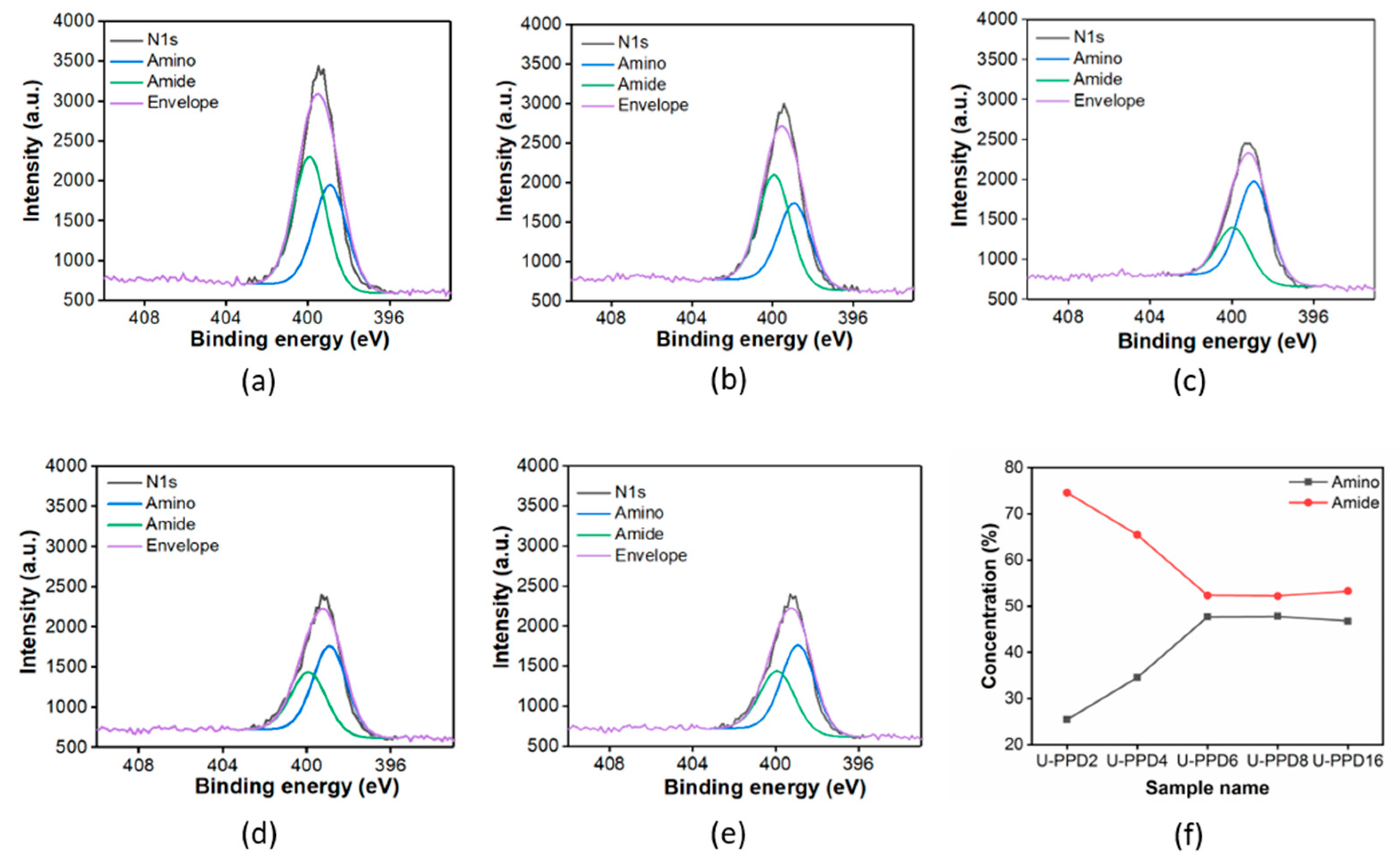
3.1. Chemical Characterization of the Multi-Emissive Optical Probes
3.2. Optical Characterization of the Multi-Emissive Pattern
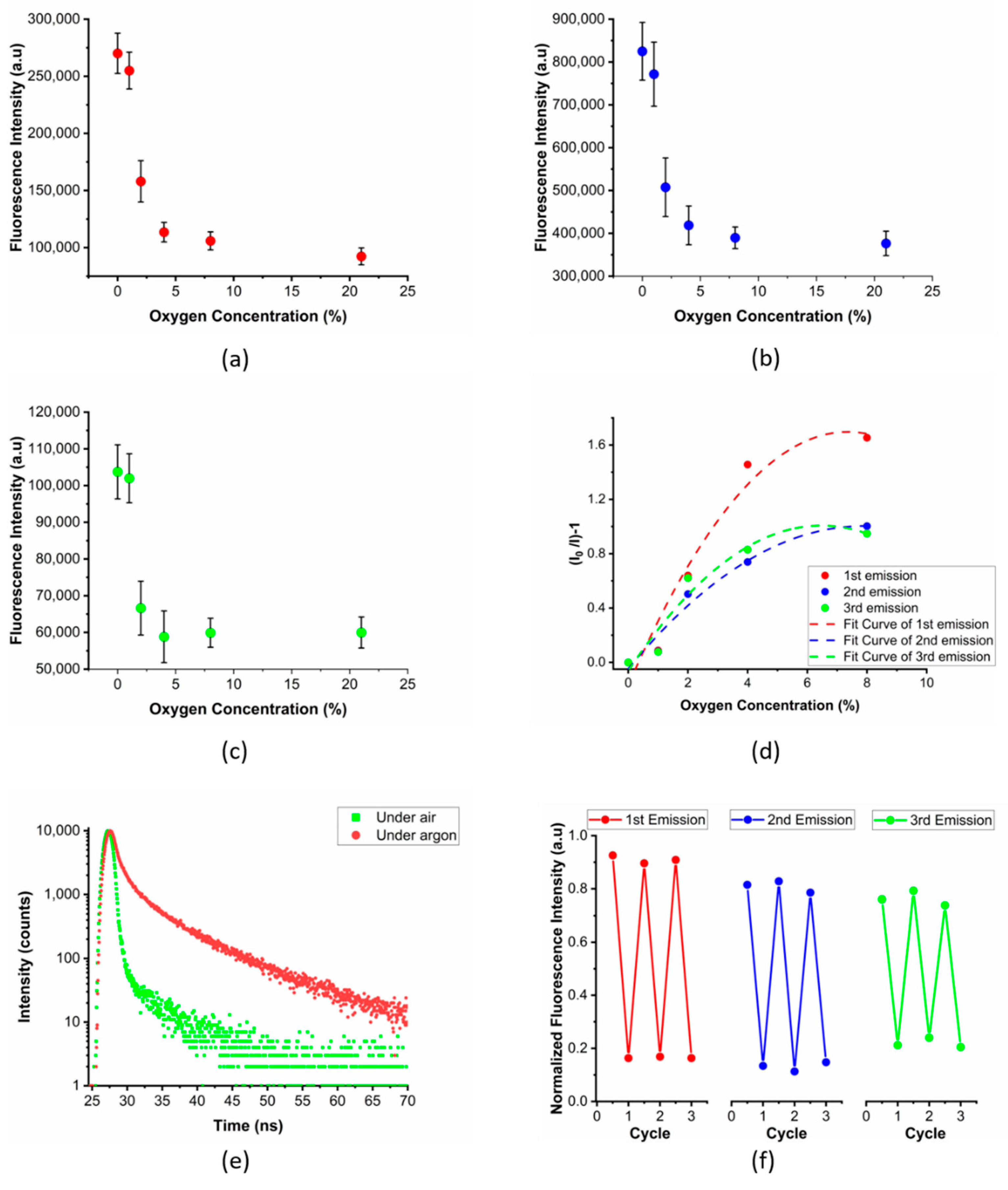
3.3. Fluorescent PVA Films and Their Multi-Emissive Oxygen Sensing Properties
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wang, X.; Wolfbeis, O.S. Optical methods for sensing and imaging oxygen: Materials, spectroscopies and applications. Chem. Soc. Rev. 2014, 43, 3666–3761. [Google Scholar] [CrossRef]
- Dmitriev, R.I.; Papkovsky, D.B. Quenched-Phosphorescence Detection of Molecular Oxygen; Detection Science; Papkovsky, D.B., Dmitriev, R.I., Eds.; The Royal Society of Chemistry: London, UK, 2018; ISBN 978-1-78801-175-4. [Google Scholar]
- Armagan, E.; Papkovsky, D.B.; Toncelli, C. Chapter 2: New Polymer-based Sensor Materials and Fabrication Technologies for Large-scale Applications. In Quenched-Phosphorescence Detection of Molecular Oxygen; The Royal Society of Chemistry: London, UK, 2018. [Google Scholar]
- Quaranta, M.; Borisov, S.M.; Klimant, I. Indicators for optical oxygen sensors. Bioanal. Rev. 2012, 4, 115–157. [Google Scholar] [CrossRef]
- Toncelli, C.; Arzhakova, O.V.; Dolgova, A.; Volynskii, A.L.; Bakeev, N.F.; Kerry, J.P.; Papkovsky, D.B. Oxygen-sensitive phosphorescent nanomaterials produced from high-density polyethylene films by local solvent-crazing. Anal. Chem. 2014, 86, 1917–1923. [Google Scholar] [CrossRef]
- Toncelli, C.; Arzhakova, O.V.; Dolgova, A.; Volynskii, A.L.; Kerry, J.P.; Papkovsky, D.B. Phosphorescent oxygen sensors produced by spot-crazing of polyphenylenesulfide films. J. Mater. Chem. C 2014, 2, 8035–8041. [Google Scholar] [CrossRef]
- Kelly, C.A.; Toncelli, C.; Kerry, J.P.; Papkovsky, D.B. Discrete O2 sensors produced by a spotting method on polyolefin fabric substrates. Sens. Actuators B Chem. 2014, 203, 935–940. [Google Scholar] [CrossRef]
- Kelly, C.A.; Toncelli, C.; Cruz-Romero, M.; Arzhakova, O.V.; Kerry, J.P.; Papkovsky, D.B. Phosphorescent O2 sensors integrated in polymeric film materials by local solvent crazing. Mater. Des. 2015, 77, 110–113. [Google Scholar] [CrossRef]
- Kwon, M.S.; Lee, D.; Seo, S.; Jung, J.; Kim, J. Tailoring Intermolecular Interactions for Efficient Room-Temperature Phosphorescence from Purely Organic Materials in Amorphous Polymer Matrices. Angew. Chem. Int. Ed. 2014, 53, 11177–11181. [Google Scholar] [CrossRef] [PubMed]
- Yuan, W.Z.; Shen, X.Y.; Zhao, H.; Lam, J.W.Y.; Tang, L.; Lu, P.; Wang, C.; Liu, Y.; Wang, Z.; Zheng, Q.; et al. Crystallization-Induced Phosphorescence of Pure Organic Luminogens at Room Temperature. J. Phys. Chem. C 2010, 114, 6090–6099. [Google Scholar] [CrossRef]
- Shimizu, M.; Shigitani, R.; Nakatani, M.; Kuwabara, K.; Miyake, Y.; Tajima, K.; Sakai, H.; Hasobe, T. Siloxy Group-Induced Highly Efficient Room Temperature Phosphorescence with Long Lifetime. J. Phys. Chem. C 2016, 120, 11631–11639. [Google Scholar] [CrossRef]
- Gong, Y.; Zhao, L.; Peng, Q.; Fan, D.; Yuan, W.Z.; Zhang, Y.; Tang, B.Z. Crystallization-induced dual emission from metal- and heavy atom-free aromatic acids and esters. Chem. Sci. 2015, 6, 4438–4444. [Google Scholar] [CrossRef]
- Yang, L.; Wang, X.; Zhang, G.; Chen, X.; Zhang, G.; Jiang, J. Aggregation-induced intersystem crossing: A novel strategy for efficient molecular phosphorescence. Nanoscale 2016, 8, 17422–17426. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Ma, X.; Wu, S.; Tian, H. A Rapidly Self-Healing Supramolecular Polymer Hydrogel with Photostimulated Room-Temperature Phosphorescence Responsiveness. Angew. Chem. Int. Ed. 2014, 53, 14149–14152. [Google Scholar] [CrossRef]
- Kwon, M.S.; Yu, Y.; Coburn, C.; Phillips, A.W.; Chung, K.; Shanker, A.; Jung, J.; Kim, G.; Pipe, K.; Forrest, S.R.; et al. Suppressing molecular motions for enhanced room-temperature phosphorescence of metal-free organic materials. Nat. Commun. 2015, 6, 8947. [Google Scholar] [CrossRef] [PubMed]
- Deng, Y.; Zhao, D.; Chen, X.; Wang, F.; Song, H.; Shen, D. Long lifetime pure organic phosphorescence based on water soluble carbon dots. Chem. Commun. (Camb.) 2013, 49, 5751–5753. [Google Scholar] [CrossRef] [PubMed]
- Tan, J.; Zhang, J.; Li, W.; Zhang, L.; Yue, D. Synthesis of amphiphilic carbon quantum dots with phosphorescence properties and their multifunctional applications. J. Mater. Chem. C 2016, 4, 10146–10153. [Google Scholar] [CrossRef]
- Tan, J.; Zou, R.; Zhang, J.; Li, W.; Zhang, L.; Yue, D. Large-scale synthesis of N-doped carbon quantum dots and their phosphorescence properties in a polyurethane matrix. Nanoscale 2016, 8, 4742–4747. [Google Scholar] [CrossRef]
- Dong, X.; Wei, L.; Su, Y.; Li, Z.; Geng, H.; Yang, C.; Zhang, Y. Efficient long lifetime room temperature phosphorescence of carbon dots in a potash alum matrix. J. Mater. Chem. C 2015, 3, 2798–2801. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, M.; Yang, Q.; Wu, Q.; Shi, J.; Gong, A.; Yang, M. Efficient room-temperature phosphorescence from nitrogen-doped carbon dots in composite matrices. Chem. Mater. 2016, 28, 8221–8227. [Google Scholar] [CrossRef]
- Tao, S.; Lu, S.; Geng, Y.; Zhu, S.; Redfern, S.A.T.; Song, Y.; Feng, T.; Xu, W.; Yang, B. Design of Metal-Free Polymer Carbon Dots: A New Class of Room-Temperature Phosphorescent Materials. Angew. Chem. 2018, 130, 2417–2422. [Google Scholar] [CrossRef]
- Lei, B.; Wang, L.; Zhang, H.; Liu, Y.; Dong, H.; Zheng, M.; Zhou, X. Luminescent carbon dots assembled SBA-15 and its oxygen sensing properties. Sens. Actuators B Chem. 2016, 230, 101–108. [Google Scholar] [CrossRef]
- Wang, L.; Zhang, H.; Zhou, X.; Liu, Y.; Lei, B. Preparation, characterization and oxygen sensing properties of luminescent carbon dots assembled mesoporous silica microspheres. J. Colloid Interface Sci. 2016, 478, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wei, J.S.; Zhang, P.; Niu, X.Q.; Zhao, W.; Zhu, Z.Y.; Ding, H.; Xiong, H.M. Red-Emissive Carbon Dots for Fingerprints Detection by Spray Method: Coffee Ring Effect and Unquenched Fluorescence in Drying Process. ACS Appl. Mater. Interfaces 2017, 9, 18429–18433. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Gao, H.; Chen, X.; Fang, G.; Yuan, S.; Yuan, Y. Excitation-Independent Dual-Color Carbon Dots: Surface-State Controlling and Solid-State Lighting. ACS Photonics 2017, 4, 2352–2358. [Google Scholar] [CrossRef]
- Jiang, K.; Zhang, L.; Lu, J.; Xu, C.; Cai, C.; Lin, H. Triple-Mode Emission of Carbon Dots: Applications for Advanced Anti-Counterfeiting. Angew. Chem. Int. Ed. 2016, 55, 7231–7235. [Google Scholar] [CrossRef]
- Cabrerizo, F.M.; Arnbjerg, J.; Denofrio, M.P.; Erra-Balsells, R.; Ogilby, P.R. Fluorescence quenching by oxygen: “Debunking” a classic rule. ChemPhysChem 2010, 11, 796–798. [Google Scholar] [CrossRef]
- Kikuchi, K.; Sato, C.; Watabe, M.; Ikeda, H.; Takahashi, Y.; Miyashi, T. New aspects of fluorescence quenching by molecular oxygen. J. Am. Chem. Soc. 1993, 115, 5180–5184. [Google Scholar] [CrossRef]
- Abdel-Shafi, A.A.; Wilkinson, F. Charge Transfer Effects on the Efficiency of Singlet Oxygen Production Following Oxygen Quenching of Excited Singlet and Triplet States of Aromatic Hydrocarbons in Acetonitrile. J. Phys. Chem. A 2000, 104, 5747–5757. [Google Scholar] [CrossRef]
- Erickson, H.P. Size and shape of protein molecules at the nanometer level determined by sedimentation, gel filtration, and electron microscopy. Biol. Proced. Online 2009, 11, 32–51. [Google Scholar] [CrossRef]
- Ding, H.; Yu, S.-B.; Wei, J.-S.; Xiong, H.-M. Full-Color Light-Emitting Carbon Dots with a Surface-State-Controlled Luminescence Mechanism. ACS Nano 2016, 10, 484–491. [Google Scholar] [CrossRef]
- Losito, I.; Palmisano, F.; Zambonin, P.G. O-Phenylenediamine Electropolymerization By Cyclic Voltammetry Combined With Electrospray Ionization-Ion Trap Mass Spectrometry. Anal. Chem. 2003, 75, 4988–4995. [Google Scholar] [CrossRef]
- Keuleers, R.; Desseyn, H.O.; Rousseau, B.; Van Alsenoy, C. Vibrational Analysis of Urea. J. Phys. Chem. A 1999, 103, 4621–4630. [Google Scholar] [CrossRef]
- Badawi, H.M.; Förner, W.; Ali, S.A. A comparative study of the infrared and Raman spectra of aniline and o-, m-, p-phenylenediamine isomers. Spectrochim. Acta Part A Mol. Biomol. Spectrosc. 2013, 112, 388–396. [Google Scholar] [CrossRef] [PubMed]
- Schaber, P.M.; Colson, J.; Higgins, S.; Thielen, D.; Anspach, B.; Brauer, J. Thermal decomposition (pyrolysis) of urea in an open reaction vessel. Thermochim. Acta 2004, 424, 131–142. [Google Scholar] [CrossRef]
- Martins, N.C.T.; Ângelo, J.; Girão, A.V.; Trindade, T.; Andrade, L.; Mendes, A. N-doped carbon quantum dots/TiO2 composite with improved photocatalytic activity. Appl. Catal. B Environ. 2016, 193, 67–74. [Google Scholar] [CrossRef]
- Chen, X.; Zhang, J.; Ma, S.; Hu, H.; Zhou, Z. Microstructure, mechanical, and tribological properties of CN xthin films prepared by reactive magnetron sputtering. Acta Metall. Sin. (Engl. Lett.) 2014, 27, 31–36. [Google Scholar] [CrossRef]
- Das, S.K.; Dickinson, C.; Lafir, F.; Brougham, D.F.; Marsili, E. Synthesis, characterization and catalytic activity of gold nanoparticles biosynthesized with Rhizopus oryzae protein extract. Green Chem. 2012, 14, 1322–1334. [Google Scholar] [CrossRef]
- Liu, J.; Zhang, T.; Wang, Z.; Dawson, G.; Chen, W. Simple pyrolysis of urea into graphitic carbon nitride with recyclable adsorption and photocatalytic activity. J. Mater. Chem. 2011, 21, 14398–14401. [Google Scholar] [CrossRef]
- Zhang, W.; Wang, S.; Ji, J.; Li, Y.; Zhang, G.; Zhang, F.; Fan, X. Primary and tertiary amines bifunctional graphene oxide for cooperative catalysis. Nanoscale 2013, 5, 6030–6033. [Google Scholar] [CrossRef]
- Wei, Y.; Yang, C.; Ding, T. Tetrahedron Leuers. Science 1996, 4039, 731–734. [Google Scholar]
- Pham, Q.L.; Haldorai, Y.; Nguyen, V.H.; Tuma, D.; Shim, J.J. Facile synthesis of poly (p-phenylenediamine)/MWCNT nanocomposites and characterization for investigation of structural effects of carbon nanotubes. Bull. Mater. Sci. 2011, 34, 37–43. [Google Scholar] [CrossRef]
- Miraqyan, N.A.; Arakelyan, R.H.; Durgaryan, N.A.; Durgaryan, A.H. Synthesis and investigation of poly(p-phenylenediamine)–poly(1,4-benzoquinonediimine-N,N-diyl-1,4-phenylene). Chem. Pap. 2018, 72, 1517–1524. [Google Scholar] [CrossRef]
- Corbett, J.F. Benzoquinone Imines. Part IV. Mechanism and Kinetics of the Formation of Bandrowski’s Base. J. Chem. Soc. B 1969, 0, 818–822. [Google Scholar] [CrossRef]
- Song, Y.; Zhu, S.; Zhang, S.; Fu, Y.; Wang, L.; Zhao, X.; Yang, B. Investigation from chemical structure to photoluminescent mechanism: A type of carbon dots from the pyrolysis of citric acid and an amine. J. Mater. Chem. C 2015, 3, 5976–5984. [Google Scholar] [CrossRef]
- He, J.; He, Y.; Chen, Y.; Zhang, X.; Hu, C.; Zhuang, J.; Lei, B.; Liu, Y. Construction and multifunctional applications of carbon dots/PVA nanofibers with phosphorescence and thermally activated delayed fluorescence. Chem. Eng. J. 2018, 347, 505–513. [Google Scholar] [CrossRef]
- Laws, W.R.; Contino, P.B. Fluorescence quenching studies: Analysis of nonlinear Stern-Volmer data. Methods Enzymol. 1992, 210, 448–463. [Google Scholar] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample Name | Reaction Atmosphere | Reaction Time (h) | Precursors | Final/Initial Mass (%) |
|---|---|---|---|---|
| U-PPD2 | Air | 2 | Urea-PPD | 71.2 |
| U-PPD4 | Air | 4 | Urea-PPD | 56.6 |
| U-PPD6 | Air | 6 | Urea-PPD | 53.8 |
| U-PPD8 | Air | 8 | Urea-PPD | 56.8 |
| U-PPD16 | Air | 16 | Urea-PPD | 57.5 |
| U16 | Air | 16 | Urea | 32.7 |
| PPD16 | Air | 16 | PPD | 51.8 |
| D-U16 | Nitrogen | 16 | Urea | N.D.a |
| D-PPD16 | Nitrogen | 16 | PPD | N.D.a |
| D-U-PPD16 | Nitrogen | 16 | Urea-PPD | N.D.a |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Armagan, E.; Thiyagarajan, S.; Wei, K.; Gursoy, A.; Fortunato, G.; Amstad, E.; Rossi, R.M.; Toncelli, C. Reversible Oxygen Sensing Based on Multi-Emission Fluorescence Quenching. Sensors 2020, 20, 477. https://doi.org/10.3390/s20020477
Armagan E, Thiyagarajan S, Wei K, Gursoy A, Fortunato G, Amstad E, Rossi RM, Toncelli C. Reversible Oxygen Sensing Based on Multi-Emission Fluorescence Quenching. Sensors. 2020; 20(2):477. https://doi.org/10.3390/s20020477
Chicago/Turabian StyleArmagan, Efe, Shankar Thiyagarajan, Kongchang Wei, Akin Gursoy, Giuseppino Fortunato, Esther Amstad, René Michel Rossi, and Claudio Toncelli. 2020. "Reversible Oxygen Sensing Based on Multi-Emission Fluorescence Quenching" Sensors 20, no. 2: 477. https://doi.org/10.3390/s20020477
APA StyleArmagan, E., Thiyagarajan, S., Wei, K., Gursoy, A., Fortunato, G., Amstad, E., Rossi, R. M., & Toncelli, C. (2020). Reversible Oxygen Sensing Based on Multi-Emission Fluorescence Quenching. Sensors, 20(2), 477. https://doi.org/10.3390/s20020477