Metabarcoding of Environmental DNA Samples to Explore the Use of Uranium Mine Containment Ponds as a Water Source for Wildlife


Abstract
1. Introduction
2. Materials and Methods
2.1. Sites and eDNA Sample Collection
2.2. eDNA Sample Processing
2.3. Library Preparation and Sequencing
2.4. Bioinformatic Analyses
2.5. Carryover Calculations
2.6. Data Accessibility
3. Results
3.1. Reads from MiSeq
3.2. Taxa Identification per Marker
3.3. Taxa Identification across Sites
3.4. Filter versus Centrifuge Processed Samples
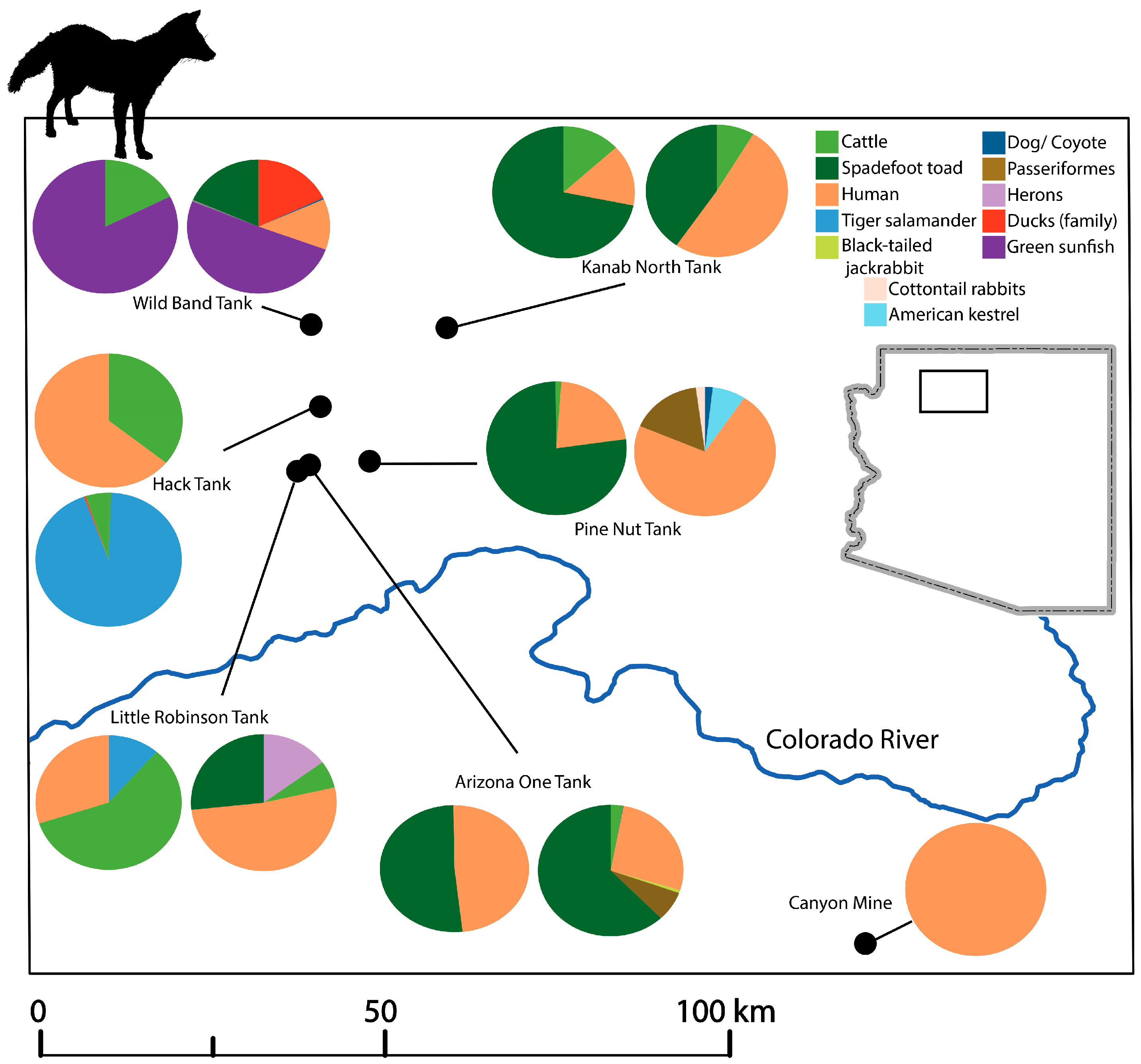
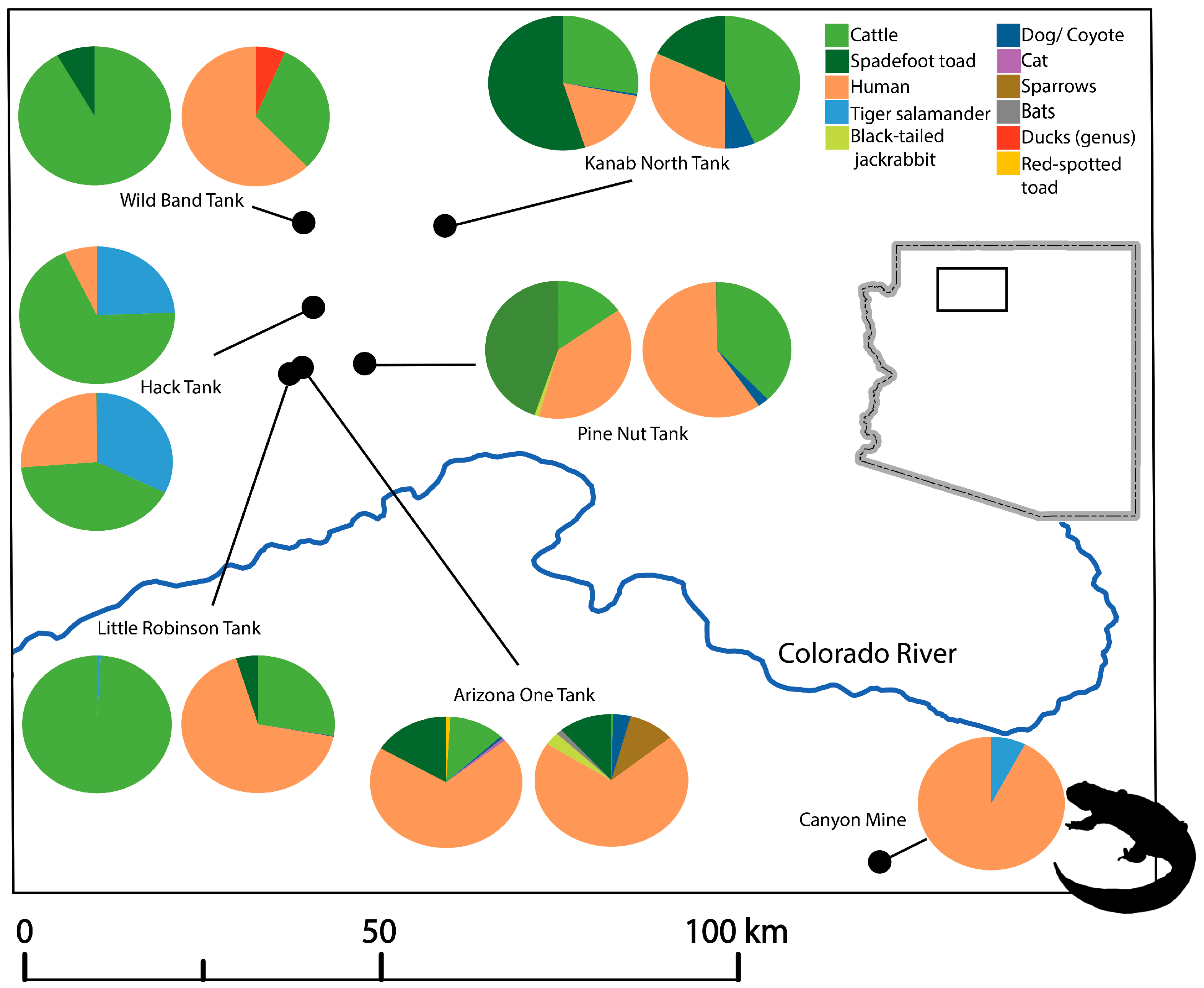
3.5. Taxa Identification per Site
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Rattner, B.A. History of wildlife toxicology. Ecotoxicology 2009, 18, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Kendall, R.J. Wildlife toxicology: Where we have been and where we are going. J. Environ. Anal. Toxicol. 2016, 6, 348. [Google Scholar] [CrossRef]
- Mackay, D.; Fraser, A. Bioaccumulation of persistent organic chemicals: Mechanisms and models. Environ. Pollut. 2000, 110, 375–391. [Google Scholar] [CrossRef]
- Inck, J.E.; Linder, G.; Darrah, A.J.; Drost, C.A.; Duniway, M.C.; Johnson, M.J.; Méndez-Harclerode, F.M.; Nowak, E.M.; Valdez, E.W.; van Riper, C., III; et al. Exposure pathways and biological receptors: Baseline data for the canyon uranium mine, Coconino County, Arizona. J. Fish Wildl. Manag. 2014, 5, 422–440. [Google Scholar] [CrossRef]
- Otton, J.K.; Van Gosen, B.S. Uranium resource availability in breccia pipes in northern Arizona. In Hydrological, Geological, and Biological Sites Characterization of Breccia Pipe Uranium Deposits in Northern Arizona; Alpine, A.E., Ed.; United States Geological Survey: Reston, VA, USA, 2010; pp. 19–42. [Google Scholar]
- U.S. Department of the Interior. Record of decision: Northern Arizona withdrawal. 2012. Available online: https://www.blm.gov/documents/national-office/blm-library/planningnepa/record-decision-northern-arizona-withdrawal-2012 (accessed on 18 May 2017).
- Hinck, J.E.; Cleveland, D.; Brumbaugh, W.G.; Linder, G.; Lankton, J. Pre-mining trace element and radiation exposure to biota from a breccia pipe uranium mine in the Grand Canyon (Arizona, USA) watershed. Environ. Monit. Assess. 2017, 189, 56. [Google Scholar] [CrossRef] [PubMed]
- Gardham, S.; Hose, G.C.; Stephenson, S.; Chariton, A.A. DNA metabarcoding meets experimental ecotoxicology: Advancing knowledge on the ecological effects of copper in freshwater ecosystems. Adv. Ecol. Res. 2014, 51, 79–104. [Google Scholar] [CrossRef]
- Bohmann, K.; Evans, A.; Gilbert, M.T.; Carvalho, G.R.; Creer, S.; Knapp, M.; Yu, D.W.; de Bruyn, M. Environmental DNA for wildlife biology and biodiversity monitoring. Trends Ecol. Evol. 2014, 29, 358–367. [Google Scholar] [CrossRef] [PubMed]
- Handley, L.L. How will the ‘molecular revolution’ contribute to biological recording? Biol. J. Linnean Soc. 2015, 115, 750–767. [Google Scholar] [CrossRef]
- Creer, S.; Deiner, K.; Frey, S.; Porazinska, D.; Taberlet, P.; Thomas, W.K.; Potter, C.; Bik, H.M.; Freckleton, R. The ecologist’s field guide to sequence-based identification of biodiversity. Methods Ecol. Evol. 2016, 7, 1008–1018. [Google Scholar] [CrossRef]
- Deiner, K.; Bik, H.M.; Machler, E.; Seymour, M.; Lacoursiere-Roussel, A.; Altermatt, F.; Creer, S.; Bista, I.; Lodge, D.M.; de Vere, N.; et al. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Mol. Ecol. 2017, 9, 16. [Google Scholar] [CrossRef] [PubMed]
- Ficetola, G.F.; Miaud, C.; Pompanon, F.; Taberlet, P. Species detection using environmental DNA from water samples. Biol. Lett. 2008, 4, 423–425. [Google Scholar] [CrossRef] [PubMed]
- Jerde, C.L.; Mahon, A.R.; Chadderton, W.L.; Lodge, D.M. “Sight-unseen” detection of rare aquatic species using environmental DNA. Conserv. Lett. 2011, 4, 150–157. [Google Scholar] [CrossRef]
- Spear, S.F.; Groves, J.D.; Williams, L.A.; Waits, L.P. Using environmental DNA methods to improve detectability in a hellbender (Cryptobranchus alleganiensis) monitoring program. Biol. Conserv. 2015, 183, 38–45. [Google Scholar] [CrossRef]
- Torresdal, J.D.; Farrell, A.D.; Goldberg, C.S. Environmental DNA detection of the golden tree frog (Phytotriades auratus) in bromeliads. PLoS ONE 2017, 12, e0168787. [Google Scholar] [CrossRef] [PubMed]
- Nichols, R.V.; Konigsson, H.; Danell, K.; Spong, G. Browsed twig environmental DNA: Diagnostic PCR to identify ungulate species. Mol. Ecol. Resour. 2012, 12, 983–989. [Google Scholar] [CrossRef] [PubMed]
- Roslin, T.; Majaneva, S. The use of DNA barcodes in food web construction-terrestrial and aquatic ecologists unite! Genome 2016, 59, 603–628. [Google Scholar] [CrossRef] [PubMed]
- Vestheim, H.; Jarman, S.N. Blocking primers to enhance PCR amplification of rare sequences in mixed samples—A case study on prey DNA in antarctic krill stomachs. Front. Zool. 2008, 5, 12. [Google Scholar] [CrossRef] [PubMed]
- Srivathsan, A.; Sha, J.C.; Vogler, A.P.; Meier, R. Comparing the effectiveness of metagenomics and metabarcoding for diet analysis of a leaf-feeding monkey (Pygathrix nemaeus). Mol. Ecol. Resour. 2015, 15, 250–261. [Google Scholar] [CrossRef] [PubMed]
- Chain, F.J.J.; Brown, E.A.; MacIsaac, H.J.; Cristescu, M.E.; Cowie, R. Metabarcoding reveals strong spatial structure and temporal turnover of zooplankton communities among marine and freshwater ports. Divers. Distrib. 2016, 22, 493–504. [Google Scholar] [CrossRef]
- Bista, I.; Carvalho, G.R.; Walsh, K.; Seymour, M.; Hajibabaei, M.; Lallias, D.; Christmas, M.; Creer, S. Annual time-series analysis of aqueous eDNA reveals ecologically relevant dynamics of lake ecosystem biodiversity. Nat. Commun. 2017, 8, 14087. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Hajibabaei, M.; Pompanon, F.; Brochmann, C.; Willerslev, E. Towards next-generation biodiversity assessment using DNA metabarcoding. Mol. Ecol. 2012, 21, 2045–2050. [Google Scholar] [CrossRef] [PubMed]
- Kelly, R.P.; Port, J.A.; Yamahara, K.M.; Crowder, L.B. Using environmental DNA to census marine fishes in a large mesocosm. PLoS ONE 2014, 9, e86175. [Google Scholar] [CrossRef] [PubMed]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [PubMed]
- Pinol, J.; Mir, G.; Gomez-Polo, P.; Agusti, N. Universal and blocking primer mismatches limit the use of high-throughput DNA sequencing for the quantitative metabarcoding of arthropods. Mol. Ecol. Resour. 2015, 15, 819–830. [Google Scholar] [CrossRef] [PubMed]
- Riaz, T.; Shehzad, W.; Viari, A.; Pompanon, F.; Taberlet, P.; Coissac, E. Ecoprimers: Inference of new DNA barcode markers from whole genome sequence analysis. Nucleic Acids Res. 2011, 39, e145. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.G. Reproducibility of Ancient DNA Sequences from Extinct Pleistocene Fauna. Mol. Biol. Evol. 1996, 13, 283–285. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Evans, N.T.; Li, Y.; Renshaw, M.A.; Olds, B.P.; Deiner, K.; Turner, C.R.; Jerde, C.L.; Lodge, D.M.; Lamberti, G.A.; Pfrender, M.E. Fish community assessment with eDNA metabarcoding: Effects of sampling design and bioinformatic filtering. Can. J. Fish. Aquat. Sci. 2017, 74, 1362–1374. [Google Scholar] [CrossRef]
- Hänfling, B.; Lawson, H.L.; Read, D.S.; Hahn, C.; Li, J.; Nichols, P.; Blackman, R.C.; Oliver, A.; Winfield, I.J. Environmental DNA metabarcoding of lake fish communities reflects long-term data from established survey methods. Mol. Ecol. 2016, 25, 3101–3119. [Google Scholar] [CrossRef] [PubMed]
- Leonard, J.A.; Shanks, O.; Hofreiter, M.; Kreuz, E.; Hodges, L.; Ream, W.; Wayne, R.K.; Fleischer, R.C. Animal DNA in PCR reagents plagues ancient DNA research. J. Archaeol. Sci. 2007, 34, 1361–1366. [Google Scholar] [CrossRef]
- Hinck, J.E.; Hossack, B.R.; Honeycutt, R.K. Amphibian Acoustic Data from the Arizona 1, Pinenut, and Canyon Breccia Pipe Uranium Mines in Arizona; U.S. Geological Survey: Reston, VA, USA, 2017.
- Greer, A.L.; Brunner, J.L.; Collins, J.P. Spatial and temporal patterns of Ambystoma tigrinum virus (atv) prevalence in tiger salamanders Ambystoma tigrinum nebulosum. Dis. Aquat. Org. 2009, 85, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Collins, J.P. Distribution, habitats, and life history variation in the Tiger Salamander, Ambystoma tigrinum, in east-central and southeast Arizona. Copeia 1981, 1981, 666–675. [Google Scholar] [CrossRef]
- Sexton, O.J.; Bizer, J.R. Life history patterns of Ambystoma tigrinum in Montane Colorado. Am. Midl. Nat. 1978, 99, 101–118. [Google Scholar] [CrossRef]
- Takahara, T.; Minamoto, T.; Yamanaka, H.; Doi, H.; Kawabata, Z. Estimation of fish biomass using environmental DNA. PLoS ONE 2012, 7, e35868. [Google Scholar] [CrossRef] [PubMed]
- Elbrecht, V.; Leese, F. Can DNA-based ecosystem assessments quantify species abundance? Testing primer bias and biomass—Sequence relationships with an innovative metabarcoding protocol. PLoS ONE 2015, 10, e0130324. [Google Scholar] [CrossRef] [PubMed]
- Leray, M.; Agudelo, N.; Mills, S.C.; Meyer, C.P. Effectiveness of annealing blocking primers versus restriction enzymes for characterization of generalist diets: Unexpected prey revealed in the gut contents of two coral reef fish species. PLoS ONE 2013, 8, e58076. [Google Scholar] [CrossRef] [PubMed]
- Boessenkool, S.; Epp, L.S.; Haile, J.; Bellemain, E.; Edwards, M.; Coissac, E.; Willerslev, E.; Brochmann, C. Blocking human contaminant DNA during PCR allows amplification of rare mammal species from sedimentary ancient DNA. Mol. Ecol. 2012, 21, 1806–1815. [Google Scholar] [CrossRef] [PubMed]
- Evans, N.T.; Olds, B.P.; Renshaw, M.A.; Turner, C.R.; Li, Y.; Jerde, C.L.; Mahon, A.R.; Pfrender, M.E.; Lamberti, G.A.; Lodge, D.M. Quantification of mesocosm fish and amphibian species diversity via environmental DNA metabarcoding. Mol. Ecol. Resour. 2016, 16, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Ushio, M.; Fukuda, H.; Inoue, T.; Makoto, K.; Kishida, O.; Sato, K.; Murata, K.; Nikaido, M.; Sado, T.; Sato, Y.; et al. Environmental DNA enables detection of terrestrial mammals from forest pond water. Mol. Ecol. Resour. 2017, 7, 11. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.M.; Williamson, C.H.; Sanchez, D.E.; Sobek, C.J.; Chambers, C.L. Species from feces: Order-wide identification of chiroptera from guano and other non-invasive genetic samples. PLoS ONE 2016, 11, e0162342. [Google Scholar] [CrossRef] [PubMed]
- Miya, M.; Sato, Y.; Fukunaga, T.; Sado, T.; Poulsen, J.Y.; Sato, K.; Minamoto, T.; Yamamoto, S.; Yamanaka, H.; Araki, H.; et al. Mifish, a set of universal PCR primers for metabarcoding environmental DNA from fishes: Detection of more than 230 subtropical marine species. R. Soc. Open Sci. 2015, 2, 150088. [Google Scholar] [CrossRef] [PubMed]
- Fahner, N.A.; Shokralla, S.; Baird, D.J.; Hajibabaei, M. Large-scale monitoring of plants through environmental DNA metabarcoding of soil: Recovery, resolution, and annotation of four DNA markers. PLoS ONE 2016, 11, e0157505. [Google Scholar] [CrossRef] [PubMed]
- Eichmiller, J.J.; Miller, L.M.; Sorensen, P.W. Optimizing techniques to capture and extract environmental DNA for detection and quantification of fish. Mol. Ecol. Resour. 2016, 16, 56–68. [Google Scholar] [CrossRef] [PubMed]
- Williams, K.E.; Huyvaert, K.P.; Piaggio, A.J. No filters, no fridges: A method for preservation of water samples for eDNA analysis. BMC Res. Notes 2016, 9, 298. [Google Scholar] [CrossRef] [PubMed]
- Cannon, M.V.; Hester, J.; Shalkhauser, A.; Chan, E.R.; Logue, K.; Small, S.T.; Serre, D. In silico assessment of primers for eDNA studies using primertree and application to characterize the biodiversity surrounding the Cuyahoga River. Sci. Rep. 2016, 6, 22908. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Sampling Site | Site Type | Latitude | Longitude | Month-Year Sampled | Number of Centrifuged Samples | Number of Filtered Samples | Total Volume Processed (mL) |
|---|---|---|---|---|---|---|---|
| Arizona One Tank | Tank | 36°29′57.95′′ N | 112°47′45.32′′ W | July-15 | 4 | 180 | |
| August-16 | 4 | 1 | 230 | ||||
| Hack Tank | Tank | 36°36′52.52′′ N | 112°50′54.59′′ W | March-15 | 4 | 180 | |
| July-15 | 4 | 180 | |||||
| Kanab North Tank | Tank | 36°42′3.53′′ N | 112°39′39.11′′ W | July-15 | 4 | 180 | |
| August-16 | 4 | 1 | 230 | ||||
| Little Robinson Tank | Tank | 36°30′0.42′′ N | 112°50′31.07′′ W | July-15 | 2 | 90 | |
| August-16 | 4 | 1 | 205 | ||||
| Pine Nut Tank | Tank | 36°30′26.77′′ N | 112°43′44.30′′ W | July-15 | 4 | 180 | |
| August-16 | 2 | 2 | 109 | ||||
| Wild Band Tank | Tank | 36°41′29.50′′ N | 112°49′18.34′′ W | July-15 | 2 | 90 | |
| August-16 | 4 | 1 | 230 | ||||
| Canyon Mine | Mine | 35°52′57.50′′ N | 112°5′44.52′′ W | June-16 | 1 | 45 |
| Site | Arizona One Tank | Kanab North Tank | Little Robinson Tank | Pine Nut Tank | Wild Band Tank | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Sample type | Filter | Tube | Filter | Tube | Filter | Tube | Filter | Tube | Filter | Tube |
| Average volume processed (mL) | 50 | 45 | 50 | 45 | 25 | 45 | 19 | 45 | 50 | 45 |
| Number of samples | 1 | 4 | 1 | 4 | 1 | 4 | 2 | 2 | 1 | 4 |
| Average number of taxa (16S marker) | 1 | 3.5 | 4 | 4.5 | 3 | 3.25 | 1 | 2.5 | 2 | 2 |
| Average number of taxa (12S marker) | 3 | 3 | 0 | 1.5 | 0 | 1.75 | 0 | 3 | 1 | 2.5 |
| Primer Name | Primer Sequence 5′–3′ |
|---|---|
| 12S-V5-Tailed-F1 | ACACTCTTTCCCTACACGACGCTCTTCCGATCTACTGGGATTAGATACCCC |
| 12S-V5-Tailed-R1 | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTTAGAACAGGCTCCTCTAG |
| Taylor_16S_DEGEN_F1_Tailed | ACACTCTTTCCCTACACGACGCTCTTCCGATCTGTTGGGGYGACYTYGGA |
| Taylor_16S_DEGEN_R1_Tailed | GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCTGCTGTTATCCCTRGRGTARC |
| Type | Resolution | Scientific Name | Common Name | Marker |
|---|---|---|---|---|
| Amphibians | Species | Ambystoma tigrinum | Tiger salamander | 12S, 16S |
| Amphibians | Species | Anaxyrus punctatus | Red-spotted toad | 16S |
| Amphibians | Genus | Spea spp. | Spadefoot toads | 12S, 16S |
| Birds | Genus | Anas spp. | Duck | 16S |
| Birds | Family | Anatidae | Duck | 12S |
| Birds | Family | Aredeidae | Heron | 12S |
| Birds | Family | Emberizidae | New world sparrows | 16S |
| Birds | Species | Falco sparverius | American kestrel | 12S |
| Birds | Order | Passeriformes | Perching birds | 12S |
| Birds | Species | Gallus gallus * | Chicken | 12S, 16S |
| Birds | Species | Meleagris gallopavo * | Turkey | 12S, 16S |
| Fish | Species | Lepomis cyanellus | Green sunfish | 12S |
| Fish | Species | Cyprinus carpio * | Common carp | 12S |
| Fish | Genus | Hypophthalmichthys spp. * | Bigheaded carps | 12S, 16S |
| Fish | Genus | Lepisosteus spp. * | Gar | 16S |
| Fish | Species | Mylopharyngodon piceus ** | Black carp | 12S, 16S |
| Fish | Family | Salmonidae * | Salmonid fish | 12S |
| Mammals | Species | Bos taurus | Cattle | 12S, 16S |
| Mammals | Genus | Canis spp. | Dog | 12S, 16S |
| Mammals | Species | Felis catus | Cat | 16S |
| Mammals | Species | Homo sapiens | Human | 12S, 16S |
| Mammals | Species | Lepus californicus | Black-tailed jackrabbit | 12S, 16S |
| Mammals | Genus | Myotis spp. | Myotis bats | 12S, 16S |
| Mammals | Genus | Sylvilagus spp. | Cottontail rabbits | 12S, 16S |
| Mammals | Species | Urocyon cinereoargenteus | Gray fox | 12S |
| Mammals | Species | Sus scrofa * | Pig | 12S, 16S |
| Reptiles | Family | Colubridae * | Colubrid snakes | 16S |
| Taxa | Total Number of 12S Reads | Percentage of Total 12S Reads | Taxa | Total Number of 16S Reads | Percentage of Total 16S Reads |
|---|---|---|---|---|---|
| Spea spp. | 1,417,797 | 35 | Homo sapiens | 2,227,802 | 47 |
| Homo sapiens | 1,286,429 | 32 | Bos Taurus | 1,314,121 | 28 |
| Bos taurus | 517,287 | 13 | Spea spp. | 889,532 | 19 |
| Ambystoma tigrinum | 477,703 | 12 | Ambystoma tigrinum | 108,565 | 2.3 |
| Lepomis cyanellus | 263,750 | 6.4 | Canis spp. | 83,389 | 1.8 |
| Passeriformes | 41,122 | 1.0 | Emberizidae | 42,153 | <1.0 |
| Aredeidae | 34,876 | <1.0 | Lepus californicus | 20,834 | <1.0 |
| Anatidae | 26,852 | <1.0 | Anas spp. | 15,055 | <1.0 |
| Lepus californicus | 3596 | <1.0 | Myotis spp. | 5351 | <1.0 |
| Falco sparverius | 1794 | <1.0 | Anaxyrus punctatus | 3371 | <1.0 |
| Canis spp. | 1576 | <1.0 | Felis catus | 2884 | <1.0 |
| Sylvilagus spp. | 1564 | <1.0 | Sylvilagus spp. | 191 | <1.0 |
| Urocyon cinereoargenteus | 310 | <1.0 | |||
| Myotis spp. | 110 | <1.0 | |||
| Total | 4,074,766 | Total | 4,713,248 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Klymus, K.E.; Richter, C.A.; Thompson, N.; Hinck, J.E. Metabarcoding of Environmental DNA Samples to Explore the Use of Uranium Mine Containment Ponds as a Water Source for Wildlife. Diversity 2017, 9, 54. https://doi.org/10.3390/d9040054
Klymus KE, Richter CA, Thompson N, Hinck JE. Metabarcoding of Environmental DNA Samples to Explore the Use of Uranium Mine Containment Ponds as a Water Source for Wildlife. Diversity. 2017; 9(4):54. https://doi.org/10.3390/d9040054
Chicago/Turabian StyleKlymus, Katy E., Catherine A. Richter, Nathan Thompson, and Jo Ellen Hinck. 2017. "Metabarcoding of Environmental DNA Samples to Explore the Use of Uranium Mine Containment Ponds as a Water Source for Wildlife" Diversity 9, no. 4: 54. https://doi.org/10.3390/d9040054
APA StyleKlymus, K. E., Richter, C. A., Thompson, N., & Hinck, J. E. (2017). Metabarcoding of Environmental DNA Samples to Explore the Use of Uranium Mine Containment Ponds as a Water Source for Wildlife. Diversity, 9(4), 54. https://doi.org/10.3390/d9040054