
Hydrophobin-Based Surface Engineering for Sensitive and Robust Quantification of Yeast Pheromones

Abstract
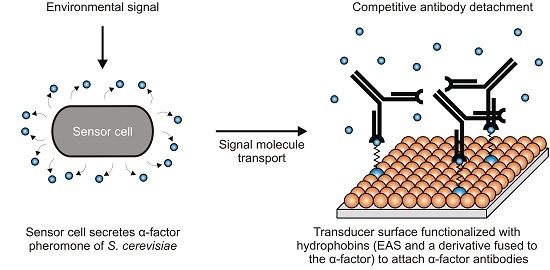
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
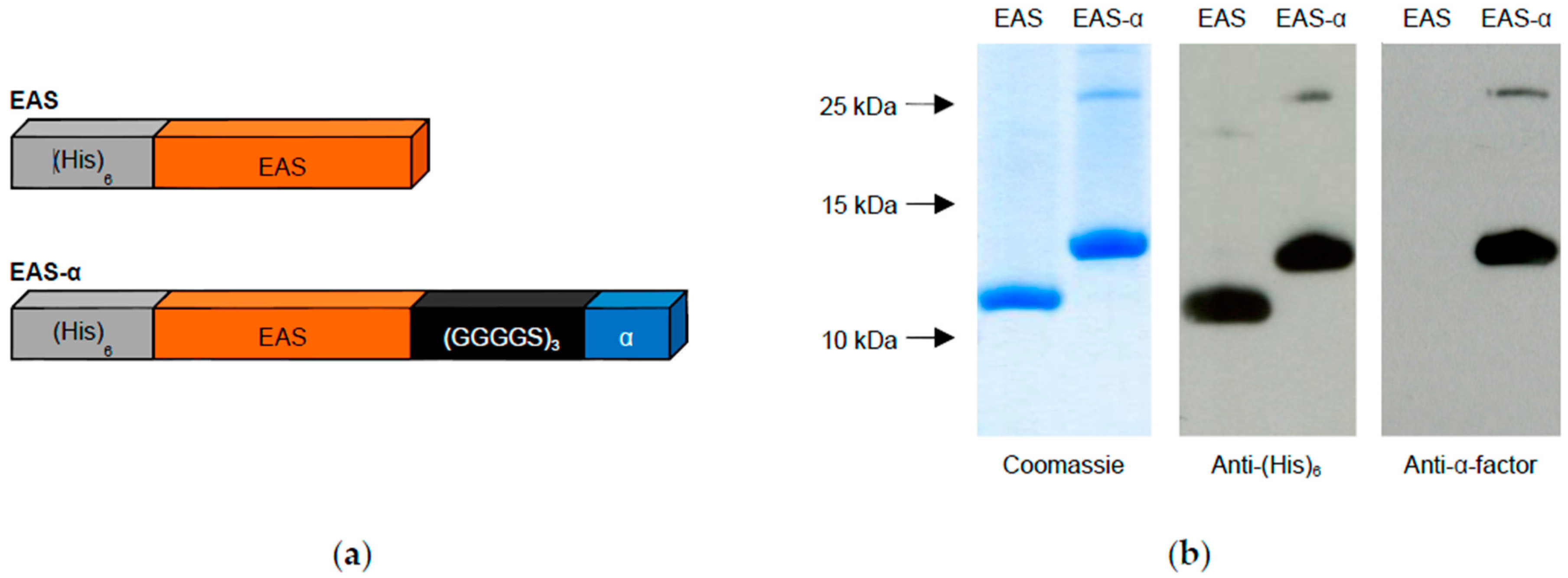
2.1. Molecular Cloning of the Hydrophobin Genes EAS and EAS-α
2.2. Expression and Purification of Hydrophobins
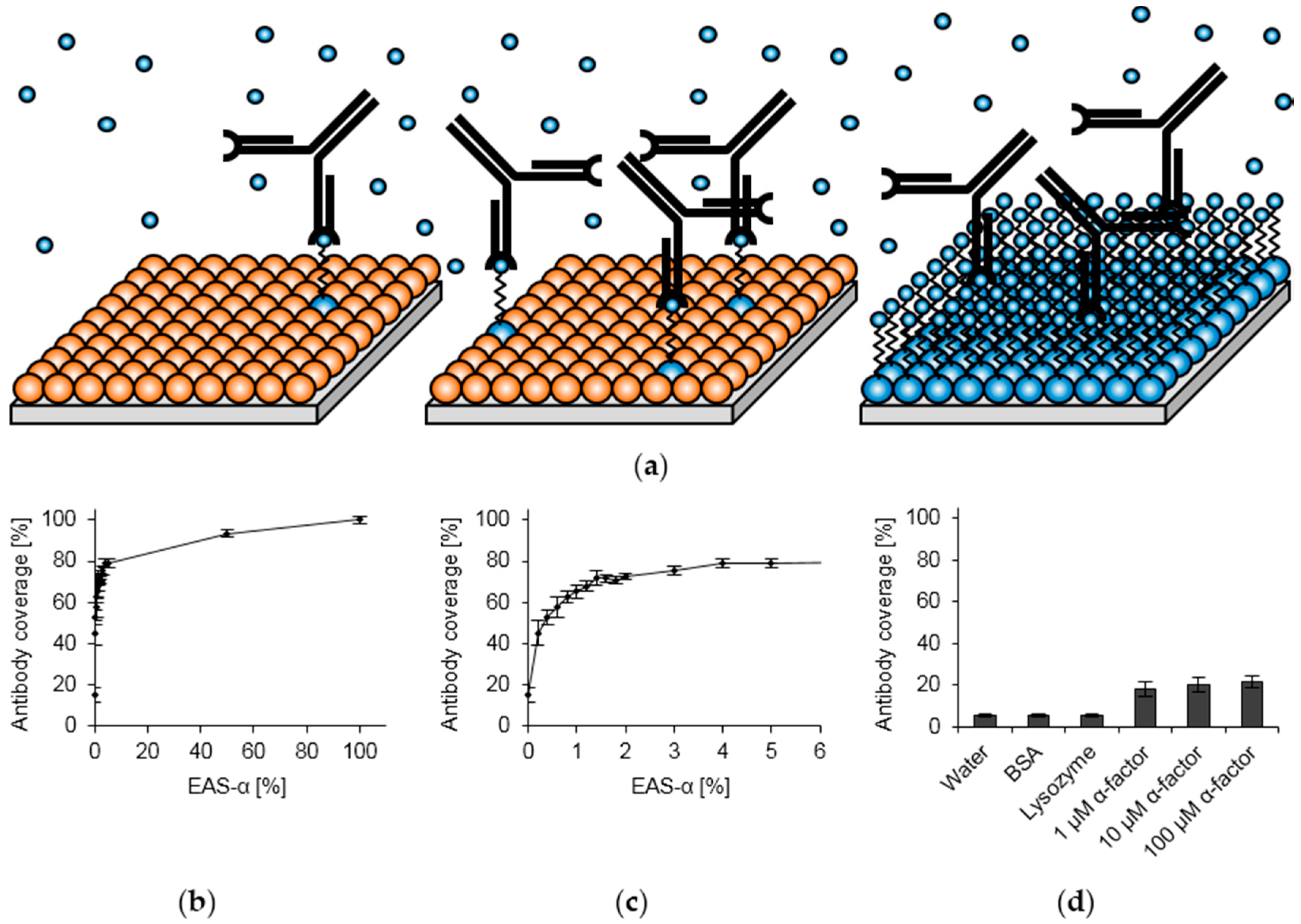
2.3. Surface Functionalization
2.4. Optimization of Hydrophobin Layer Composition
2.5. Competitive Enzyme-Linked Immunosorbent Assay (ELISA)
2.6. Inverse ELISA
2.7. Yeast Strains, Cultivation and Transformation
2.8. Measurement of α-Factor Concentrations in Yeast Culture Supernatants
3. Results and Discussion
3.1. Hydrophobin Design and Purification
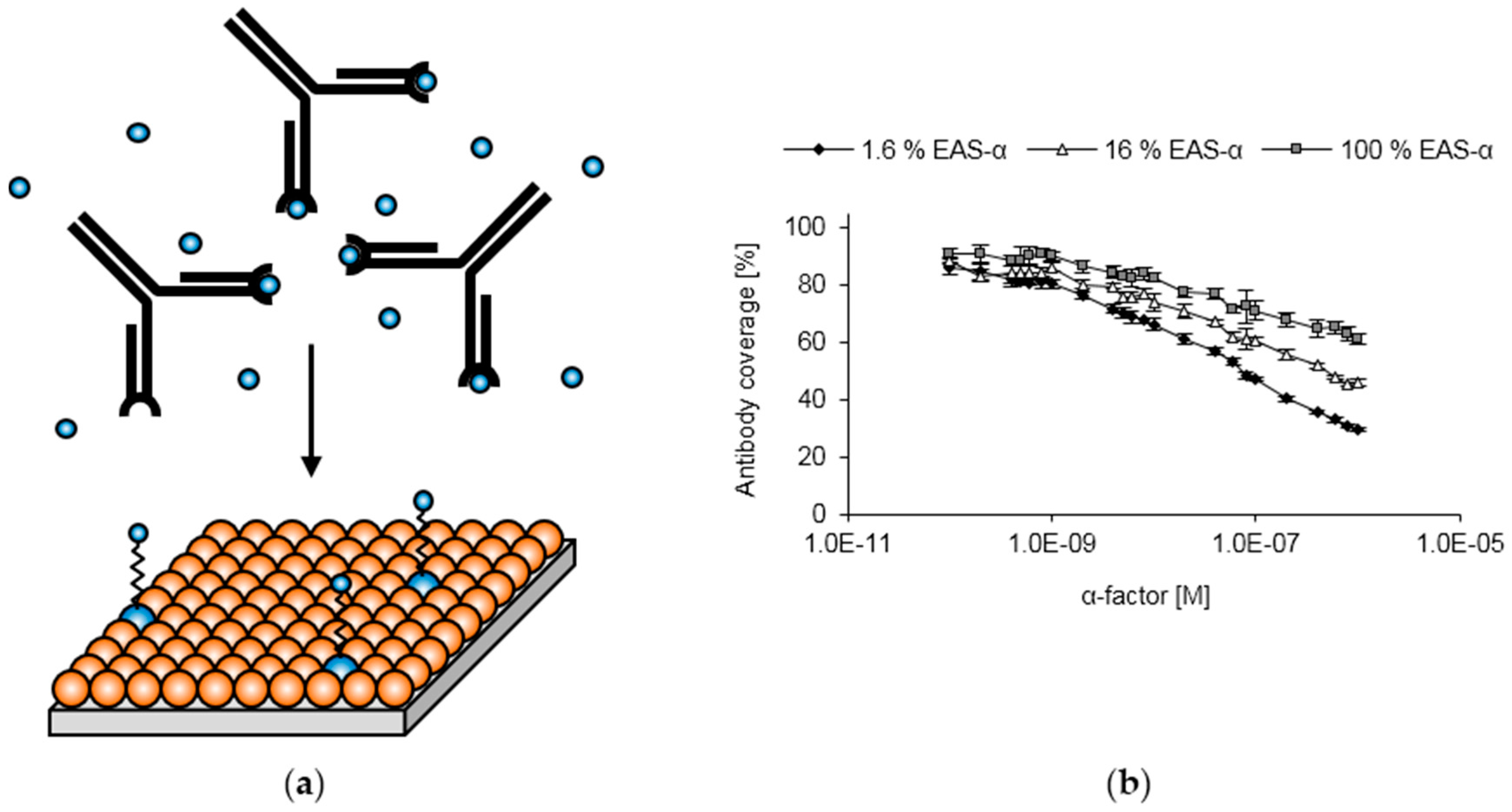
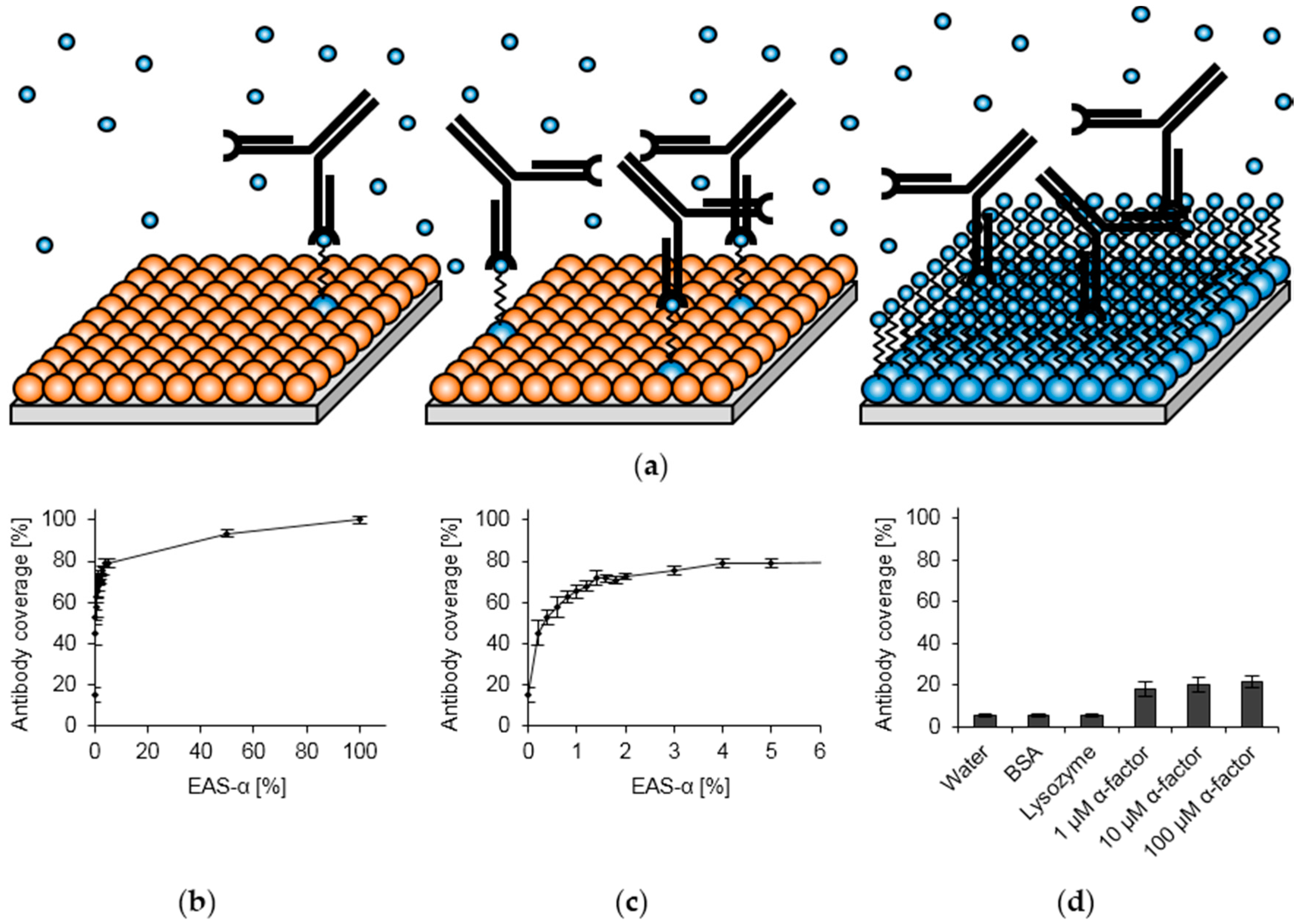
3.2. Surface Functionalization with Recombinant Hydrophobins
3.3. Optimization of Hydrophobin Layer Composition
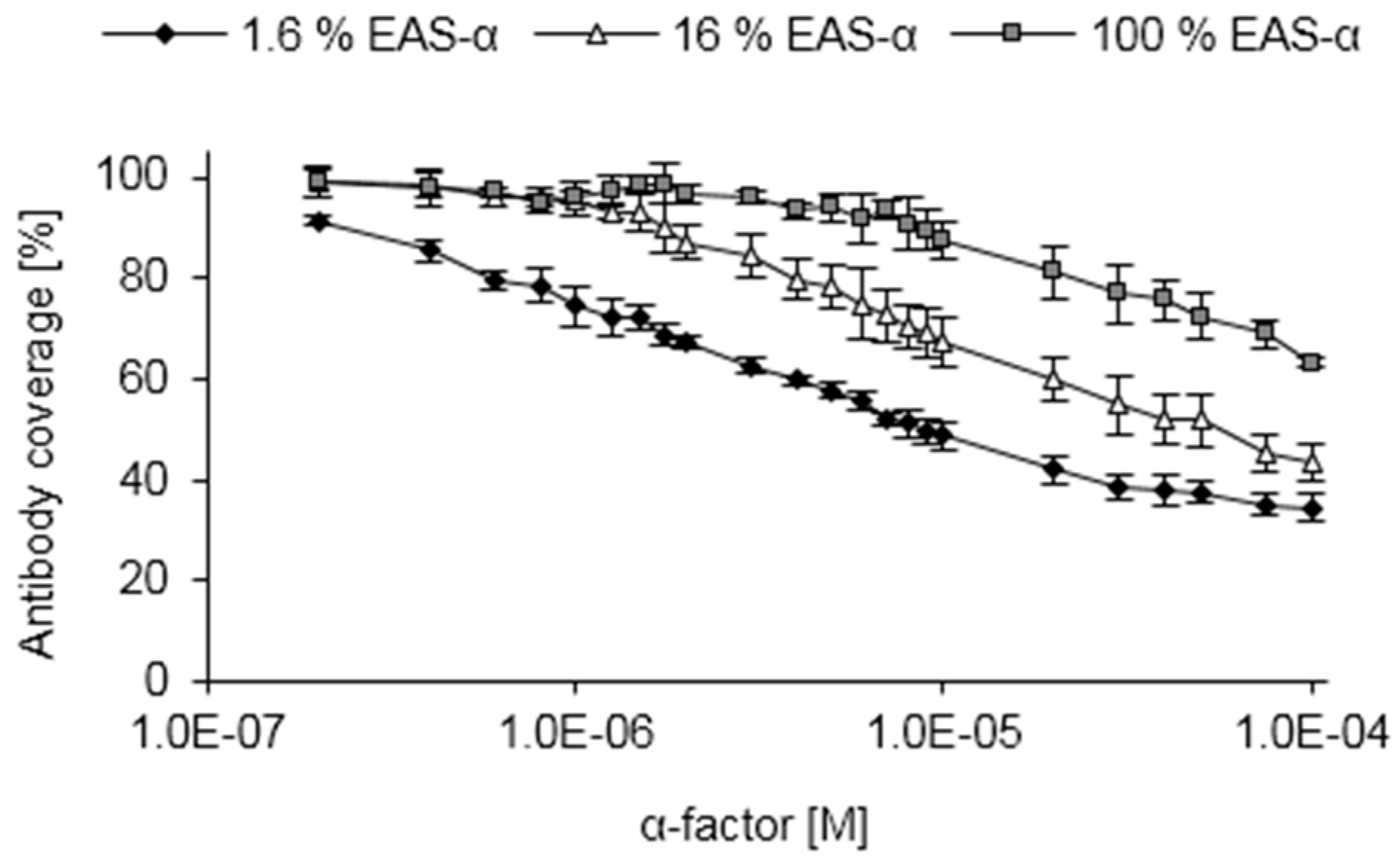
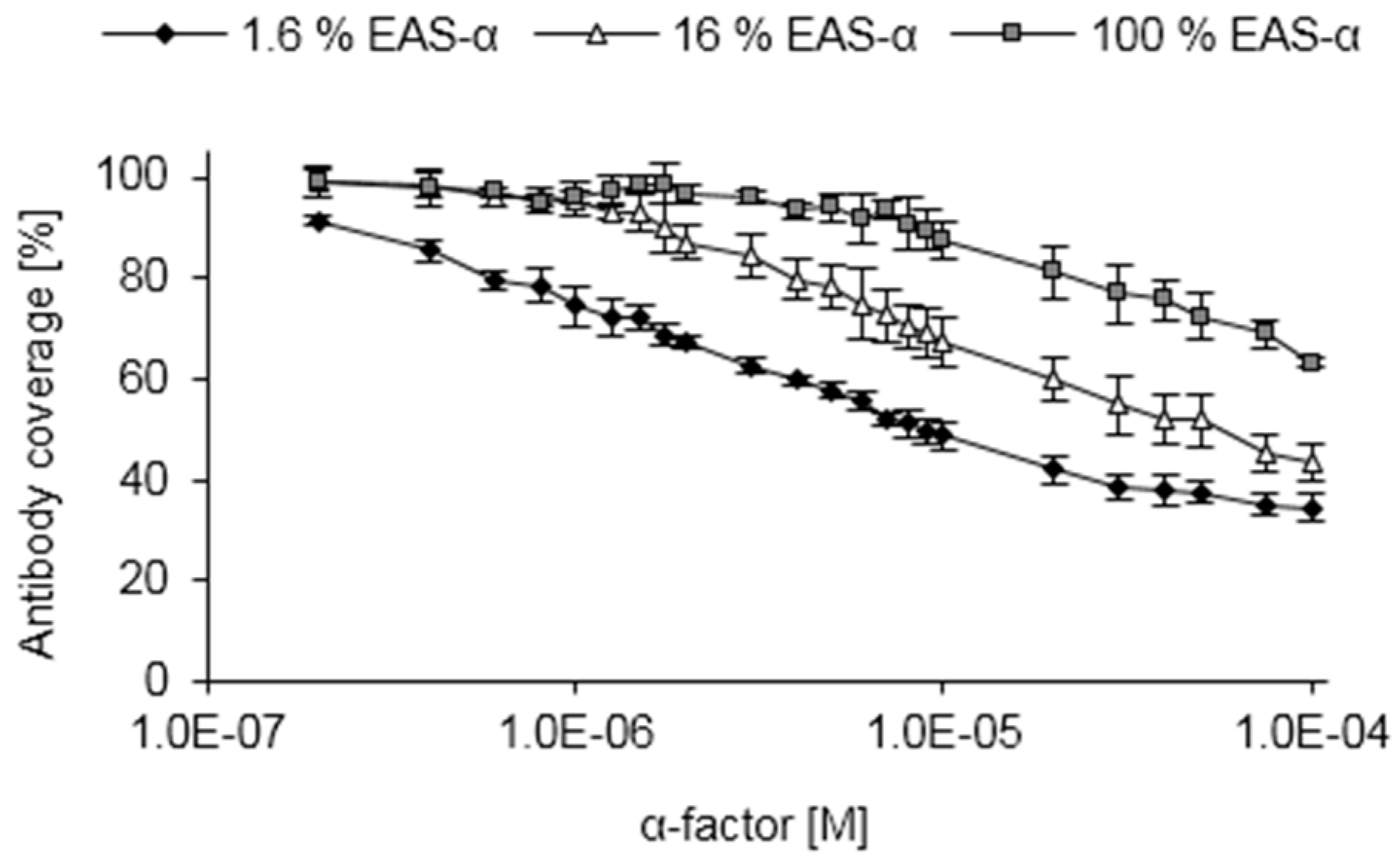
3.4. Competitive ELISA
3.5. Performing the Competitive ELISA Under Varying Conditions
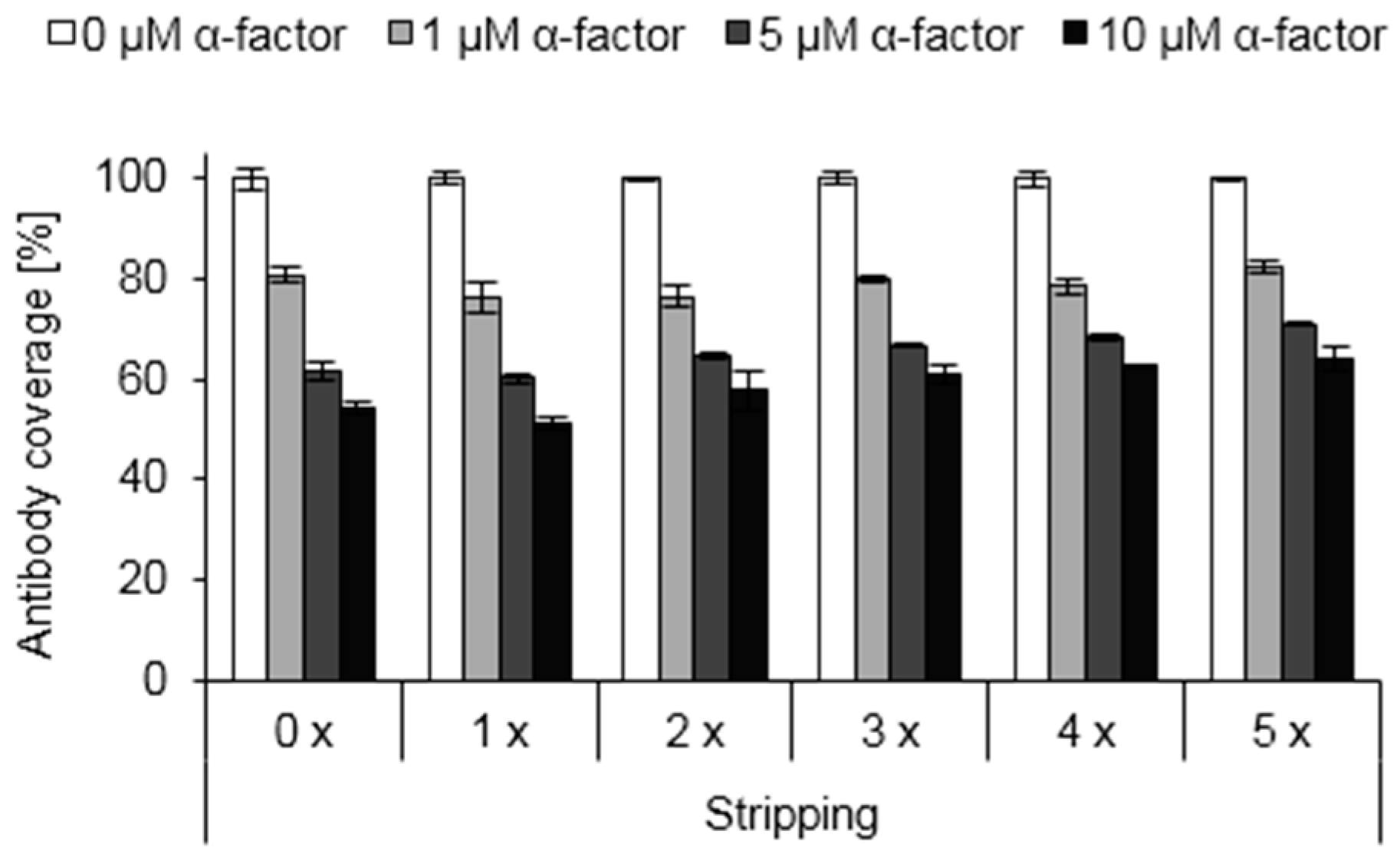
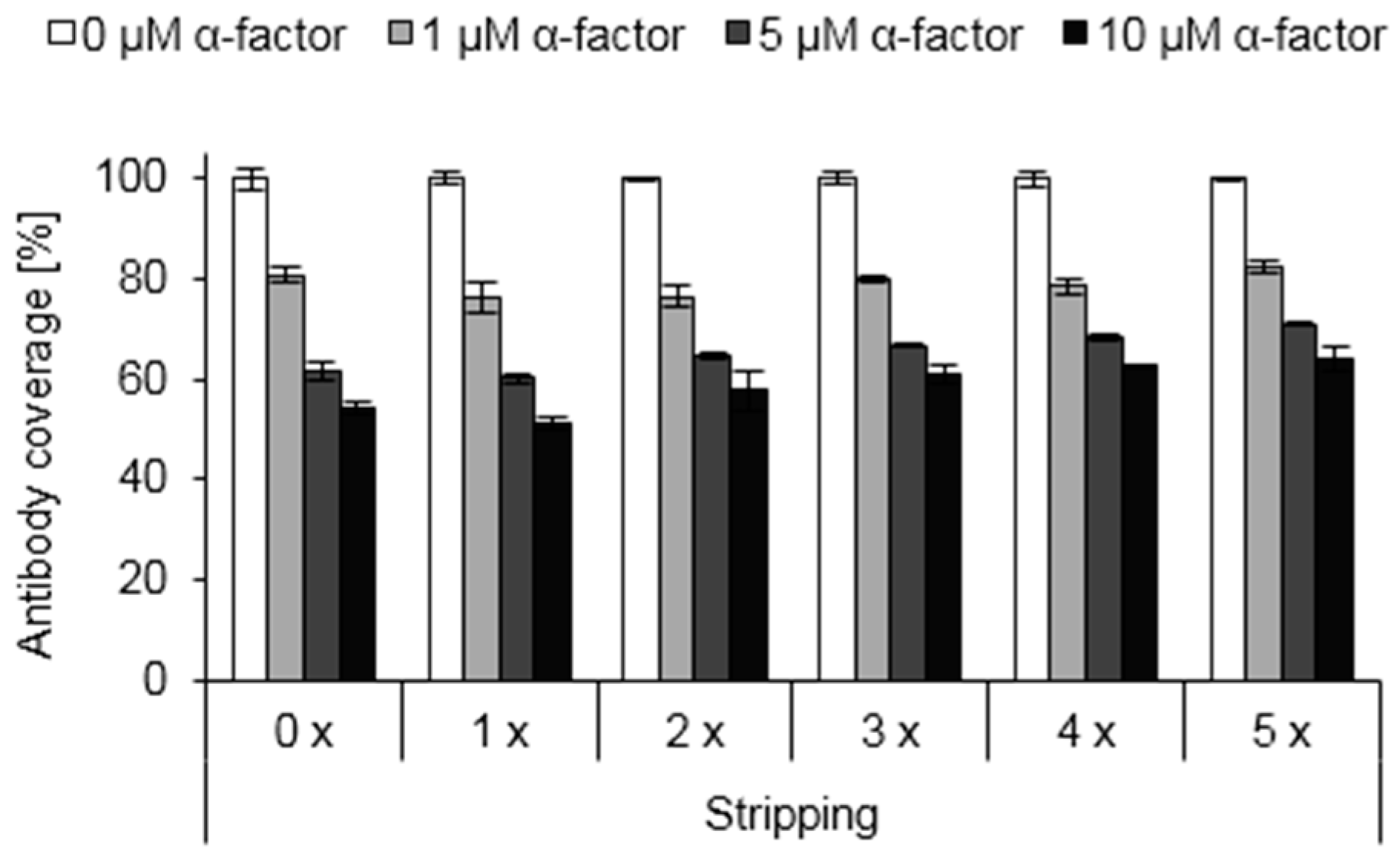
3.6. Reusability of Functionalized Surfaces
3.7. Inverse ELISA
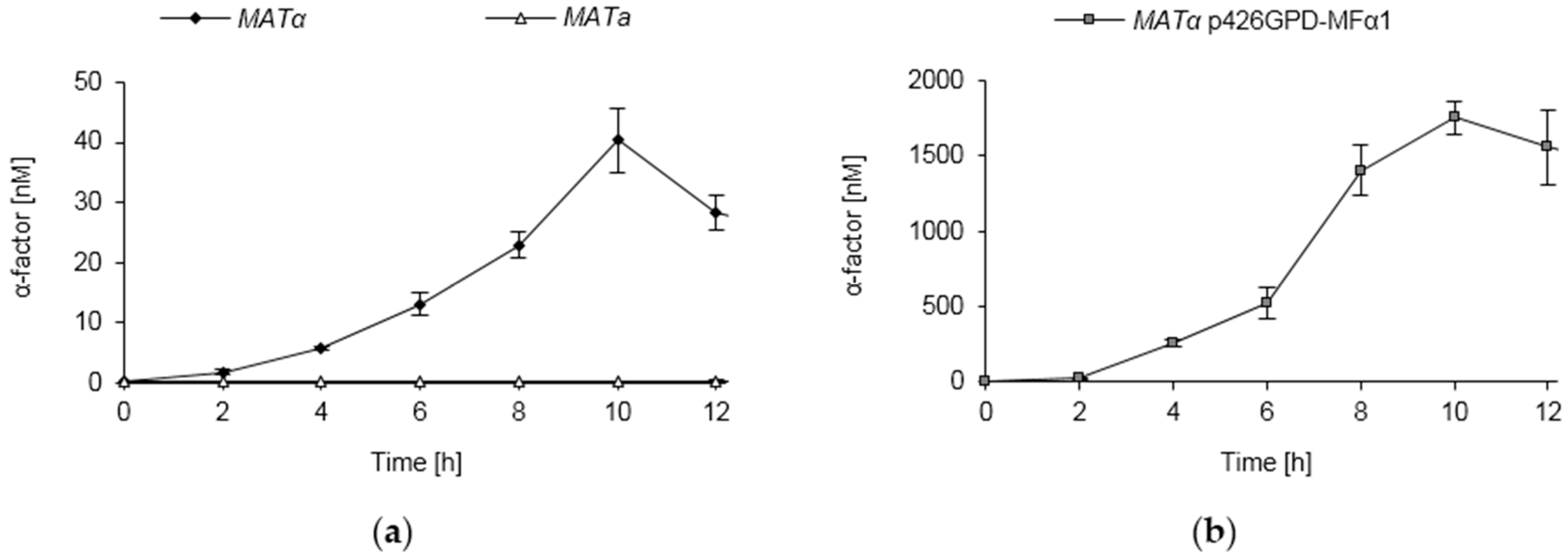
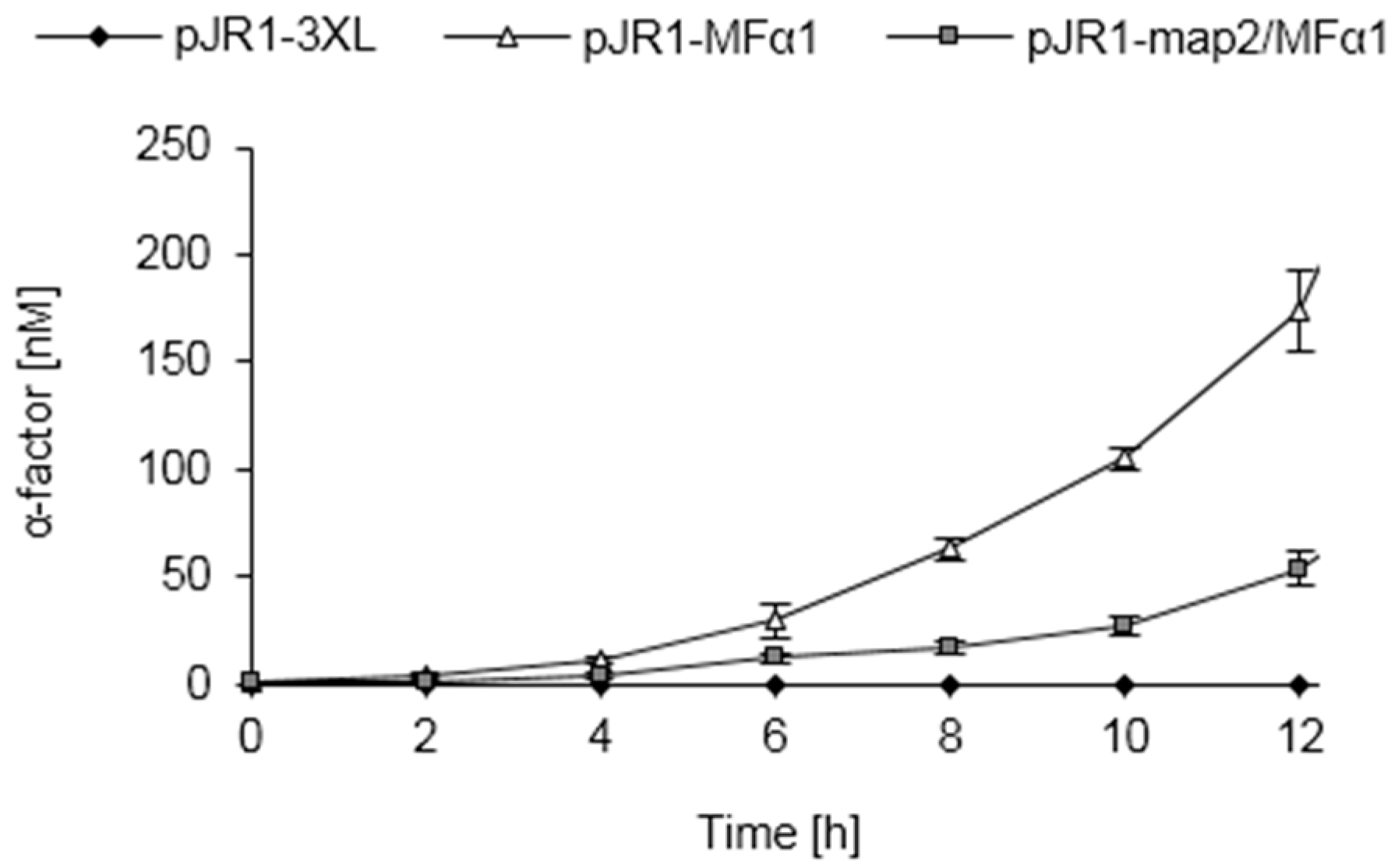
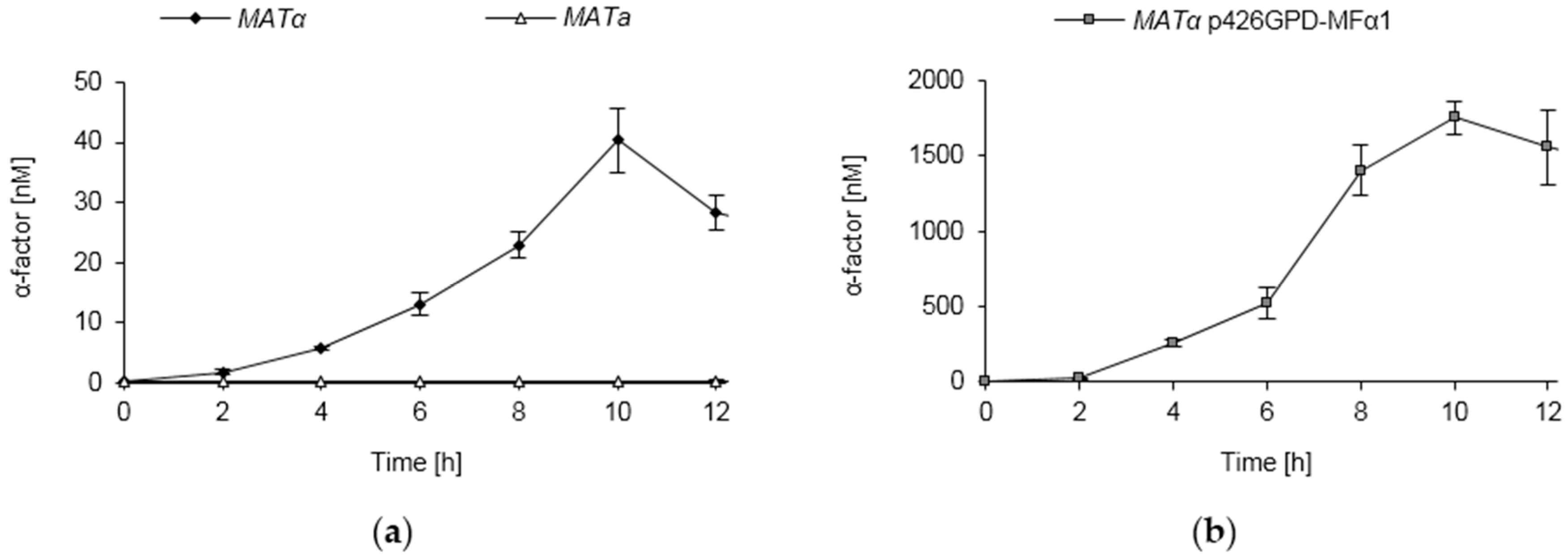
3.8. Pheromone Secretion of S. cerevisiae Strains
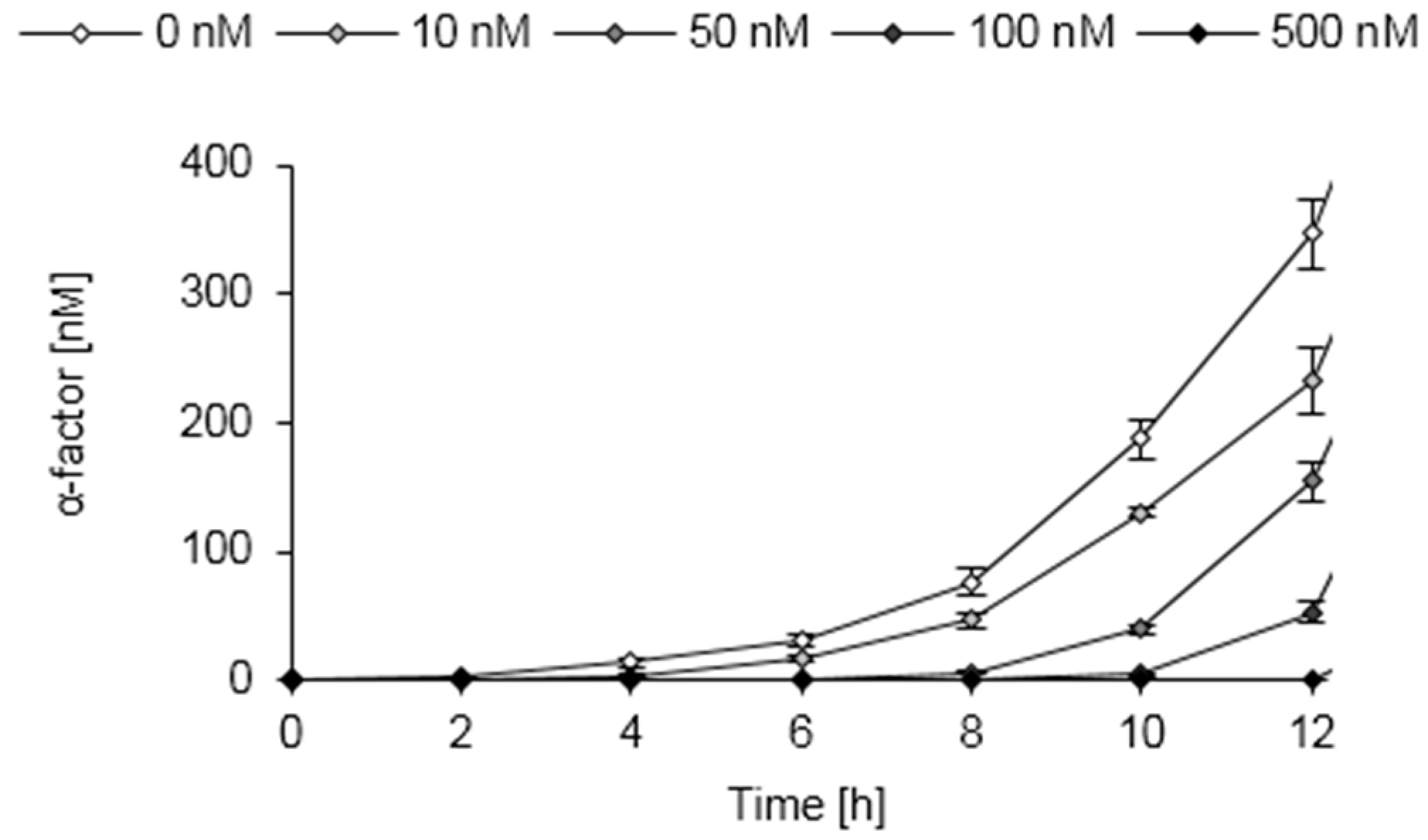
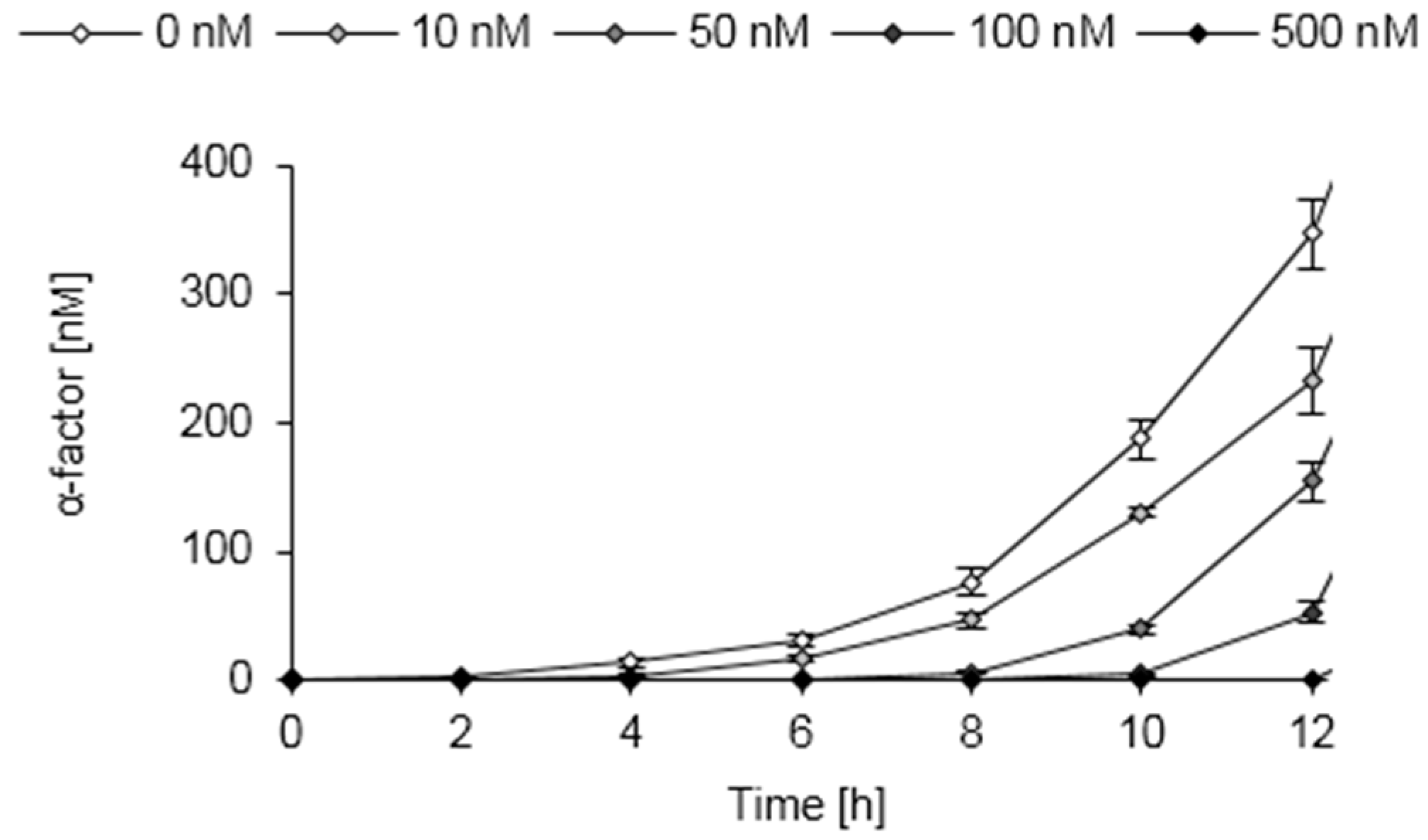
3.9. Pheromone Secretion of Engineered S. pombe Cells
3.10. Establishment of a Functional Whole-Cell Biosensor
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| S. cerevisiae | Saccharomyces cerevisiae |
| S. pombe | Schizosaccharomyces pombe |
| E. coli | Escherichia coli |
| IPTG | isopropyl-β-d-thiogalactoside |
| SDS | sodium dodecyl sulfate |
| PAGE | polyacrylamide gel electrophoresis |
| PVDF | polyvinylidene fluoride |
| PBST | phosphate-buffered saline containing 0.01% (v/v) Tween-20 |
| BSA | bovine serum albumin |
| HRP | horseradish peroxidase |
| ELISA | enzyme-linked immunosorbent assay |
| EMM | Edinburgh Minimal Medium |
| EGTA | ethylene glycol tetra-acetic acid |
| MBP | maltose binding protein |
| HA | haemagglutinin |
References
- Linder, M.B.; Szilvay, G.R.; Nakari-Setälä, T.; Penttilä, M.E. Hydrophobins: The protein-amphiphiles of filamentous fungi. FEMS Microbiol. Rev. 2005, 29, 877–896. [Google Scholar] [CrossRef] [PubMed]
- Ren, Q.; Kwan, A.H.; Sunde, M. Two forms and two faces, multiple states and multiple uses: Properties and applications of the self-assembling fungal hydrophobins. Biopolymers 2013, 100, 601–612. [Google Scholar] [CrossRef] [PubMed]
- Sunde, M.; Kwan, A.H.; Templeton, M.D.; Beever, R.E.; Mackay, J.P. Structural analysis of hydrophobins. Micron 2008, 39, 773–784. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.B.; de Vocht, M.L. Hydrophobins, the fungal coat unravelled. Biochim. Biophys. Acta 2000, 1469, 79–86. [Google Scholar] [CrossRef]
- Zampieri, F.; Wösten, H.A.B.; Scholtmeijer, K. Creating surface properties using a palette of hydrophobins. Materials 2010, 3, 4607–4625. [Google Scholar] [CrossRef]
- Kershaw, M.J.; Talbot, N.J. Hydrophobins and repellents: Proteins with fundamental roles in fungal morphogenesis. Fungal Genet. Biol. 1998, 23, 18–33. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.B. Hydrophobins: Multipurpose proteins. Annu. Rev. Microbiol. 2001, 55, 625–646. [Google Scholar] [CrossRef] [PubMed]
- Wösten, H.A.B.; Wessels, J.G.H. Hydrophobins, from molecular structure to multiple functions in fungal development. Mycoscience 1997, 38, 363–374. [Google Scholar] [CrossRef]
- Hektor, H.J.; Scholtmeijer, K. Hydrophobins: Proteins with potential. Curr. Opin. Biotechnol. 2005, 16, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Janssen, M.I.; van Leeuwen, M.B.; Scholtmeijer, K.; van Kooten, T.G.; Dijkhuizen, L.; Wösten, H.A.B. Coating with genetic engineered hydrophobin promotes growth of fibroblasts on a hydrophobic solid. Biomaterials 2002, 23, 4847–4854. [Google Scholar] [CrossRef]
- Janssen, M.I.; van Leeuwen, M.B.; van Kooten, T.G.; de Vries, J.; Dijkhuizen, L.; Wösten, H.A.B. Promotion of fibroblast activity by coating with hydrophobins in the beta-sheet end state. Biomaterials 2004, 25, 2731–2739. [Google Scholar] [CrossRef] [PubMed]
- Misra, R.; Li, J.; Cannon, G.C.; Morgan, S.E. Nanoscale reduction in surface friction of polymer surfaces modified with Sc3 hydrophobin from Schizophyllum commune. Biomacromolecules 2006, 7, 1463–1470. [Google Scholar] [CrossRef] [PubMed]
- Hakala, T.J.; Laaksonen, P.; Saikko, V.; Ahlroos, T.; Helle, A.; Mahlberg, R.; Hähl, H.; Jacobs, K.; Kuosmanen, P.; Linder, M.B.; Holmberg, K. Adhesion and tribological properties of hydrophobin proteins in aqueous lubrication on stainless steel surfaces. RSC Adv. 2012, 2, 9867–9872. [Google Scholar] [CrossRef]
- Lee, S.; Røn, T.; Pakkanen, K.I.; Linder, M.B. Hydrophobins as aqueous lubricant additive for a soft sliding contact. Coll. Surf. B Biointerfaces 2015, 125, 264–269. [Google Scholar] [CrossRef] [PubMed]
- Bilewicz, R.; Witomski, J.; Van der Heyden, A.; Tagu, D.; Palin, B.; Rogalska, E. Modification of electrodes with self-assembled hydrophobin layers. J. Phys. Chem. B 2001, 105, 9772–9777. [Google Scholar] [CrossRef]
- Zhao, Z.X.; Qiao, M.Q.; Yin, F.; Shao, B.; Wu, B.Y.; Wang, Y.Y.; Wang, X.S.; Qin, X.; Li, S.; Yu, L.; et al. Amperometric glucose biosensor based on self-assembly hydrophobin with high efficiency of enzyme utilization. Biosens. Bioelectron. 2007, 22, 3021–3027. [Google Scholar] [CrossRef] [PubMed]
- Hou, S.; Li, X.; Li, X.; Feng, X.Z.; Wang, R.; Wang, C.; Yu, L.; Qiao, M.Q. Surface modification using a novel type I hydrophobin HGFI. Anal. Bioanal. Chem. 2009, 394, 783–789. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Hou, S.; Feng, X.; Yu, Y.; Ma, J.; Li, L. Patterning of neural stem cells on poly(lactic-co-glycolic acid) film modified by hydrophobin. Coll. Surf. B Biointerfaces 2009, 74, 370–374. [Google Scholar] [CrossRef] [PubMed]
- Asakawa, H.; Tahara, S.; Nakamichi, M.; Takehara, K.; Ikeno, S.; Linder, M.B.; Haruyama, T. The amphiphilic protein HFBII as a genetically taggable molecular carrier for the formation of a self-organized functional protein layer on a solid surface. Langmuir 2009, 25, 8841–8844. [Google Scholar] [CrossRef] [PubMed]
- Kostiainen, M.A.; Szilvay, G.R.; Smith, D.K.; Linder, M.B.; Ikkala, O. Multivalent dendrons for high-affinity adhesion of proteins to DNA. Angew. Chem. Int. Ed. Engl. 2006, 45, 3538–3542. [Google Scholar] [CrossRef] [PubMed]
- Kurppa, K.; Hytönen, V.P.; Nakari-Setälä, T.; Kulomaa, M.S.; Linder, M.B. Molecular engineering of avidin and hydrophobin for functional self-assembling interfaces. Coll. Surf. B Biointerfaces 2014, 120, 102–109. [Google Scholar] [CrossRef] [PubMed]
- Takatsuji, Y.; Yamasaki, R.; Iwanaga, A.; Lienemann, M.; Linder, M.B.; Haruyama, T. Solid-support immobilization of a “swing” fusion protein for enhanced glucose oxidase catalytic activity. Coll. Surf. B Biointerfaces 2013, 112, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Huang, Y.; Li, S.; Xu, H.; Linder, M.B.; Qiao, M. Hydrophilic modification of polystyrene with hydrophobin for time-resolved immunofluorometric assay. Biosens. Bioelectron. 2010, 26, 1074–1079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Wang, Z.; Wang, Z.; Feng, S.; Xu, H.; Zhao, Q.; Wang, S.; Fang, J.; Qiao, M.; Kong, D. Immobilization of anti-CD31 antibody on electrospun poly(ɛ-caprolactone) scaffolds through hydrophobins for specific adhesion of endothelial cells. Coll. Surf. B Biointerfaces 2011, 85, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Bell-Pedersen, D.; Dunlap, J.C.; Loros, J.J. The Neurospora circadian clock-controlled gene, ccg-2, is allelic to eas and encodes a fungal hydrophobin required for formation of the conidial rodlet layer. Genes Dev. 1992, 6, 2382–2394. [Google Scholar] [CrossRef] [PubMed]
- Lauter, F.R.; Russo, V.E.; Yanofsky, C. Developmental and light regulation of eas, the structural gene for the rodlet protein of Neurospora. Genes Dev. 1992, 6, 2373–2381. [Google Scholar] [CrossRef] [PubMed]
- Merlini, L.; Dudin, O.; Martin, S.G. Mate and fuse: How yeast cells do it. Open Biol. 2013, 3, 130008. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Rödel, G.; Ostermann, K. Artificial cell-cell communication as an emerging tool in synthetic biology applications. J. Biol. Eng. 2015, 9, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.; Rödel, G.; Ostermann, K. Application of the yeast pheromone system for controlled cell-cell communication and signal amplification. Lett. Appl. Microbiol. 2011, 52, 521–526. [Google Scholar] [CrossRef] [PubMed]
- Adeniran, A.; Sherer, M.; Tyo, K.E. Yeast-based biosensors: Design and applications. FEMS Yeast Res. 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Baronian, K.H. The use of yeast and moulds as sensing elements in biosensors. Biosens. Bioelectron. 2004, 19, 953–962. [Google Scholar] [CrossRef] [PubMed]
- Davey, J.; Davis, K.; Hughes, M.; Ladds, G.; Powner, D. The processing of yeast pheromones. Semin Cell. Dev. Biol. 1998, 9, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Gibson, G.E.; Hirsch, J.A.; Fonzetti, P.; Jordan, B.D.; Cirio, R.T.; Elder, J. Vitamin B1 (thiamine) and dementia. Ann. N. Y. Acad Sci. 2016. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.; Mehta, R.; Al-Ani, M.; Hill, J.A.; Winchester, D.E. Determining the role of thiamine deficiency in systolic heart failure: A meta-analysis and systematic review. J. Card Fail. 2015, 21, 1000–1007. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Ausubel, F.M., Brent, R., Kingston, R.E., Moore, D.D., Seidman, J.G., Smith, J.A., Struhl, K., Eds.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1989. [Google Scholar]
- Maloy, S.; Stewart, V.; Taylor, R. Genetic Analysis of Pathogenic bacteria: A Laboratory Manual; Cold Spring Harbor Laboratory Press: New York, NY, USA, 1996. [Google Scholar]
- Gruner, L.J.; Ostermann, K.; Rödel, G. Layer thickness of hydrophobin films leads to oscillation in wettability. Langmuir 2012, 28, 6942–6949. [Google Scholar] [CrossRef] [PubMed]
- Kwan, A.H.; Winefield, R.D.; Sunde, M.; Matthews, J.M.; Haverkamp, R.G.; Templeton, M.D.; Mackay, J.P. Structural basis for rodlet assembly in fungal hydrophobins. Proc. Natl Acad. Sci. USA 2006, 103, 3621–3626. [Google Scholar] [CrossRef] [PubMed]
- Morris, V.K.; Ren, Q.; Macindoe, I.; Kwan, A.H.; Byrne, N.; Sunde, M. Recruitment of class I hydrophobins to the air: Water interface initiates a multi-step process of functional amyloid formation. J. Biol. Chem. 2011, 286, 15955–15963. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976, 72, 248–254. [Google Scholar] [CrossRef]
- Schägger, H. Tricine-SDS-PAGE. Nat. Protoc. 2006, 1, 16–22. [Google Scholar] [CrossRef] [PubMed]
- Rieder, A.; Ladnorg, T.; Wöll, C.; Obst, U.; Fischer, R.; Schwartz, T. The impact of recombinant fusion-hydrophobin coated surfaces on E. coli and natural mixed culture biofilm formation. Biofouling 2011, 27, 1073–1085. [Google Scholar] [CrossRef] [PubMed]
- Heim, L. Construction of an h+S strain of Schizosaccharomyces pombe. Curr. Genet. 1990, 17, 13–19. [Google Scholar] [CrossRef] [PubMed]
- Forsburg, S.L.; Rhind, N. Basic methods for fission yeast. Yeast 2006, 23, 173–183. [Google Scholar] [CrossRef] [PubMed]
- Gietz, R.D.; Woods, R.A. Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol. 2002, 350, 87–96. [Google Scholar] [PubMed]
- Kershaw, M.J.; Thornton, C.R.; Wakley, G.E.; Talbot, N.J. Four conserved intramolecular disulphide linkages are required for secretion and cell wall localization of a hydrophobin during fungal morphogenesis. Mol. Microbiol. 2005, 56, 117–125. [Google Scholar] [CrossRef] [PubMed]
- De Vocht, M.L.; Reviakine, I.; Wösten, H.A.B.; Brisson, A.; Wessels, J.G.H.; Robillard, G.T. Structural and functional role of the disulfide bridges in the hydrophobin SC3. J. Biol. Chem. 2000, 275, 28428–28432. [Google Scholar] [CrossRef] [PubMed]
- Paris, S.; Debeaupuis, J.P.; Crameri, R.; Carey, M.; Charlès, F.; Prévost, M.C.; Schmitt, C.; Philippe, B.; Latgé, J.P. Conidial hydrophobins of Aspergillus fumigatus. Appl. Environ. Microbiol. 2003, 69, 1581–1588. [Google Scholar] [CrossRef] [PubMed]
- Scherrer, S.; De Vries, O.M.; Dudler, R.; Wessels, J.G.H.; Honegger, R. Interfacial self-assembly of fungal hydrophobins of the lichen-forming ascomycetes Xanthoria parietina and X. ectaneoides. Fungal Genet. Biol. 2000, 30, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Trembley, M.L.; Ringli, C.; Honegger, R. Hydrophobins DGH1, DGH2, and DGH3 in the lichen-forming basidiomycete Dictyonema glabratum. Fungal Genet. Biol. 2002, 35, 247–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Zhang, B.; Szilvay, G.R.; Sun, R.; Jänis, J.; Wang, Z.; Feng, S.; Xu, H.; Linder, M.B.; Qiao, M. Protein HGFI from the edible mushroom Grifola. frondosa is a novel 8 kDa class I hydrophobin that forms rodlets in compressed monolayers. Microbiology 2008, 154, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Kottmeier, K.; Günther, T.J.; Weber, J.; Kurtz, S.; Ostermann, K.; Rödel, G.; Bley, T. Constitutive expression of hydrophobin HFB1 from Trichoderma reesei in Pichia pastoris and its pre-purification by foam separation during cultivation. Eng. Life Sci. 2012, 12, 162–170. [Google Scholar] [CrossRef]
- Kwan, A.H.; Macindoe, I.; Vukasin, P.V.; Morris, V.K.; Kass, I.; Gupte, R.; Mark, A.E.; Templeton, M.D.; Mackay, J.P.; Sunde, M. The Cys3-Cys4 loop of the hydrophobin EAS is not required for rodlet formation and surface activity. J. Mol. Biol. 2008, 382, 708–720. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Ren, Q.; Wu, Y.N.; Morris, V.K.; Rey, A.A.; Braet, F.; Kwan, A.H.; Sunde, M. Surface functionalization of carbon nanomaterials by self-assembling hydrophobin proteins. Biopolymers 2013, 99, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Boeuf, S.; Throm, T.; Gutt, B.; Strunk, T.; Hoffmann, M.; Seebach, E.; Mühlberg, L.; Brocher, J.; Gotterbarm, T.; Wenzel, W.; et al. Engineering hydrophobin DewA to generate surfaces that enhance adhesion of human but not bacterial cells. Acta Biomater. 2012, 8, 1037–1047. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, M.H.; Borodina, I.; Moresco, J.L.; Svendsen, W.E.; Frisvad, J.C.; Søndergaard, I. High-yield production of hydrophobins RodA and RodB from Aspergillus fumigatus in Pichia pastoris. Appl. Microbiol. Biotechnol. 2011, 90, 1923–1932. [Google Scholar] [CrossRef] [PubMed]
- Schmoll, M.; Seibel, C.; Kotlowski, C.; Wöllert Genannt Vendt, F.; Liebmann, B.; Kubicek, C.P. Recombinant production of an Aspergillus nidulans class I hydrophobin (DewA) in Hypocrea. jecorina (Trichoderma reesei) is promoter-dependent. Appl. Microbiol. Biotechnol. 2010, 88, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Wohlleben, W.; Subkowski, T.; Bollschweiler, C.; von Vacano, B.; Liu, Y.; Schrepp, W.; Baus, U. Recombinantly produced hydrophobins from fungal analogues as highly surface-active performance proteins. Eur. Biophys. J. 2010, 39, 457–468. [Google Scholar] [CrossRef] [PubMed]
- De Stefano, L.; Rea, I.; Armenante, A.; Giardina, P.; Giocondo, M.; Rendina, I. Self-assembled biofilm of hydrophobins protects the silicon surface in the KOH wet etch process. Langmuir 2007, 23, 7920–7922. [Google Scholar] [CrossRef] [PubMed]
- De Vocht, M.L.; Scholtmeijer, K.; van der Vegte, E.W.; de Vries, O.M.; Sonveaux, N.; Wösten, H.A.B.; Ruysschaert, J.M.; Hadziloannou, G.; Wessels, J.G.; Robillard, G.T. Structural characterization of the hydrophobin SC3, as a monomer and after self-assembly at hydrophobic/hydrophilic interfaces. Biophys. J. 1998, 74, 2059–2068. [Google Scholar] [CrossRef]
- Gunning, A.P.; De Groot, P.W.J.; Visserb, J.; Morris, V.J. Atomic force microscopy of a hydrophobin protein from the edible mushroom Agaricus bisporus. J. Coll. Interf. Sci. 1998, 201, 118–126. [Google Scholar] [CrossRef]
- Wösten, H.A.B.; Schuren, F.H.J.; Wessels, J.G.H. Interfacial self-assembly of a hydrophobin into an amphipathic protein membrane mediates fungal attachment to hydrophobic surfaces. EMBO J. 1994, 13, 5848–5854. [Google Scholar] [PubMed]
- Iwanaga, A.; Asakawa, H.; Fukuma, T.; Nakamichi, M.; Shigematsu, S.; Linder, M.B.; Haruyama, T. Ordered nano-structure of a stamped self-organized protein layer on a HOPG surface using a HFB carrier. Coll. Surf. B Biointerfaces 2011, 84, 395–399. [Google Scholar] [CrossRef] [PubMed]
- Askolin, S.; Linder, M.B.; Scholtmeijer, K.; Tenkanen, M.; Penttilä, M.; de Vocht, M.L.; Wösten, H.A.B. Interaction and comparison of a class I hydrophobin from Schizophyllum. commune and class II hydrophobins from Trichoderma reesei. Biomacromolecules 2006, 7, 1295–1301. [Google Scholar] [CrossRef] [PubMed]
- Silverton, E.W.; Navia, M.A.; Davies, D.R. Three-dimensional structure of an intact human immunoglobulin. Proc. Natl Acad. Sci. USA 1977, 74, 5140–5144. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Lienemann, M.; Qiau, M.; Linder, M.B. Mechanisms of protein adhesion on surface films of hydrophobin. Langmuir 2010, 26, 8491–8496. [Google Scholar] [CrossRef] [PubMed]
- Rocha, L.B.; Luz, D.E.; Moraes, C.T.; Caravelli, A.; Fernandes, I.; Guth, B.E.; Horton, D.S.; Piazza, R.M. Interaction between Shiga toxin and monoclonal antibodies: Binding characteristics and in vitro neutralizing abilities. Toxins 2012, 4, 729–747. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Drăguşanu, M.; Petre, B.A.; Rempel, D.L.; Przybylski, M.; Gross, M.L. Protein-peptide affinity determination using an h/d exchange dilution strategy: Application to antigen-antibody interactions. J. Am. Soc. Mass Spectrom. 2010, 21, 1660–1667. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Feng, Y.; Zhu, Z.; Dimitrov, D.S. Human monoclonal antibody fragments binding to insulin-like growth factors I and II with picomolar affinity. Mol. Cancer Ther. 2011, 10, 1677–1685. [Google Scholar] [CrossRef] [PubMed]
- Sarlin, T.; Nakari-Setälä, T.; Linder, M.B.; Penttilä, M.; Haikara, A. Fungal hydrophobins as predictors of the gushing activity of malt. J. Inst. Brew. 2005, 111, 105–111. [Google Scholar] [CrossRef]
- Diener, C.; Schreiber, G.; Giese, W.; del Rio, G.; Schröder, A.; Klipp, E. Yeast mating and image-based quantification of spatial pattern formation. PLoS Comput. Biol. 2014, 10, e1003690. [Google Scholar] [CrossRef] [PubMed]
- Jin, M.; Errede, B.; Behar, M.; Mather, W.; Nayak, S.; Hasty, J.; Dohlman, H.G.; Elston, T.C. Yeast dynamically modify their environment to achieve better mating efficiency. Sci. Signal. 2011, 4, 277. [Google Scholar] [CrossRef] [PubMed]
- Moore, T.I.; Chou, C.S.; Nie, Q.; Jeon, N.L.; Yi, T.M. Robust spatial sensing of mating pheromone gradients by yeast cells. PLoS ONE 2008, 3, e3865. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.C.; Nielsen, L.K.; Vickers, C.E. Engineered quorum sensing using pheromone-mediated cell-to-cell communication in Saccharomyces cerevisiae. ACS Synth. Biol. 2013, 2, 136–149. [Google Scholar] [CrossRef] [PubMed]
- Rogers, D.W.; McConnell, E.; Greig, D. Molecular quantification of Saccharomyces cerevisiae α-pheromone secretion. FEMS Yeast Res. 2012, 12, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Bener Aksam, E.; Pinkse, M.W.; Verhaert, P.D. Molecular characterization of Saccharomyces cerevisiae α-pheromone by mass spectrometry-based peptidomics. FEMS Yeast Res. 2013, 13, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Sprague, G.F., Jr.; Herskowitz, I. Control of yeast cell type by the mating type locus. I. Identification and control of expression of the a-specific gene BAR1. J. Mol. Biol. 1981, 153, 305–321. [Google Scholar] [CrossRef]
- Jensen, N.B.; Strucko, T.; Kildegaard, K.R.; David, F.; Maury, J.; Mortensen, U.H.; Forster, J.; Nielsen, J.; Borodina, I. EasyClone: Method for iterative chromosomal integration of multiple genes in Saccharomyces cerevisiae. FEMS Yeast Res. 2014, 14, 238–248. [Google Scholar] [CrossRef] [PubMed]
- Böhm, J.; Hoff, B.; O′Gorman, C.M.; Wolfers, S.; Klix, V.; Binger, D.; Zadra, I.; Kürnsteiner, H.; Pöggeler, S.; Dyer, P.S.; et al. Sexual reproduction and mating-type-mediated strain development in the penicillin-producing fungus Penicillium chrysogenum. Proc. Natl Acad. Sci. USA 2013, 110, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Fowler, T.J.; DeSimone, S.M.; Mitton, M.F.; Kurjan, J.; Raper, C.A. Multiple sex pheromones and receptors of a mushroom-producing fungus elicit mating in yeast. Mol. Biol. Cell. 1999, 10, 2559–2572. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves-Sá, J.; Murray, A. Asymmetry in sexual pheromones is not required for ascomycete mating. Curr. Biol. 2011, 21, 1337–1346. [Google Scholar] [CrossRef] [PubMed]
- Hennig, S.; Clemens, A.; Rödel, G.; Ostermann, K. A yeast pheromone-based inter-species communication system. Appl. Microbiol. Biotechnol. 2015, 99, 1299–1308. [Google Scholar] [CrossRef] [PubMed]
- Mayrhofer, S.; Pöggeler, S. Functional characterization of an alpha-factor-like Sordaria macrospora peptide pheromone and analysis of its interaction with its cognate receptor in Saccharomyces cerevisiae. Eukaryot. Cell 2005, 4, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Olesnicky, N.S.; Brown, A.J.; Dowell, S.J.; Casselton, L.A. A constitutively active G-protein-coupled receptor causes mating self-compatibility in the mushroom Coprinus. EMBO J. 1999, 18, 2756–2763. [Google Scholar] [CrossRef] [PubMed]
- Maundrell, K. nmt1 of fission yeast. A highly transcribed gene completely repressed by thiamine. J. Biol. Chem. 1990, 265, 10857–10864. [Google Scholar] [PubMed]
- Javerzat, J.P.; Cranston, G.; Allshire, R.C. Fission yeast genes which disrupt mitotic chromosome segregation when overexpressed. Nucleic Acid Res. 1996, 24, 4676–4683. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Arai, A.; Takebe, Y.; Masuda, M. A chemical compound for controlled expression of nmt1-driven gene in the fission yeast Schizosaccharomyces pombe. Anal. Biochem. 2011, 412, 159–164. [Google Scholar] [CrossRef] [PubMed]
- Akyilmaz, E.; Yaşa, I.; Dinçkaya, E. Whole cell immobilized amperometric biosensor based on Saccharomyces cerevisiae for selective determination of vitamin B1 (thiamine). Anal. Biochem. 2006, 354, 78–84. [Google Scholar] [CrossRef] [PubMed]
- Akyilmaz, E.; Yorganci, E. A novel biosensor based on activation effect of thiamine on the activity of pyruvate oxidase. Biosens. Bioelectron. 2008, 23, 1874–1877. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; de Vocht, M.L.; de Jonge, J.; Poolman, B.; Robillard, G.T. Structural changes and molecular interactions of hydrophobin SC3 in solution and on a hydrophobic surface. Protein Sci. 2002, 11, 1172–1181. [Google Scholar] [CrossRef] [PubMed]




© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hennig, S.; Rödel, G.; Ostermann, K. Hydrophobin-Based Surface Engineering for Sensitive and Robust Quantification of Yeast Pheromones. Sensors 2016, 16, 602. https://doi.org/10.3390/s16050602
Hennig S, Rödel G, Ostermann K. Hydrophobin-Based Surface Engineering for Sensitive and Robust Quantification of Yeast Pheromones. Sensors. 2016; 16(5):602. https://doi.org/10.3390/s16050602
Chicago/Turabian StyleHennig, Stefan, Gerhard Rödel, and Kai Ostermann. 2016. "Hydrophobin-Based Surface Engineering for Sensitive and Robust Quantification of Yeast Pheromones" Sensors 16, no. 5: 602. https://doi.org/10.3390/s16050602
APA StyleHennig, S., Rödel, G., & Ostermann, K. (2016). Hydrophobin-Based Surface Engineering for Sensitive and Robust Quantification of Yeast Pheromones. Sensors, 16(5), 602. https://doi.org/10.3390/s16050602