Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network




Abstract
1. Introduction
2. SPR Principles, Methods and Analysis
2.1. SPR Principles
2.2. Immobilization Strategies
2.3. Kinetic Experiments and Data Analysis
3. Kinetics of Protein—Protein Interactions within the p53 Network by SPR
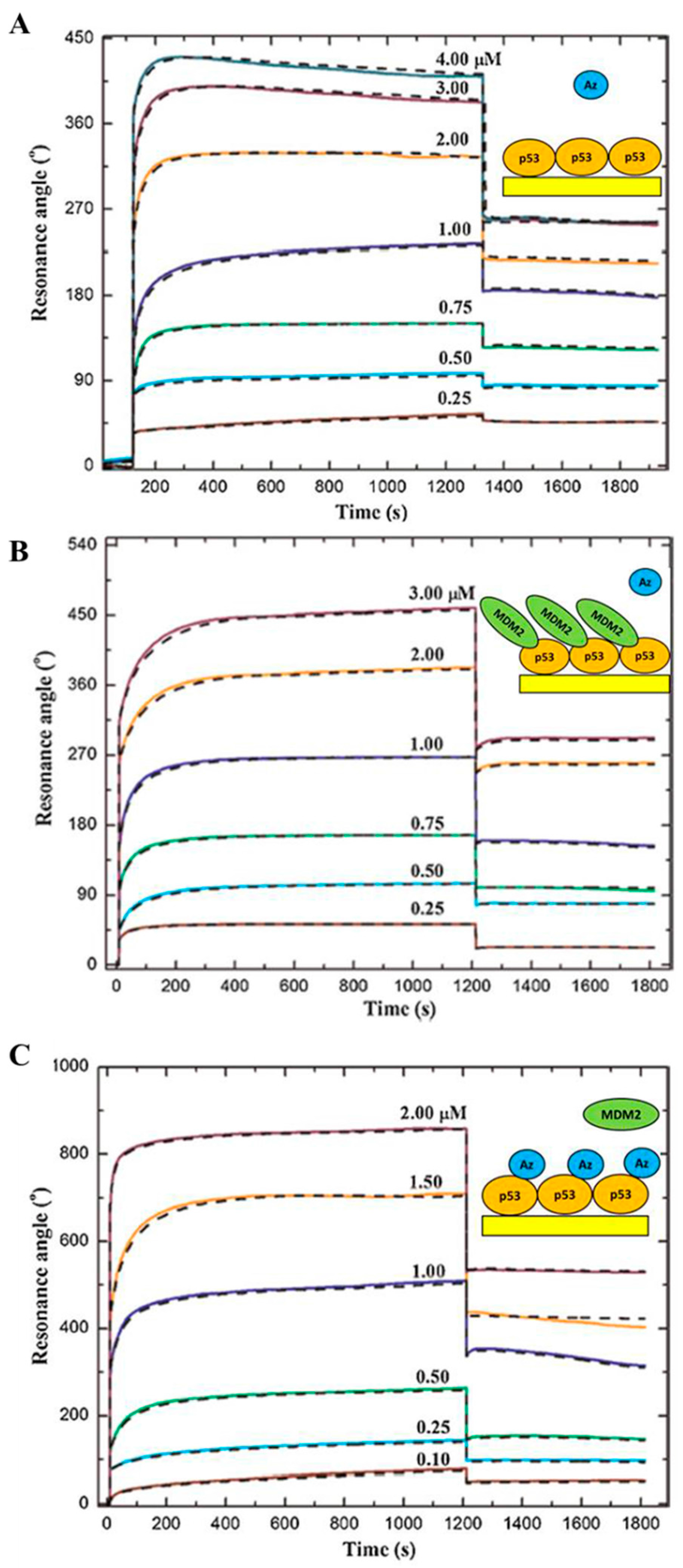
3.1. The Interaction between p53 and its Main Down-Regulator, MDM2
3.2. The Interaction between MDM2 and MDM4
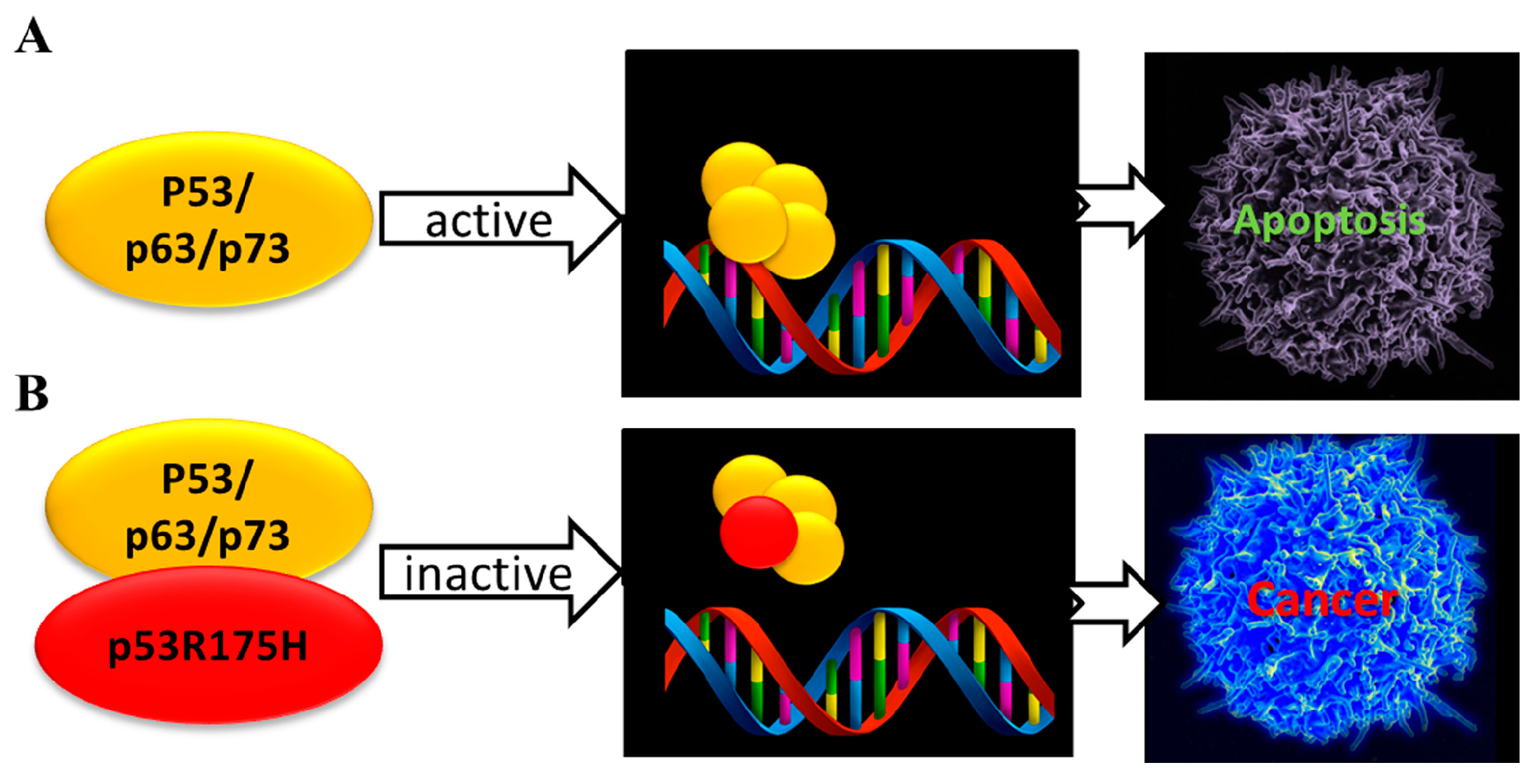
3.3. The Interaction of p53 Family Members with the Oncogenic Mutant p53R175H
3.4. The Interaction of p53 with the Anticancer Molecule Azurin and the Effect on the p53-MDM2 Binding
3.5. The Interaction of the Anticancer Drug p28 with p53
3.6. The Interaction of Mutants of DBDp53 with p28
3.7. The Interaction of p53 Family with Azurin and p28
4. Conclusions and Perspectives
Acknowledgements
Author Contributions
Conflicts of Interest
References
- Nguyen, H.H.; Park, J.; Kang, S.; Kim, M. Surface Plasmon Resonance: A Versatile Technique for Biosensor Applications. Sensors 2015, 15, 10481–10510. [Google Scholar] [CrossRef] [PubMed]
- Homola, J.; Yee, S.S.; Gauglitz, G. Surface plasmon resonance sensors: Review. Sens. Actuators B Chem. 1999, 54, 3–15. [Google Scholar] [CrossRef]
- Cooper, M.A. Label-free screening of bio-molecular interactions. Anal. Bioanal. Chem. 2003, 377, 834–842. [Google Scholar] [CrossRef] [PubMed]
- Patching, S.G. Surface plasmon resonance spectroscopy for characterisation of membrane protein—Ligand interactions and its potential for drug discovery. Biochim. Biophys. Acta 2014, 1838, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Mariani, S.; Minunni, M. Surface plasmon resonance applications in clinical analysis. Anal. Bioanal. Chem. 2014, 406, 2303–2323. [Google Scholar] [CrossRef] [PubMed]
- Ro, H.-S.; Koh, B.H.; Jung, S.O.; Park, H.K.; Shin, Y.-B.; Kim, M.-G.; Chung, B.H. Surface plasmon resonance imaging protein arrays for analysis of triple protein interactions of HPV, E6, E6AP, and p53. Proteomics 2006, 6, 2108–2111. [Google Scholar] [CrossRef] [PubMed]
- Aykul, S.; Martinez-Hackert, E. Determination of half-maximal inhibitory concentration using biosensor-based protein interaction analysis. Anal. Biochem. 2016, 508, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.; Levine, A. p53 Research: The past thirty years and the next thirty years. Cold Spring Harb. Perspect. Biol. 2010, 2, a000893. [Google Scholar] [CrossRef] [PubMed]
- Bothner, B.; Lewis, W.S.; DiGiammarino, E.L.; Weber, J.D.; Bothner, S.J.; Kriwacki, R.W. Defining the molecular basis of Arf and Hdm2 interactions. J. Mol. Biol. 2001, 314, 263–277. [Google Scholar] [CrossRef] [PubMed]
- Burch, L.R.; Midgley, C.A.; Currie, R.A.; Lane, D.P.; Hupp, T.R. Mdm2 binding to a conformationally sensitive domain on p53 can be modulated by RNA. FEBS Lett. 2000, 472, 93–98. [Google Scholar] [CrossRef]
- Chen, C.; Gorlatova, N.; Kelman, Z.; Herzberg, O. Structures of p63 DNA binding domain in complexes with half-site and with spacer-containing full response elements. Proc. Natl. Acad. Sci. USA 2011, 108, 6456–6461. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Gohain, N.; Zhan, C.; Lu, W.-Y.; Pazgier, M.; Lu, W. Structural basis of how stress-induced MDMX phosphorylation activates p53. Oncogene 2016, 35, 1919–1925. [Google Scholar] [CrossRef] [PubMed]
- Kashuba, E.; Yurchenko, M.; Yenamandra, S.P.; Snopok, B.; Szekely, L.; Bercovich, B.; Ciechanover, A.; Klein, G. Epstein-Barr virus-encoded EBNA-5 forms trimolecular protein complexes with MDM2 and p53 and inhibits the transactivating function of p53. Int. J. Cancer 2011, 128, 817–825. [Google Scholar] [CrossRef] [PubMed]
- Knappskog, S.; Lønning, P.E. Effects of the MDM2 promoter SNP285 and SNP309 on Sp1 transcription factor binding and cancer risk. Transcription 2011, 2, 207–210. [Google Scholar] [CrossRef] [PubMed]
- Lambert, B.; Buckle, M. Characterisation of the interface between nucleophosmin (NPM) and p53: Potential role in p53 stabilisation. FEBS Lett. 2006, 580, 345–350. [Google Scholar] [CrossRef] [PubMed]
- Lyakhovich, A.; Shekhar, M.P.V. Supramolecular Complex Formation between Rad6 and Proteins of the p53 Pathway during DNA Damage-Induced Response. Mol. Cell. Biol. 2003, 23, 2463–2475. [Google Scholar] [CrossRef] [PubMed]
- Qin, D.; Lee, H.; Yuan, C.; Ju, Y.; Tsai, M.-D. Identification of potential binding sites for the FHA domain of human Chk2 by in vitro binding studies. Biochem. Biophys. Res. Commun. 2003, 311, 803–808. [Google Scholar] [CrossRef] [PubMed]
- Savchenko, A.; Yurchenko, M.; Snopok, B.; Kashuba, E. Study on the spatial architecture of p53, MDM2, and p14ARF containing complexes. Mol. Biotechnol. 2009, 41, 270–277. [Google Scholar] [CrossRef] [PubMed]
- Tomita, Y.; Marchenko, N.; Erster, S.; Nemajerova, A.; Dehner, A.; Klein, C.; Pan, H.; Kessler, H.; Pancoska, P.; Moll, U.M. WT p53, but not tumor-derived mutants, bind to Bcl2 via the DNA binding domain and induce mitochondrial permeabilization. J. Biol. Chem. 2006, 281, 8600–8606. [Google Scholar] [CrossRef] [PubMed]
- Van Dieck, J.; Lum, J.K.; Teufel, D.P.; Fersht, A.R. S100 proteins interact with the N-terminal domain of MDM2. FEBS Lett. 2010, 584, 3269–3274. [Google Scholar] [CrossRef] [PubMed]
- Xia, N.; Liu, L.; Yi, X.; Wang, J. Studies of interaction of tumor suppressor p53 with apo-MT using surface plasmon resonance. Anal. Bioanal. Chem. 2009, 395, 2569–2575. [Google Scholar] [CrossRef] [PubMed]
- Domenici, F.; Frasconi, M.; Mazzei, F.; D’Orazi, G.; Bizzarri, A.R.; Cannistraro, S. Azurin modulates the association of Mdm2 with p53: SPR evidence from interaction of the full-length proteins. J. Mol. Recognit. 2011, 24, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Moscetti, I.; Teveroni, E.; Moretti, F.; Bizzarri, A.R.; Cannistraro, S. MDM2–MDM4 molecular interaction investigated by atomic force spectroscopy and surface plasmon resonance. Int. J. Nanomed. 2016, 11, 4221–4229. [Google Scholar] [CrossRef]
- Moscetti, I.; Bizzarri, A.R.; Cannistraro, S. Binding kinetics of mutant p53R175H with wild type p53 and p63: A Surface Plasmon Resonance and Atomic Force Spectroscopy study. Biophys. Chem. 2017, 228, 55–61. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Di Agostino, S.; Coppari, E.; Bizzarri, A.R.; Blandino, G.; Cannistraro, S. Interaction of mutant p53 with p73: A Surface Plasmon Resonance and Atomic Force Spectroscopy study. Biochim. Biophys. Acta 2014, 1840, 1958–1964. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Philip, P.A.; Wang, Z.; Banerjee, S.; Zafar, S.F.; Goustin, A.-S.; Almhanna, K.; Yang, D.; Wang, S.; Sarkar, F.H.; Mohammad, R.M. Reactivation of p53 by Novel MDM2 Inhibitors: Implications for Pancreatic Cancer Therapy. Curr. Cancer Drug Targets 2010, 10, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Bechill, J.; Zhong, R.; Zhang, C.; Solomaha, E.; Spiotto, M.T. A High-Throughput Cell-Based Screen Identified a 2-[(E)-2-Phenylvinyl]-8-Quinolinol Core Structure That Activates p53. PLoS ONE 2016, 11. [Google Scholar] [CrossRef] [PubMed]
- Danelius, E.; Pettersson, M.; Bred, M.; Min, J.; Waddell, M.B.; Guy, R.K.; Grøtli, M.; Erdelyi, M. Flexibility is important for inhibition of the MDM2/p53 protein-protein interaction by cyclic β-hairpins. Org. Biomol. Chem. 2016, 14, 10386–10393. [Google Scholar] [CrossRef] [PubMed]
- Fry, D.C.; Wartchow, C.; Graves, B.; Janson, C.; Lukacs, C.; Kammlott, U.; Belunis, C.; Palme, S.; Klein, C.; Vu, B. Deconstruction of a Nutlin: Dissecting the Binding Determinants of a Potent Protein–Protein Interaction Inhibitor. ACS Med. Chem. Lett. 2013, 4, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Huang, A.; Zhou, L.; Zhang, D.; Yao, J.; Zhang, Y. Molecular design and validation of halogen bonding orthogonal to hydrogen bonding in breast cancer MDM2-peptide complex. J. Mol. Graph. Model. 2016, 70, 40–44. [Google Scholar] [CrossRef] [PubMed]
- Kikuchi, J.; Furukawa, Y.; Hayashi, N. Identification of novel p53-binding proteins by biomolecular interaction analysis combined with tandem mass spectrometry. Mol. Biotechnol. 2003, 23, 203–212. [Google Scholar] [CrossRef]
- Lv, C.; Hong, Y.; Miao, L.; Li, C.; Xu, G.; Wei, S.; Wang, B.; Huang, C.; Jiao, B. Wentilactone A as a novel potential antitumor agent induces apoptosis and G2/M arrest of human lung carcinoma cells, and is mediated by HRas-GTP accumulation to excessively activate the Ras/Raf/ERK/p53-p21 pathway. Cell Death Dis. 2013, 4, e952. [Google Scholar] [CrossRef] [PubMed]
- Okuda, Y.; Nakamura, H.K.; Kuwata, K. Novel anti-cancer compounds: Structure-based discovery of chemical chaperons for p53. Oncol. Rep. 2009, 22, 739–744. [Google Scholar] [PubMed]
- Pazgier, M.; Liu, M.; Zou, G.; Yuan, W.; Li, C.; Li, C.; Li, J.; Monbo, J.; Zella, D.; Tarasov, S.G.; Lu, W. Structural basis for high-affinity peptide inhibition of p53 interactions with MDM2 and MDMX. Proc. Natl. Acad. Sci. USA 2009, 106, 4665–4670. [Google Scholar] [CrossRef] [PubMed]
- Roxburgh, P.; Hock, A.K.; Dickens, M.P.; Mezna, M.; Fischer, P.M.; Vousden, K.H. Small molecules that bind the Mdm2 RING stabilize and activate p53. Carcinogenesis 2012, 33, 791–798. [Google Scholar] [CrossRef] [PubMed]
- Smith, J.M.; Frost, J.R.; Fasan, R. Designer macrocyclic organo-peptide hybrids inhibit the interaction between p53 and HDM2/X by accommodating a functional α-helix. Chem. Commun. Camb. Engl. 2014, 50, 5027–5030. [Google Scholar] [CrossRef] [PubMed]
- Signorelli, S.; Santini, S.; Yamada, T.; Bizzarri, A.R.; Beattie, C.W.; Cannistraro, S. Binding of Amphipathic Cell Penetrating Peptide p28 to Wild Type and Mutated p53 as studied by Raman, Atomic Force and Surface Plasmon Resonance spectroscopies. Biochim. Biophys. Acta 2017, 1861, 910–921. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Cannistraro, S. The application of atomic force spectroscopy to the study of biological complexes undergoing a biorecognition process. Chem. Soc. Rev. 2010, 39, 734–749. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Cannistraro, S. Atomic Force Spectroscopy in Biological Complex Formation: Strategies and Perspectives. J. Phys. Chem. B 2009, 113, 16449–16464. [Google Scholar] [CrossRef] [PubMed]
- Hinterdorfer, P.; Dufrêne, Y.F. Detection and localization of single molecular recognition events using atomic force microscopy. Nat. Methods 2006, 3, 347–355. [Google Scholar] [CrossRef] [PubMed]
- Van Der Merwe, P.A. Surface Plasmon Resonance; Oxford University Press: Oxford, UK, 2001; ISBN 0-19-963746-6. [Google Scholar]
- Schasfoort, R.B.M. Handbook of Surface Plasmon Resonance; Royal Society of Chemistry: London, UK, 2017; ISBN 978-1-78801-139-6. [Google Scholar]
- Karlsson, R.; Katsamba, P.S.; Nordin, H.; Pol, E.; Myszka, D.G. Analyzing a kinetic titration series using affinity biosensors. Anal. Biochem. 2006, 349, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Björquist, P.; Boström, S. Determination of the kinetic constants of tissue factor/factor VII/factor VIIA and antithrombin/heparin using surface plasmon resonance. Thromb. Res. 1997, 85, 225–236. [Google Scholar] [CrossRef]
- O’Shannessy, D.J.; Brigham-Burke, M.; Soneson, K.K.; Hensley, P.; Brooks, I. Determination of rate and equilibrium binding constants for macromolecular interactions using surface plasmon resonance: Use of nonlinear least squares analysis methods. Anal. Biochem. 1993, 212, 457–468. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.W. Antigen-antibody binding and mass transport by convection and diffusion to a surface: A two-dimensional computer model of binding and dissociation kinetics. Anal. Biochem. 1993, 213, 152–161. [Google Scholar] [CrossRef] [PubMed]
- Morton, T.A.; Myszka, D.G.; Chaiken, I.M. Interpreting complex binding kinetics from optical biosensors: A comparison of analysis by linearization, the integrated rate equation, and numerical integration. Anal. Biochem. 1995, 227, 176–185. [Google Scholar] [CrossRef] [PubMed]
- Myszka, D.G.; Morton, T.A. CLAMP: A biosensor kinetic data analysis program. Trends Biochem. Sci. 1998, 23, 149–150. [Google Scholar] [CrossRef]
- Shangary, S.; Wang, S. Targeting the MDM2-p53 Interaction for Cancer Therapy. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2008, 14, 5318–5324. [Google Scholar] [CrossRef] [PubMed]
- Funari, G.; Domenici, F.; Nardinocchi, L.; Puca, R.; D’Orazi, G.; Bizzarri, A.R.; Cannistraro, S. Interaction of p53 with Mdm2 and azurin as studied by atomic force spectroscopy. J. Mol. Recognit. 2010, 23, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Zhou, J.; Qin, L.; Chen, Y.; Huang, Y.; Liu, H.; Su, Z. A Fusion Protein of the p53 Transaction Domain and the p53-Binding Domain of the Oncoprotein MdmX as an Efficient System for High-Throughput Screening of MdmX Inhibitors. Biochemistry (Moscow) 2017, 56, 3273–3282. [Google Scholar] [CrossRef] [PubMed]
- Dawson, R.; Müller, L.; Dehner, A.; Klein, C.; Kessler, H.; Buchner, J. The N-terminal domain of p53 is natively unfolded. J. Mol. Biol. 2003, 332, 1131–1141. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Jiang, X. Mdm2 and MdmX partner to regulate p53. FEBS Lett. 2012, 586, 1390–1396. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Yan, Z.; Liao, X.; Li, Y.; Yang, J.; Wang, Z.-G.; Zuo, Y.; Kawai, H.; Shadfan, M.; Ganapathy, S.; Yuan, Z.-M. The p53 inhibitors MDM2/MDMX complex is required for control of p53 activity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 12001–12006. [Google Scholar] [CrossRef] [PubMed]
- Pant, V.; Xiong, S.; Iwakuma, T.; Quintás-Cardama, A.; Lozano, G. Heterodimerization of Mdm2 and Mdm4 is critical for regulating p53 activity during embryogenesis but dispensable for p53 and Mdm2 stability. Proc. Natl. Acad. Sci. USA 2011, 108, 11995–12000. [Google Scholar] [CrossRef] [PubMed]
- Wade, M.; Li, Y.-C.; Wahl, G.M. MDM2, MDMX and p53 in oncogenesis and cancer therapy. Nat. Rev. Cancer 2013, 13, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Sharp, D.A.; Kratowicz, S.A.; Sank, M.J.; George, D.L. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J. Biol. Chem. 1999, 274, 38189–38196. [Google Scholar] [CrossRef] [PubMed]
- Tanimura, S.; Ohtsuka, S.; Mitsui, K.; Shirouzu, K.; Yoshimura, A.; Ohtsubo, M. MDM2 interacts with MDMX through their RING finger domains. FEBS Lett. 1999, 447, 5–9. [Google Scholar] [CrossRef]
- Collavin, L.; Lunardi, A.; Del Sal, G. p53-family proteins and their regulators: Hubs and spokes in tumor suppression. Cell Death Differ. 2010, 17, 901–911. [Google Scholar] [CrossRef] [PubMed]
- Deyoung, M.P.; Ellisen, L.W. p63 and p73 in human cancer: Defining the network. Oncogene 2007, 26, 5169–5183. [Google Scholar] [CrossRef] [PubMed]
- Petitjean, A.; Mathe, E.; Kato, S.; Ishioka, C.; Tavtigian, S.V.; Hainaut, P.; Olivier, M. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: Lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007, 28, 622–629. [Google Scholar] [CrossRef] [PubMed]
- Di Como, C.J.; Gaiddon, C.; Prives, C. p73 function is inhibited by tumor-derived p53 mutants in mammalian cells. Mol. Cell. Biol. 1999, 19, 1438–1449. [Google Scholar] [CrossRef] [PubMed]
- Gaiddon, C.; Lokshin, M.; Ahn, J.; Zhang, T.; Prives, C. A subset of tumor-derived mutant forms of p53 down-regulate p63 and p73 through a direct interaction with the p53 core domain. Mol. Cell. Biol. 2001, 21, 1874–1887. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Munarriz, E.; Rossi, M.; Cristofanelli, B.; Shaul, Y.; Castagnoli, L.; Levine, A.J.; Sacchi, A.; Cesareni, G.; Oren, M.; et al. Physical and functional interaction between p53 mutants and different isoforms of p73. J. Biol. Chem. 2000, 275, 29503–29512. [Google Scholar] [CrossRef] [PubMed]
- Strano, S.; Fontemaggi, G.; Costanzo, A.; Rizzo, M.G.; Monti, O.; Baccarini, A.; Del Sal, G.; Levrero, M.; Sacchi, A.; Oren, M.; et al. Physical interaction with human tumor-derived p53 mutants inhibits p63 activities. J. Biol. Chem. 2002, 277, 18817–18826. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Reumers, J.; Couceiro, J.R.; De Smet, F.; Gallardo, R.; Rudyak, S.; Cornelis, A.; Rozenski, J.; Zwolinska, A.; Marine, J.-C.; et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat. Chem. Biol. 2011, 7, 285–295. [Google Scholar] [CrossRef] [PubMed]
- Service, R.F. Rescuing the guardian of the genome. Science 2016, 354, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Kehrloesser, S.; Osterburg, C.; Tuppi, M.; Schäfer, B.; Vousden, K.H.; Dötsch, V. Intrinsic aggregation propensity of the p63 and p73 TI domains correlates with p53R175H interaction and suggests further significance of aggregation events in the p53 family. Cell Death Differ. 2016, 23, 1952–1960. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.; Fersht, A.R. Propagation of aggregated p53: Cross-reaction and coaggregation vs. seeding. Proc. Natl. Acad. Sci. USA 2015, 112, 2443–2448. [Google Scholar] [CrossRef] [PubMed]
- Billant, O.; Léon, A.; Le Guellec, S.; Friocourt, G.; Blondel, M.; Voisset, C. The dominant-negative interplay between p53, p63 and p73: A family affair. Oncotarget 2016. [Google Scholar] [CrossRef] [PubMed]
- Rajagopalan, S.; Huang, F.; Fersht, A.R. Single-Molecule characterization of oligomerization kinetics and equilibria of the tumor suppressor p53. Nucleic Acids Res. 2011, 39, 2294–2303. [Google Scholar] [CrossRef] [PubMed]
- Gaglia, G.; Guan, Y.; Shah, J.V.; Lahav, G. Activation and control of p53 tetramerization in individual living cells. Proc. Natl. Acad. Sci. USA 2013, 110, 15497–15501. [Google Scholar] [CrossRef] [PubMed]
- Punj, V.; Bhattacharyya, S.; Saint-Dic, D.; Vasu, C.; Cunningham, E.A.; Graves, J.; Yamada, T.; Constantinou, A.I.; Christov, K.; White, B.; et al. Bacterial cupredoxin azurin as an inducer of apoptosis and regression in human breast cancer. Oncogene 2004, 23, 2367–2378. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Goto, M.; Punj, V.; Zaborina, O.; Chen, M.L.; Kimbara, K.; Majumdar, D.; Cunningham, E.; Das Gupta, T.K.; Chakrabarty, A.M. Bacterial redox protein azurin, tumor suppressor protein p53, and regression of cancer. Proc. Natl. Acad. Sci. USA 2002, 99, 14098–14103. [Google Scholar] [CrossRef] [PubMed]
- Taranta, M.; Bizzarri, A.R.; Cannistraro, S. Probing the interaction between p53 and the bacterial protein azurin by single molecule force spectroscopy. J. Mol. Recognit. 2008, 21, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Gabellieri, E.; Bucciantini, M.; Stefani, M.; Cioni, P. Does azurin bind to the transactivation domain of p53? A Trp phosphorescence study. Biophys. Chem. 2011, 159, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Apiyo, D.; Wittung-Stafshede, P. Unique complex between bacterial azurin and tumor-suppressor protein p53. Biochem. Biophys. Res. Commun. 2005, 332, 965–968. [Google Scholar] [CrossRef] [PubMed]
- Kussie, P.H.; Gorina, S.; Marechal, V.; Elenbaas, B.; Moreau, J.; Levine, A.J.; Pavletich, N.P. Structure of the MDM2 Oncoprotein Bound to the P53 Tumor Suppressor Transactivation Domain. Science 1996, 948–953. [Google Scholar]
- Chi, S.-W.; Lee, S.-H.; Kim, D.-H.; Ahn, M.-J.; Kim, J.-S.; Woo, J.-Y.; Torizawa, T.; Kainosho, M.; Han, K.-H. Structural details on mdm2-p53 interaction. J. Biol. Chem. 2005, 280, 38795–38802. [Google Scholar] [CrossRef] [PubMed]
- De Grandis, V.; Bizzarri, A.R.; Cannistraro, S. Docking study and free energy simulation of the complex between p53 DNA-binding domain and azurin. J. Mol. Recognit. 2007, 20, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Di Agostino, S.; Andolfi, L.; Cannistraro, S. A combined atomic force microscopy imaging and docking study to investigate the complex between p53 DNA binding domain and Azurin. J. Mol. Recognit. 2009, 22, 506–515. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Mehta, R.R.; Lekmine, F.; Christov, K.; King, M.L.; Majumdar, D.; Shilkaitis, A.; Green, A.; Bratescu, L.; Beattie, C.W.; et al. A peptide fragment of azurin induces a p53-mediated cell cycle arrest in human breast cancer cells. Mol. Cancer Ther. 2009, 8, 2947–2958. [Google Scholar] [CrossRef] [PubMed]
- Lulla, R.R.; Goldman, S.; Yamada, T.; Beattie, C.W.; Bressler, L.; Pacini, M.; Pollack, I.F.; Fisher, P.G.; Packer, R.J.; Dunkel, I.J.; et al. Phase I trial of p28 (NSC745104), a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in pediatric patients with recurrent or progressive central nervous system tumors: A Pediatric Brain Tumor Consortium Study. Neuro-Oncology 2016, 18, 1319–1325. [Google Scholar] [CrossRef] [PubMed]
- Warso, M.A.; Richards, J.M.; Mehta, D.; Christov, K.; Schaeffer, C.; Rae Bressler, L.; Yamada, T.; Majumdar, D.; Kennedy, S.A.; Beattie, C.W.; et al. A first-in-class, first-in-human, phase I trial of p28, a non-HDM2-mediated peptide inhibitor of p53 ubiquitination in patients with advanced solid tumours. Br. J. Cancer 2013, 108, 1061–1070. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Christov, K.; Shilkaitis, A.; Bratescu, L.; Green, A.; Santini, S.; Bizzarri, A.R.; Cannistraro, S.; Gupta, T.K.D.; Beattie, C.W. p28, a first in class peptide inhibitor of cop1 binding to p53. Br. J. Cancer 2013, 108, 2495–2504. [Google Scholar] [CrossRef] [PubMed]
- Yamada, T.; Das Gupta, T.K.; Beattie, C.W. p28, an anionic cell-penetrating peptide, increases the activity of wild type and mutated p53 without altering its conformation. Mol. Pharm. 2013, 10, 3375–3383. [Google Scholar] [CrossRef] [PubMed]
- Bizzarri, A.R.; Santini, S.; Coppari, E.; Bucciantini, M.; Di Agostino, S.; Yamada, T.; Beattie, C.W.; Cannistraro, S. Interaction of an anticancer peptide fragment of azurin with p53 and its isolated domains studied by atomic force spectroscopy. Int. J. Nanomed. 2011, 6, 3011–3019. [Google Scholar] [CrossRef] [PubMed]
- Santini, S.; Bizzarri, A.R.; Cannistraro, S. Modelling the interaction between the p53 DNA-binding domain and the p28 peptide fragment of Azurin. J. Mol. Recognit. 2011, 24, 1043–1055. [Google Scholar] [CrossRef] [PubMed]
- Coppari, E.; Yamada, T.; Bizzarri, A.R.; Beattie, C.W.; Cannistraro, S. A nanotechnological, molecular-modeling, and immunological approach to study the interaction of the anti-tumorigenic peptide p28 with the p53 family of proteins. Int. J. Nanomed. 2014, 9, 1799–1813. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein-Protein Interaction | kon (M−1 s−1) | koff (s−1) | KD (M) | τ (s) | Ref. |
|---|---|---|---|---|---|
| MDM2/p53 | (0.8 ± 0.3) × 106 | (2.1 ± 0.2) | (0.4 ± 0.1) × 10−6 | (0.5 ± 0.1) | [22] |
| MDM2/MDM4 | (4.3 ± 0.3) × 103 | (1.7 ± 0.7) × 10−3 | (3.9 ± 0.1) × 10−7 | (6 ± 2) × 102 | [23], unp |
| p53R175H/p53 | (1.28 ± 0.04) × 104 | (4.6 ± 0.4) × 10−5 | (3.6 ± 0.3) × 10−9 | (2.17 ± 0.02) × 104 | [24] |
| p53R175H/p63 | (1.52 ± 0.04) × 104 | (5.3 ± 0.4) × 10−5 | (3.5 ± 0.3) × 10−9 | (1.89 ± 0.01) × 104 | [24] |
| p53R175H/p73 | (6.4 ± 0.5) × 103 | (3.1 ± 1.8) × 10−3 | (4.9 ± 0.6) × 10−7 | (3.2 ± 1.9) × 102 | [25] |
| Az/p53 | (8.2 ± 0.5) × 104 | (9.4 ± 0.7) × 10−2 | (1.2 ± 0.1) × 10−6 | (1.0 ± 0.1) × 10 | [22] |
| Az/(p53-MDM2) | (6.8 ± 0.5) × 104 | (9.0 ± 0.3) × 10−2 | (1.3 ± 0.1) × 10−6 | (1.0 ± 0.1) × 10 | [22] |
| MDM2/(p53-Az) | (5.0 ± 0.3) × 105 | (0.6 ± 0.2) | (1.2 ± 0.4) × 10−6 | (1.60 ± 0.02) | [22] |
| p28/p53 | (2.4 ± 0.4) × 102 | (3.0 ± 0.1) × 10−3 | (1.2 ± 0.1) × 10−5 | (3.3 ± 0.3) × 102 | unp. |
| p28/CTDp53 | - | - | N.I. | - | unp. |
| p28/DBDp53 | (2.4 ± 0.5) × 102 | (2.0 ± 0.1) × 10−5 | (8.8 ± 1.9) × 10−8 | (5.0 ± 0.3) × 104 | [37], unp. |
| p28/DBDp53-K164E | - | - | 7.3 × 10−8 | - | [37] |
| p28/DBDp53-R273H | - | - | N.I. | - | [37] |
| p28/DBDp53-P223L/V274F | - | - | 2.4 × 10−4 | - | [37] |
| Az/DBDp63 | (4.6 ± 0.5) × 102 | (4.2 ± 0.3) × 10−4 | (8.2 ± 0.3) × 10−6 | (2.4 ± 0.1) × 103 | unp. |
| p28/DBDp63 | (9.2 ± 0.7) | (3.7 ± 0.3) × 10−5 | (4.2 ± 0.4) × 10−6 | (2.7 ± 0.2) × 104 | unp. |
| p28/p73 | (9.3 ± 0.5) × 104 | (1.4 ± 0.1) × 10−3 | (1.5 ± 0.4) × 10−8 | (7.1 ± 0.5) × 102 | unp. |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moscetti, I.; Cannistraro, S.; Bizzarri, A.R. Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network. Sensors 2017, 17, 2680. https://doi.org/10.3390/s17112680
Moscetti I, Cannistraro S, Bizzarri AR. Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network. Sensors. 2017; 17(11):2680. https://doi.org/10.3390/s17112680
Chicago/Turabian StyleMoscetti, Ilaria, Salvatore Cannistraro, and Anna Rita Bizzarri. 2017. "Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network" Sensors 17, no. 11: 2680. https://doi.org/10.3390/s17112680
APA StyleMoscetti, I., Cannistraro, S., & Bizzarri, A. R. (2017). Surface Plasmon Resonance Sensing of Biorecognition Interactions within the Tumor Suppressor p53 Network. Sensors, 17(11), 2680. https://doi.org/10.3390/s17112680