454 Pyrosequencing Analysis of Fungal Assemblages from Geographically Distant, Disparate Soils Reveals Spatial Patterning and a Core Mycobiome




Abstract
:
1. Introduction
2. Results and Discussion
2.1. Results
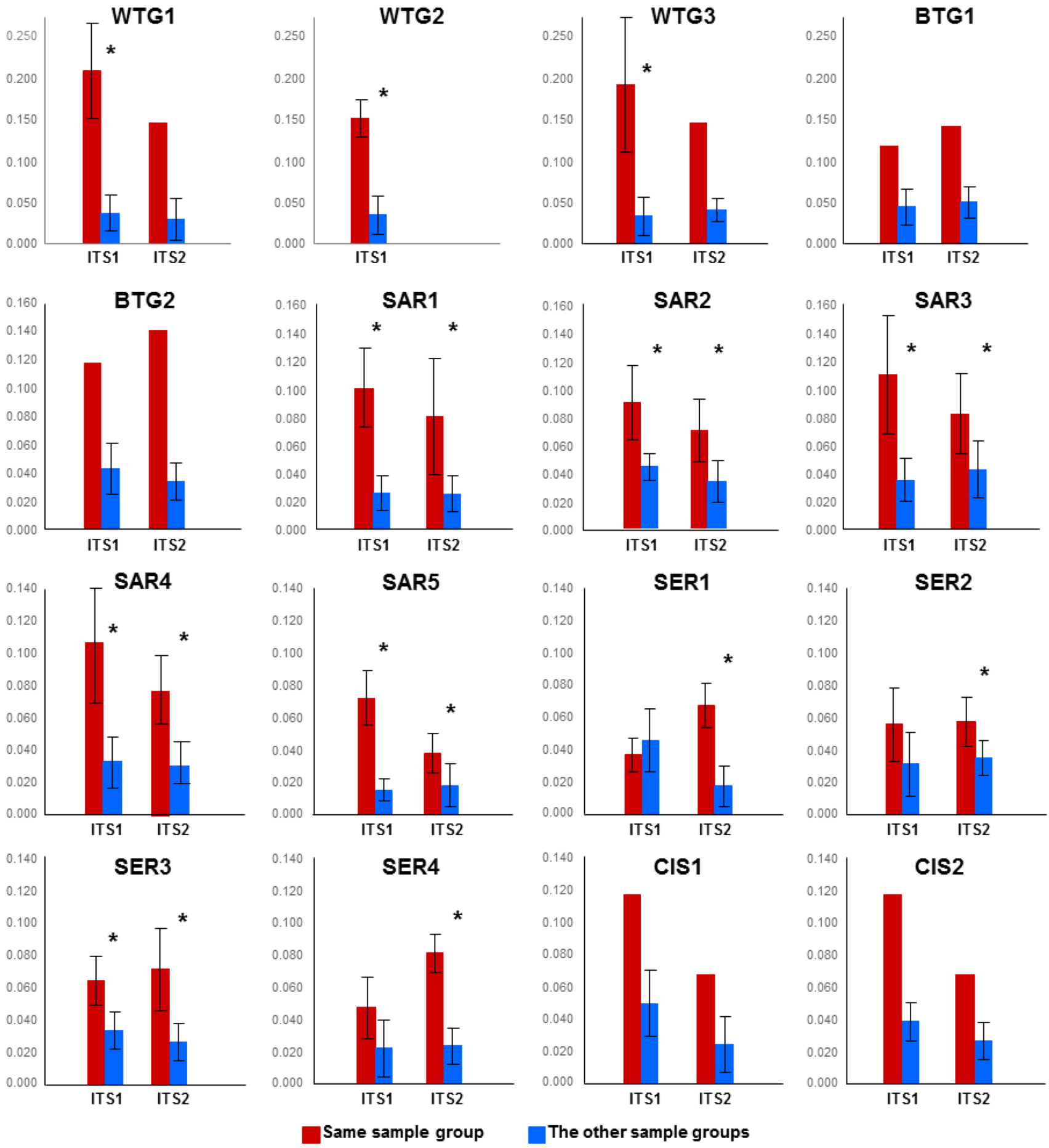
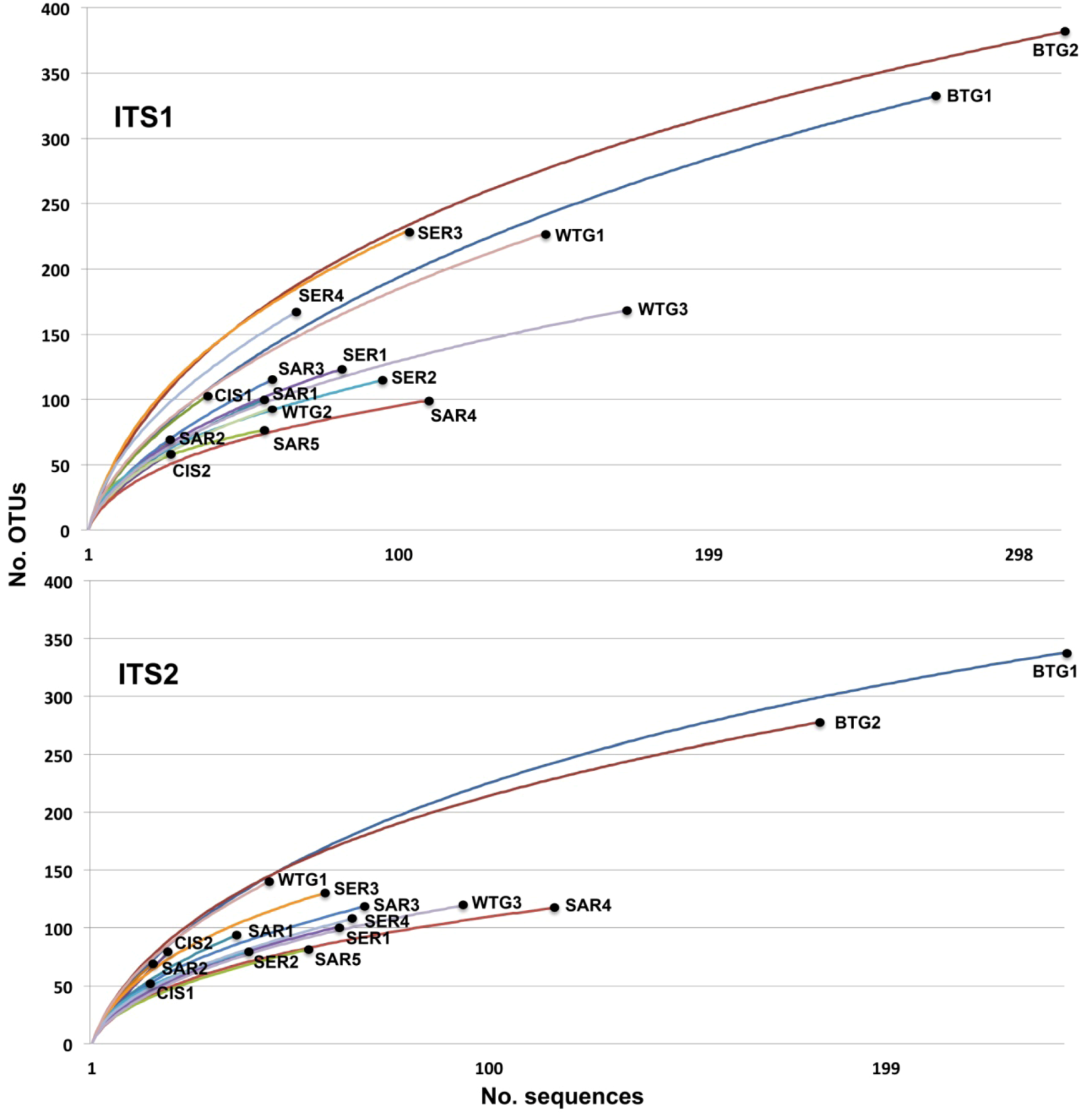
2.1.1. Comparison of the Mycobiomes in the Different Soil Samples

{kind=link}
{kind=link}
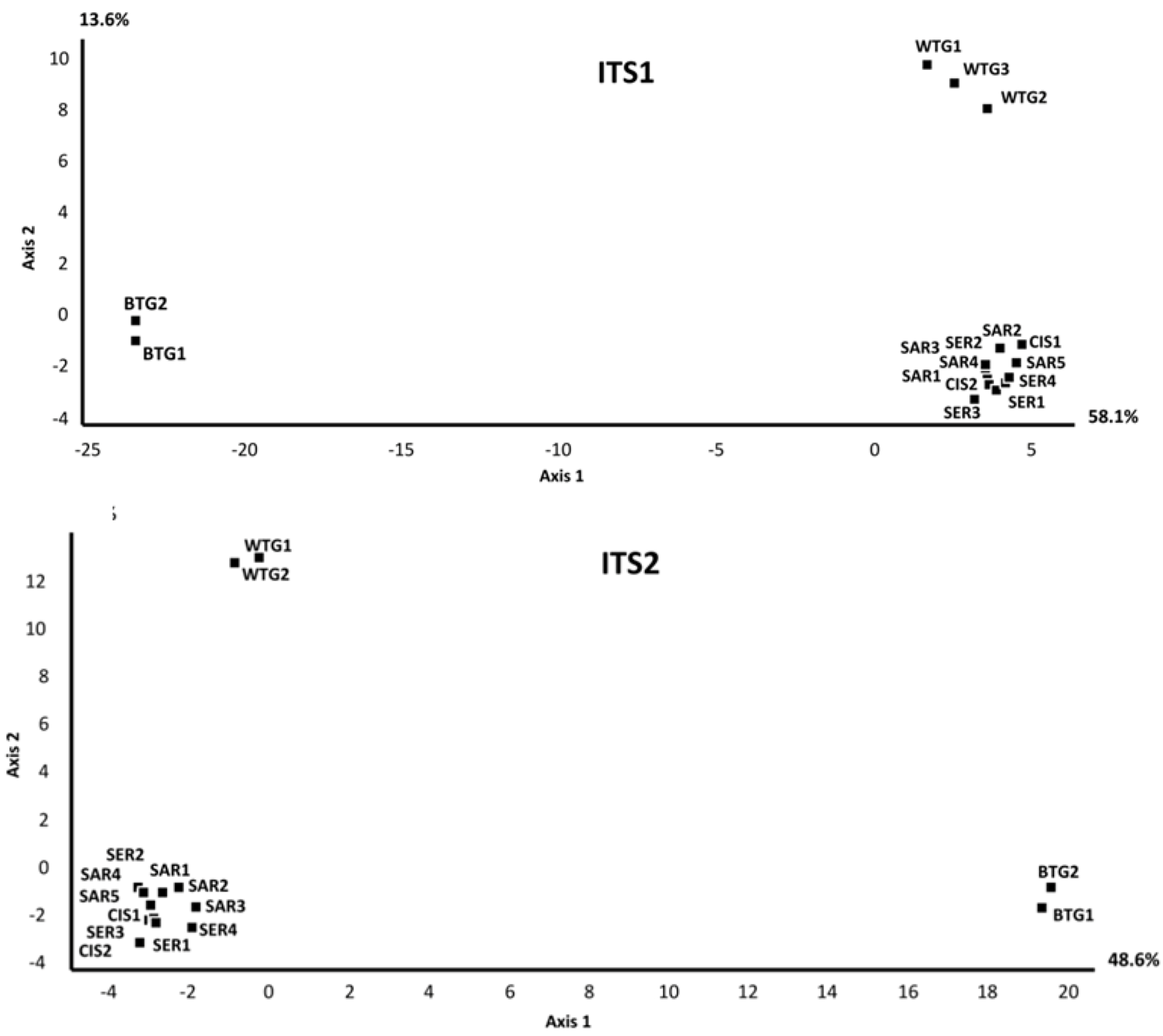{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locality | Acronym for locality/sample | Substrate | Management | Dominant plant functional type | Bioclimatic region [45] | Biogeographical region [46] | References |
| Montemagno, Piedmont, Italy | WTG 1 | White truffle (Tuber magnatum) grounds | — | ||||
| WTG1 | Alluvial | natural productive area | ECM/non-ECM6 | Temperate | Continental | ||
| WTG2 | Alluvial | non-productive area | ECM/non-ECM | Temperate | Continental | ||
| WTG3 | Alluvial | natural productive area | ECM/non-ECM | Temperate | Continental | ||
| BTG 2 | Black truffle (Tuber melanosporum) grounds | [39] | |||||
| BTG1 | Limestone | “brûlé” (burned) area | ECM/non-ECM | Temperate | Alpine | ||
| BTG2 | Limestone | non-“brûlé” (unburned) area | ECM/non-ECM | Temperate | Alpine | ||
| Berchidda, Sardinia, Italy | SAR 3 | Mediterranean agro-silvo-pastoral system | [43] | ||||
| SAR1 | Granite | tilled vineyard | non-ECM | Mediterranean | Mediterranean | ||
| SAR2 | Granite | covered vineyard | non-ECM | Mediterranean | Mediterranean | ||
| SAR3 | Granite | managed meadow | non-ECM | Mediterranean | Mediterranean | ||
| SAR4 | Granite | wooded pasture | ECM/ non-ECM | Mediterranean | Mediterranean | ||
| SAR5 | Granite | cork-oak forest | ECM | Mediterranean | Mediterranean | ||
| Piedmont, Italy | SER 4 | Soil and rock debris | [41] | ||||
| SER1 | Serpentine | disused mine | sparse vegetation | Temperate | Alpine | ||
| SER2 | Serpentine | anthropized area (road side) | sparse vegetation | Temperate | Alpine | ||
| SER3 | Serpentine | disused mine | sparse vegetation | Temperate | Alpine | ||
| SER4 | Serpentine | anthropized area (road side) | sparse vegetation | Temperate | Alpine | ||
| CIS 5 | Contaminated industrial soil | — | |||||
| CIS1 | Alluvial | abandoned industrial area | sparse vegetation | Temperate | Continental | ||
| CIS2 | Alluvial | abandoned industrial area | sparse vegetation | Temperate | Continental |
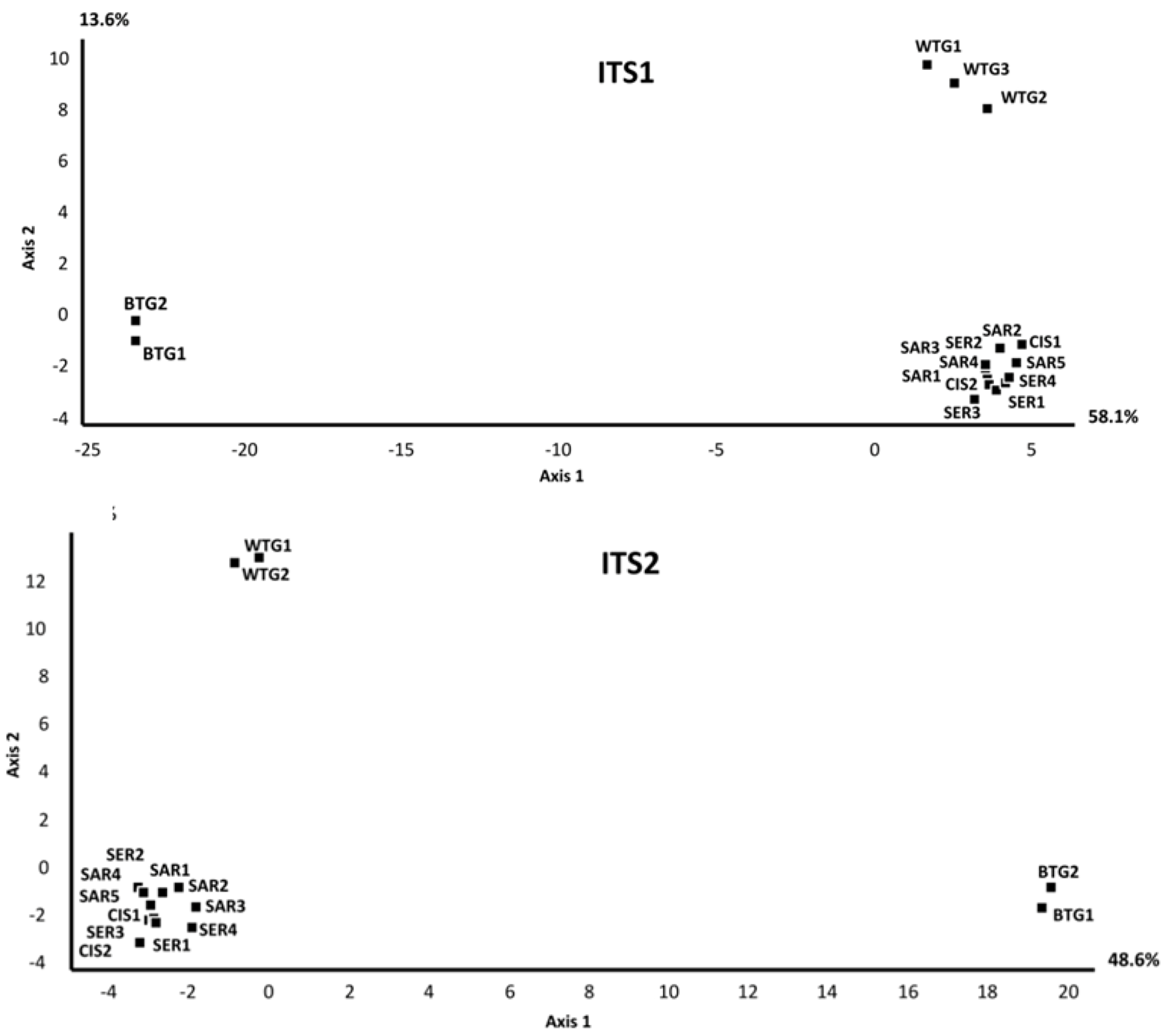
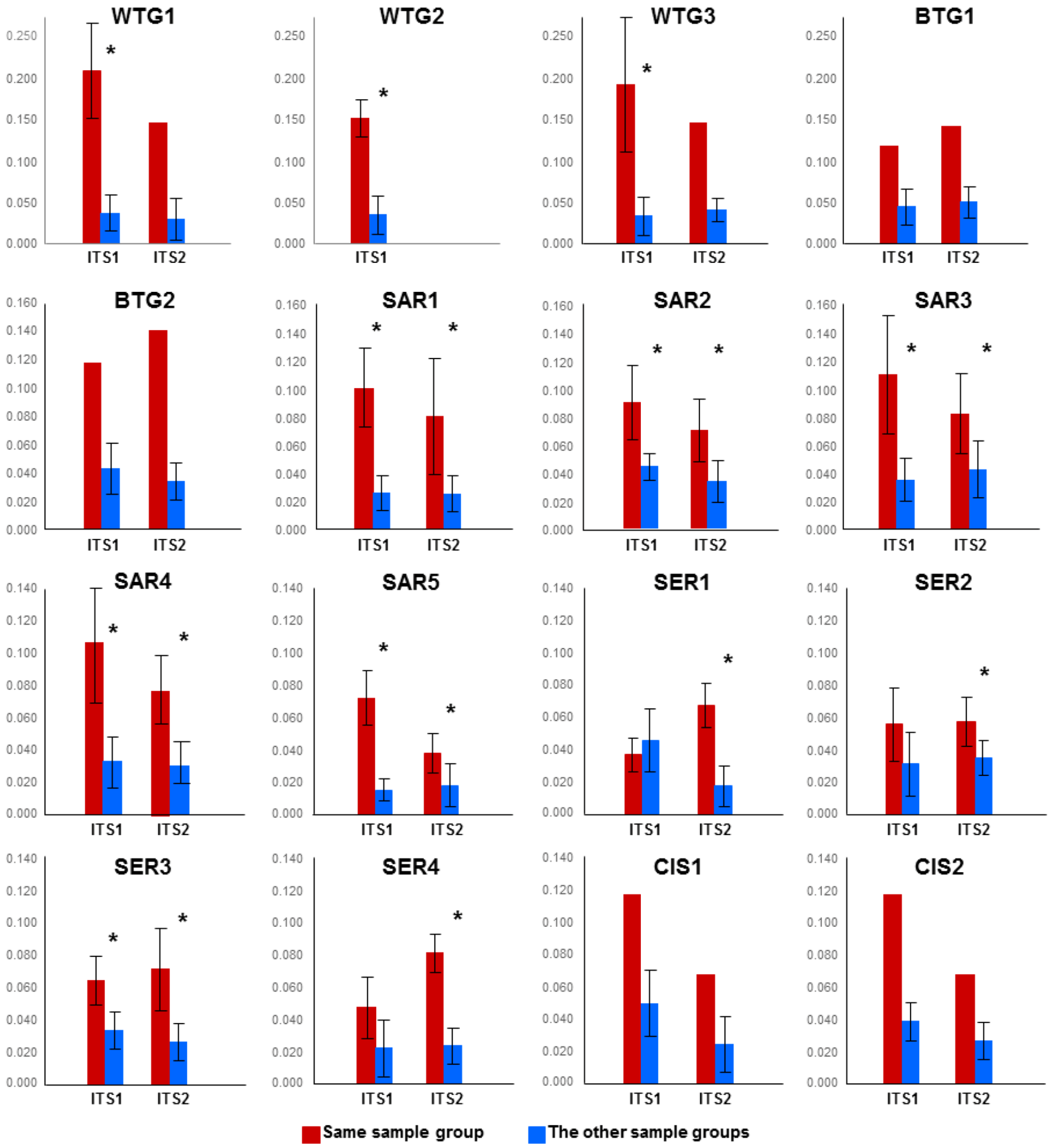
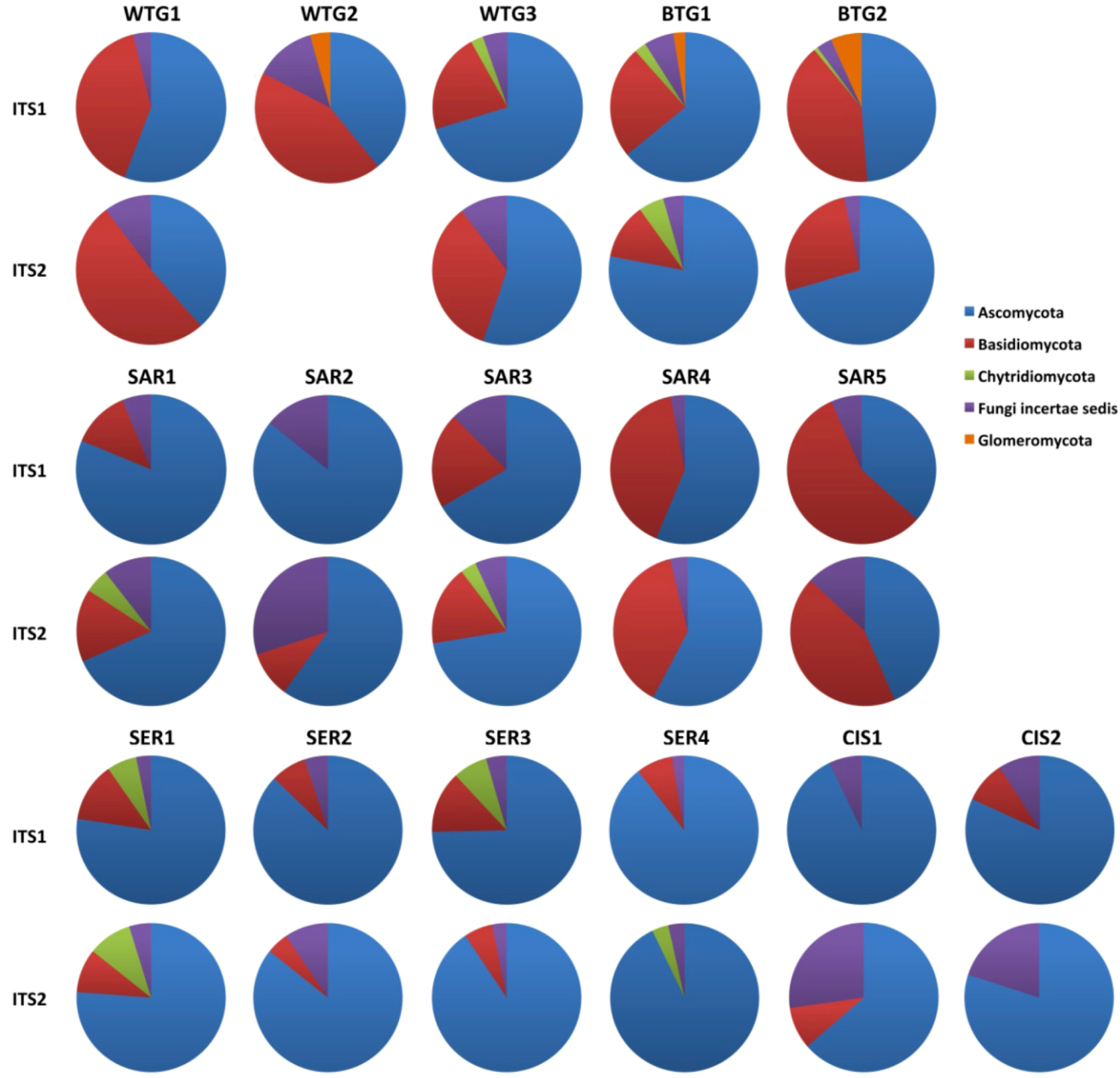
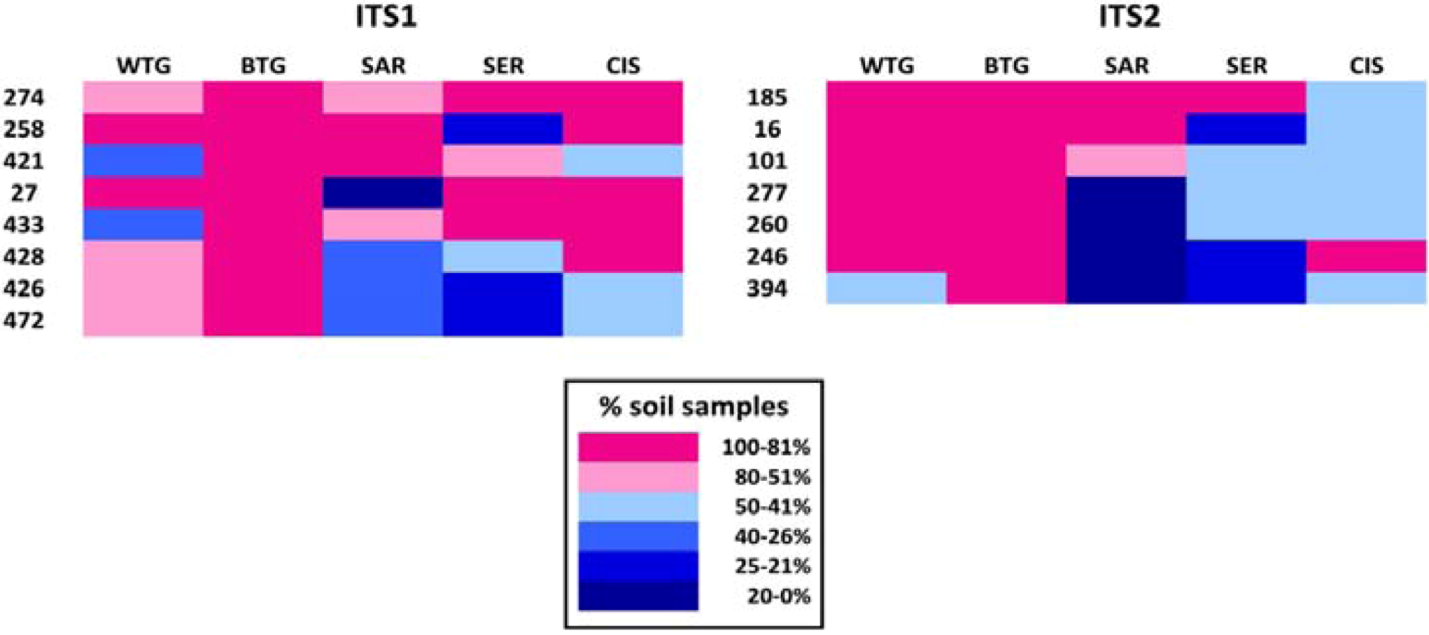
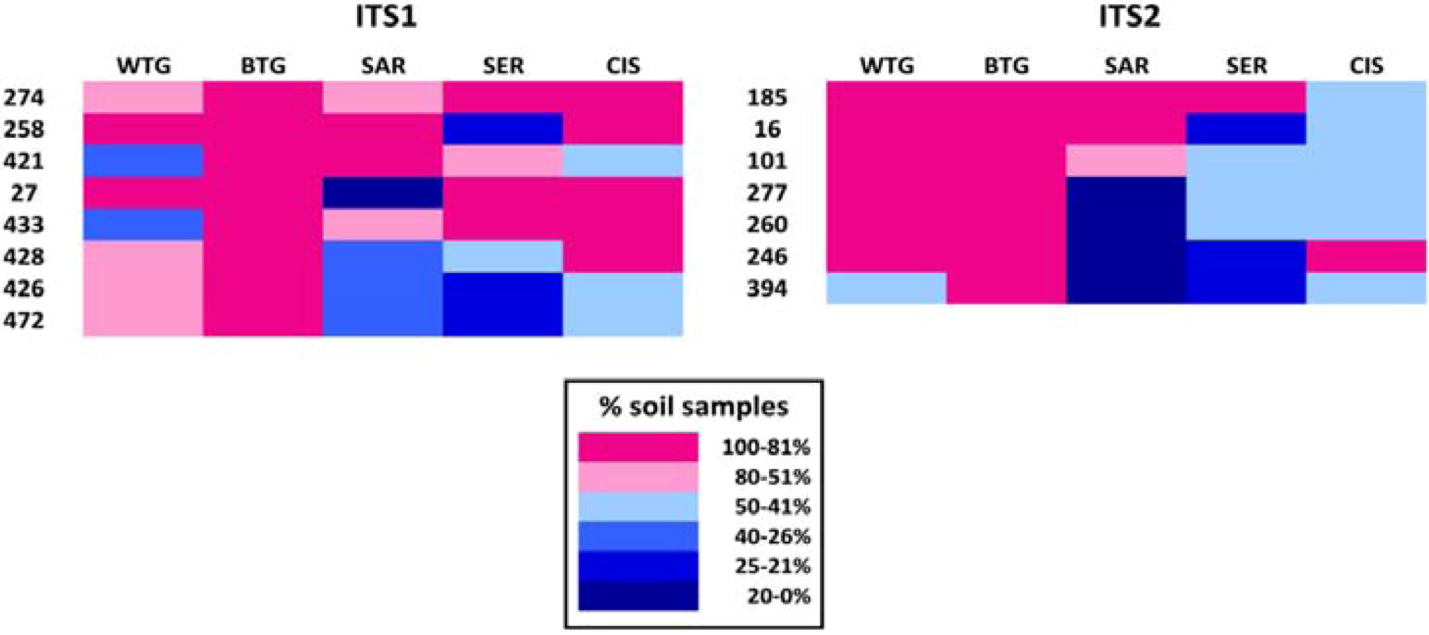
2.1.2. Fungal OTUs Shared by the Different Sample Groups
| OTU ID | No. soil samples | No. soils | No. samples | Tot. reads | Taxonomic assignment | ||||
|---|---|---|---|---|---|---|---|---|---|
| WTG | BTG | SAR | SER | CIS | |||||
| 274 | 2 | 2 | 4 | 4 | 2 | 5 | 14 | 130 | Cladosporium sp. |
| 258 | 3 | 2 | 5 | 1 | 2 | 5 | 13 | 712 | Fusarium sp. |
| 421 | 1 | 2 | 5 | 3 | 1 | 5 | 12 | 559 | Mortierella sp. |
| 27 | 3 | 2 | 1 | 4 | 2 | 5 | 12 | 285 | Mortierella alpina |
| 433 | 1 | 2 | 3 | 4 | 2 | 5 | 12 | 56 | Alternaria sp. |
| 428 | 2 | 2 | 2 | 2 | 2 | 5 | 10 | 50 | Fusarium sp. |
| 426 | 2 | 2 | 2 | 1 | 1 | 5 | 8 | 112 | Zygomycota sp. |
| 472 | 2 | 2 | 2 | 1 | 1 | 5 | 8 | 102 | Bionectria ochroleuca |
| 468 | 3 | 2 | - | 4 | 2 | 4 | 11 | 101 | Ilyonectria radicicola |
| 425 | 3 | 2 | 3 | 1 | - | 4 | 9 | 179 | Basidiomycota sp. |
| 463 | 2 | 2 | - | 3 | 1 | 4 | 8 | 145 | Mortierella sp. |
| 416 | 1 | 2 | 2 | 2 | - | 4 | 7 | 27 | Periconia macrospinosa |
| 570 | 3 | 1 | 1 | 1 | - | 4 | 6 | 32 | Tetracladium marchalianum |
| 429 | 3 | 1 | - | 1 | 1 | 4 | 6 | 44 | Truncatella angustata |
| 465 | 1 | 2 | 1 | 1 | - | 4 | 5 | 63 | Helotiales sp. |
| 701 | 1 | 2 | 1 | 1 | - | 4 | 5 | 10 | Penicillium sp. |
| 464 | - | 2 | 1 | 1 | 1 | 4 | 5 | 74 | Ascomycota sp. |
| 431 | - | 2 | 1 | 1 | 1 | 4 | 5 | 43 | Basidiomycota sp. |
| 325 | - | - | 3 | 3 | 2 | 3 | 8 | 99 | Epicoccum nigrum |
| 420 | 3 | 2 | - | 3 | - | 3 | 8 | 1279 | Tuber melanosporum |
| 162 | - | - | 1 | 4 | 1 | 3 | 6 | 154 | Fusarium sp. |
| 275 | - | 2 | 2 | 1 | - | 3 | 5 | 28 | Thelebolaceae sp. |
| 461 | - | 2 | 2 | 1 | - | 3 | 5 | 23 | Ascomycota sp. |
| 304 | - | 2 | 2 | - | 1 | 3 | 5 | 19 | Unknown fungus |
| 485 | - | 2 | 1 | 2 | - | 3 | 5 | 17 | Ascomycota sp. |
| 412 | - | - | 1 | 2 | 2 | 3 | 5 | 42 | Cladophialophora carrionii |
| 28 | 3 | - | 1 | 1 | - | 3 | 5 | 110 | Fusarium sp. |
| 447 | 3 | 1 | 1 | - | - | 3 | 5 | 21 | Olpidiaceae sp. |
| 442 | 3 | - | 1 | - | 1 | 3 | 5 | 39 | Gibellulopsis nigrescens |
| 613 | 3 | - | 1 | - | 1 | 3 | 5 | 27 | Tuber sp. |
| 432 | 1 | - | - | 3 | 1 | 3 | 5 | 44 | Chroogomphus rutilus |
| 435 | - | 1 | - | 2 | 2 | 3 | 5 | 51 | Ulurua sp. |
| 477 | 2 | 2 | - | 1 | - | 3 | 5 | 13 | Pleosporales sp. |
| 475 | 2 | - | - | 1 | 2 | 3 | 5 | 9 | Acremonium persicinum |
| 470 | 2 | 2 | - | - | 1 | 3 | 5 | 91 | Mortierellales sp. |
| 502 | - | 2 | 1 | 1 | - | 3 | 4 | 15 | Devriesia pseudoamericana |
| 790 | - | 2 | 1 | 1 | - | 3 | 4 | 5 | Tuber indicum |
| 430 | 1 | 2 | - | 1 | - | 3 | 4 | 49 | Cadophora sp. |
| 446 | - | 2 | - | 1 | 1 | 3 | 4 | 17 | Unknown fungus |
| 511 | 1 | 1 | - | - | 2 | 3 | 4 | 8 | Pueraria montana |
| OTU ID | No. soil samples | No. soils | No. samples | Tot. reads | Taxonomic assignment | ||||
| WTG | BTG | SAR | SER | CIS | |||||
| 185 | 2 | 2 | 5 | 4 | 1 | 5 | 14 | 139 | Cladosporium sp. |
| 16 | 2 | 2 | 5 | 1 | 1 | 5 | 11 | 475 | Fusarium sp. |
| 101 | 2 | 2 | 3 | 2 | 1 | 5 | 10 | 65 | Fusarium sp. |
| 277 | 2 | 2 | 1 | 2 | 1 | 5 | 8 | 131 | Tetracladium sp. |
| 260 | 2 | 2 | 1 | 2 | 1 | 5 | 8 | 48 | Mortierella alpina |
| 246 | 2 | 2 | 1 | 1 | 2 | 5 | 8 | 38 | Fusarium sp. |
| 394 | 1 | 2 | 1 | 1 | 1 | 5 | 6 | 24 | Paraphoma chrysanthemicola |
| 478 | - | 2 | 4 | 4 | 1 | 4 | 11 | 206 | Phoma sp. |
| 399 | 2 | 2 | 3 | 2 | - | 4 | 9 | 100 | Helotiales sp. |
| 431 | - | 1 | 3 | 4 | 1 | 4 | 9 | 50 | Alternaria sp. |
| 384 | - | 2 | 2 | 2 | 2 | 4 | 8 | 15 | Penicillium sp. |
| 485 | 2 | 2 | 1 | - | 1 | 4 | 6 | 417 | Tuber sp. |
| 481 | 2 | 2 | 1 | - | 1 | 4 | 6 | 144 | Bionectria ochroleuca |
| 415 | - | 2 | 2 | 1 | 1 | 4 | 6 | 23 | Podospora miniglutinans |
| 402 | 2 | - | 1 | 1 | 1 | 4 | 5 | 19 | Gibellulopsis nigrescens |
| 418 | 2 | 1 | 1 | 1 | - | 4 | 5 | 14 | Exophiala sp. |
| 447 | 2 | 2 | 5 | - | - | 3 | 9 | 98 | Cryptococcus aerius |
| 53 | 1 | 2 | 4 | - | - | 3 | 7 | 311 | Chaetomium sp. |
| 433 | - | 1 | 3 | 4 | - | 3 | 8 | 38 | Fusarium sp. |
| 449 | 2 | - | 2 | 1 | - | 3 | 5 | 58 | Penicillium sp. |
| 440 | 2 | 2 | 2 | - | - | 3 | 6 | 44 | Metarhizium anisopliae |
| 132 | 2 | 1 | 2 | - | - | 3 | 5 | 34 | Mortierella sp. |
| 98 | 1 | 2 | 2 | - | - | 3 | 5 | 27 | Preussia sp. |
| 374 | - | 2 | 1 | 1 | - | 3 | 4 | 10 | Ascomycota sp. |
| 521 | 1 | 2 | 1 | - | - | 3 | 4 | 5 | Hypocreales sp. |
| 3 | 2 | 1 | - | 3 | - | 3 | 6 | 93 | Mortierella sp. |
| 516 | - | 2 | - | 1 | 1 | 3 | 4 | 16 | Aureobasidium pullulans |
| 68 | 1 | 2 | - | - | 1 | 3 | 4 | 47 | Mortierella sp. |
2.2. Discussion
2.2.1. Soil Mycobiomes Are Spatially Structured Over a Broad Scale
2.2.2. Core Soil Fungi Likely Exhibit Nutritional Versatility
3. Experimental Section
3.1. Study Sites
3.2. Sampling, DNA Extraction, PCR and Pyrosequencing
3.3. Processing of Pyrosequencing Data
3.4. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
References
- Shade, A.; Handelsman, J. Beyond the Venn diagram: the hunt for a core microbiome. Environ. Microbiol. 2012, 14, 4–12. [Google Scholar] [CrossRef]
- Curtis, T.P.; Sloan, W.T. Exploring microbial diversity—A vast below. Science 2005, 309, 1331–1333. [Google Scholar] [CrossRef]
- Vogel, T.M.; Simonet, P.; Jansson, J.K.; Hirsch, P.R.; Tiedje, J.M.; van Elsas, J.D.; Bailey, M.J.; Nalin, R.; Philippot, L. TerraGenome: a consortium for the sequencing of a soil metagenome. Nat.Rev. Microbiol. 2009, 7, 252–252. [Google Scholar] [CrossRef]
- Green, J.; Bohannan, B.J.M. Spatial scaling of microbial biodiversity. Trends Ecol. Evol. 2006, 21, 501–507. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Bohannan, B.J.M.; Brown, J.H.; Colwell, R.K.; Fuhrman, J.A.; Green, J.L.; Horner-Devine, M.C.; Kane, M.; Krumins, J.A.; Kuske, C.R.; et al. Microbial biogeography: putting microorganisms on the map. Nature Rev. Microbiol. 2006, 4, 102–112. [Google Scholar]
- Prosser, J.I.; Bohannan, B.J.M.; Curtis, T.P.; Ellis, R.J.; Firestone, M.K.; Freckleton, R.P.; Green, J.L.; Green, L.E.; Killham, K.; Lennon, J.J.; et al. The role of ecological theory in microbial ecology. Nature Rev. Microbiol. 2007, 5, 384–392. [Google Scholar] [CrossRef]
- Hanson, C.A.; Fuhrman, J.A.; Horner-Devine, M.C.; Martiny, J.B.H. Beyond biogeographic patterns: processes shaping the microbial landscape. Nat. Rev. Microbiol. 2012, 10, 497–506. [Google Scholar]
- Lindström, E.S.; Langenheder, S. Local and regional factors influencing bacterial community assembly. Environ. Microbiol. Rep. 2012, 4, 1–9. [Google Scholar] [CrossRef]
- Dighton, J. Fungi in Ecosystem Processes; Mycology Series; Marcel Dekker Inc.: New York & Basel, USA & Switzerland, 2003. [Google Scholar]
- Coleman, M. From Another Kingdom. The amazing world of Fungi; Royal Botanic Garden: Edinburgh, UK, 2010; p. 176. [Google Scholar]
- Gianinazzi, S.; Gollotte, A.; Binet, M.N.; van Tuinen, D.; Redecker, D.; Wipf, D. Agroecology: the key role of arbuscular mycorrhizas in ecosystem services. Mycorrhiza 2010, 20, 519–530. [Google Scholar] [CrossRef]
- Jeffery, S.; Gardi, C.; Jones, A.; Montanarella, L.; Marmo, L.; Miko, L.; Ritz, K.; Peres, G.; Römbke, J.; van der Putten, W.H. European Atlas of Soil Biodiversity; European Commission, Publications Office of the European Union: Luxembourg, Luxembourg, 2010; p. 128. [Google Scholar]
- Fisher, M.C.; Henk, D.A.; Briggs, C.J.; Brownstein, J.S.; Madoff, L.C.; McCraw, S.L.; Gurr, S.J. Emerging fungal threats to animal, plant and ecosystem health. Nature 2012, 484, 186–194. [Google Scholar]
- Bissett, A.; Richardson, A.E.; Baker, G.; Wakelin, S.; Thrall, P.H. Life history determines biogeographical patterns of soil bacterial communities over multiple spatial scales. Mol. Ecol. 2010, 19, 4315–4327. [Google Scholar] [CrossRef]
- Ragon, M.; Fontaine, M.C.; Moreira, D.; López-García, P. Different biogeographic patterns of prokaryotes and microbial eukaryotes in epilithic biofilms. Mol. Ecol. 2012, 21, 3852–3868. [Google Scholar] [CrossRef]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef]
- Lauber, C.L.; Hamady, M.; Knight, R.; Fierer, N. Pyrosequencing-based assessment of soil pH as a predictor of soil bacterial community composition at the continental scale. Appl. Environ. Microbiol. 2009, 75, 5111–5120. [Google Scholar] [CrossRef]
- Philippot, L.; Cuhel, J.; Saby, N.P.A.; Chèneby, D.; Chronáková, A.; Bru, D.; Arrouays, D.; Martin-Laurent, F.; Simek, M. Mapping fine-scale spatial patterns of size and activity of the denitrifier community. Environ. Microbiol. 2009, 11, 1518–1526. [Google Scholar] [CrossRef]
- Dequiedt, S.; Saby, N.P.A.; Lelievre, M.; Jolivet, C.; Thioulouse, J.; Toutain, B.; Arrouays, D.; Bispo, A.; Lemanceau, P.; Ranjard, L. Biogeographical patterns of soil molecular microbial biomass as influenced by soil characteristics and management. Global Ecol. Biogeogr. 2011, 20, 641–652. [Google Scholar] [CrossRef]
- Griffiths, R.I.; Thomson, B.C.; James, P.; Bell, T.; Bailey, M.; Whiteley, A.S. The bacterial biogeography of British soils. Environ. Microbiol. 2011, 13, 1642–1654. [Google Scholar] [CrossRef]
- Rousk, J.; Baath, E.; Brookes, P.C.; Lauber, C.L.; Lozupone, C.; Caporaso, J.G.; Knight, R.; Fierer, N. Soil bacterial and fungal communities across a pH gradient in an arable soil. ISME J. 2010, 4, 1340–1351. [Google Scholar] [CrossRef]
- De Vries, F.T.; Bååth, E.; Kuyper, T.W.; Bloem, J. High turnover of fungal hyphae in incubation experiments. FEMS Microbiol. Ecol. 2009, 67, 389–396. [Google Scholar]
- Martin, J.P.; Haider, K. Biodegradation of 14C-labeled model and cornstalk lignins, phenols, model phenolase humic polymers, and fungal melanins as influenced by a readily available carbon source and soil. Appl. Environ. Microbiol. 1979, 38, 283–289. [Google Scholar]
- Amelung, W.; Lobe, I.; Du Preez, C.C. Fate of microbial residues in sandy soils of the South African highveld as influenced by prolonged arable cropping. Eur. J. Soil Sci. 2002, 53, 29–35. [Google Scholar] [CrossRef]
- Strickland, M.S.; Rousk, J. Considering fungal:bacterial dominance in soils—Methods, controls, and ecosystem implications. Soil Biol. Biochem. 2010, 42, 1385–139. [Google Scholar] [CrossRef]
- De Vries, F.T.; van Groenigen, J.W.; Hoffland, E.; Jaap, B. Nitrogen losses from two grassland soils with different fungal biomass. Soil Biol. Biochem. 2011, 43, 997–1005. [Google Scholar] [CrossRef]
- Kaspari, M.; Stevenson, B.S.; Shik, J.; Kerekes, J.F. Scaling community structure: how bacteria, fungi, and ant taxocenes differentiate along a tropical forest floor. Ecology 2010, 91, 2221–2226. [Google Scholar] [CrossRef]
- Hovatter, S.R.; Dejelo, C.; Case, A.L.; Blackwood, C.B. Metacommunity organization of soil microorganisms depends on habitat defined by presence of Lobelia siphilitica plants. Ecology 2011, 92, 57–65. [Google Scholar] [CrossRef]
- Ranjard, L.; Lejon, D.P.H.; Mougel, C.; Schehrer, L.; Merdinoglu, D.; Chaussod, R. Sampling strategy in molecular microbial ecology: Influence of soil sample size on DNA fingerprinting analysis of fungal and bacterial communities. Environ. Microbiol. 2003, 5, 1111–1120. [Google Scholar] [CrossRef]
- Gihring, T.M.; Green, S.J.; Schadt, C.W. Massively parallel rRNA gene sequencing exacerbates the potential for biased community diversity comparisons due to variable library sizes. Environ. Microbiol. 2012, 14, 285–290. [Google Scholar] [CrossRef]
- Davison, J.; Opik, M.; Zobel, M.; Vasar, M.; Metsis, M.; Moora, M. Communities of arbuscular mycorrhizal fungi detected in forest soil are spatially heterogeneous but do not vary throughout the growing season. Plos One 2012, 7, e41938. [Google Scholar]
- pik, M.; Metsis, M.; Daniell, T.J.; Zobel, M.; Moora, M. Large-scale parallel 454 sequencing reveals host ecological group specificity of arbuscular mycorrhizal fungi in a boreonemoral forest. New Phytol. 2009, 184, 424–437. [Google Scholar] [CrossRef]
- Lumini, E.; Orgiazzi, A.; Borriello, R.; Bonfante, P.; Bianciotto, V. Disclosing arbuscular mycorrhizal fungal biodiversity in soil through a land-use gradient using a pyrosequencing approach. Environ. Microbiol. 2010, 12, 2165–2179. [Google Scholar]
- Dumbrell, A.J.; Ashton, P.D.; Aziz, N.; Feng, G.; Nelson, M.; Dytham, C.; Fitter, A.H.; Helgason, T. Distinct seasonal assemblages of arbuscular mycorrhizal fungi revealed by massively parallel pyrosequencing. New Phytol. 2011, 190, 794–804. [Google Scholar] [CrossRef]
- Buée, M.; Reich, M.; Murat, C.; Morin, E.; Nilsson, R.H.; Uroz, S.; Martin, F. 454 pyrosequencing analyses of forest soils reveal an unexpected high fungal diversity. New Phytol. 2009, 184, 449–456. [Google Scholar] [CrossRef]
- Lim, Y.W.; Kim, B.K.; Kim, C.; Jung, H.S.; Kim, B.S.; Lee, J.H.; Chun, J. Assessment of soil fungal communities using pyrosequencing. J. Microbiol. 2010, 48, 284–289. [Google Scholar] [CrossRef]
- Tedersoo, L.; Nilsson, R.H.; Abarenkov, K.; Jairus, T.; Sadam, A.; Saar, I.; Bahram, M.; Bechem, E.; Chuyong, G.; Koljalg, U. 454 Pyrosequencing and Sanger sequencing of tropical mycorrhizal fungi provide similar results but reveal substantial methodological biases. New Phytol. 2010, 188, 291–301. [Google Scholar] [CrossRef]
- Lentendu, G.; Zinger, L.; Manel, S.; Coissac, E.; Choler, P.; Geremia, R.A.; Melodelima, C. Assessment of soil fungal diversity in different alpine tundra habitats by means of pyrosequencing. Fungal Divers. 2011, 49, 113–123. [Google Scholar] [CrossRef]
- Mello, A.; Napoli, C.; Murat, C.; Morin, E.; Marceddu, G.; Bonfante, P. ITS-1 versus ITS-2 pyrosequencing: a comparison of fungal populations in truffle grounds. Mycologia 2011, 103, 1184–1193. [Google Scholar] [CrossRef]
- Baldrian, P.; Kolarik, M.; Stursova, M.; Kopecky, J.; Valaskova, V.; Vetrovsky, T. Active and total microbial communities in forest soil are largely different and highly stratified during decomposition. ISME J. 2012, 6, 248–258. [Google Scholar] [CrossRef]
- Daghino, S.; Murat, C.; Sizzano, E.; Girlanda, M.; Perotto, S. Fungal diversity is not determined by mineral and chemical differences in serpentine substrates. Plos One 2012, 7, e44233. [Google Scholar]
- Danielsen, L.; Thürmer, A.; Meinicke, P.; Buée, M.; Morin, E.; Martin, F.; Pilate, G.; Daniel, R.; Polle, A.; Reich, M. Fungal soil communities in a young transgenic poplar plantation form a rich reservoir for fungal root communities. Ecol. Evol. 2012, 2, 1935–1948. [Google Scholar] [CrossRef]
- Orgiazzi, A.; Lumini, E.; Nilsson, R.H.; Girlanda, M.; Vizzini, A.; Bonfante, P.; Bianciotto, V. Unravelling soil fungal communities from different Mediterranean land-use backgrounds. Plos One 2012, 7, e34847. [Google Scholar]
- Xu, L.H.; Ravnskov, S.; Larsen, J.; Nilsson, R.H.; Nicolaisen, M. Soil fungal community structure along a soil health gradient in pea fields examined using deep amplicon sequencing. Soil Biol. Biochem. 2012, 46, 6–32. [Google Scholar]
- Rivas-Martínez, S.; Penas, A.; Díaz, T.E. Bioclimatic Map of Europe Cartographic Service. University of León: Spain, 2004. Available online: http://www.globalbioclimatic.org/ (accessed on 18 February 2013).
- European Commission. Council Directive 92/43/EEC on the conservation of natural habitats and of wild fauna and flora. Eur. Commun. Gazette 1992, 206, 1–50.
- Bellemain, E.; Carlsen, T.; Brochmann, C.; Coissac, E.; Taberlet, P.; Kauserud, H. ITS as an environmental DNA barcode for fungi: an in silico approach reveals potential PCR biases. BMC Microbiol. 2010, 10, 189–197. [Google Scholar] [CrossRef]
- Martiny, J.B.H.; Eisen, J.A.; Penn, K.; Allison, S.D.; Horner-Devine, M.C. Drivers of bacterial β-diversity depend on spatial scale. Proc. Natl. Acad. Sci. USA 2011, 108, 7850–7854. [Google Scholar]
- Wardle, D.A.; Bardgett, R.D.; Klironomos, J.N.; Setälä, H.; van der Putten, W.H.; Wall, D.H. Ecological linkages between aboveground and belowground biota. Science 2004, 304, 1629–1633. [Google Scholar] [CrossRef]
- Christensen, M. Species diversity and dominance in fungal communities. In The Fungal community: Its Organization and Role in the Ecosystem; Wicklow, D.T., Carroll, G.C., Eds.; Marcel Dekker: New York, NY, USA, 1981; pp. 201–232. [Google Scholar]
- Nielsen, U.N.; Osler, G.H.R.; Campbell, C.D.; Burslem, D.F.R.P.; van der Wal, R. The influence of vegetation type, soil properties and precipitation on the composition of soil mite and microbial communities at the landscape scale. J. Biogeogr. 2010, 37, 1317–1328. [Google Scholar] [CrossRef]
- Bahram, M.; Polme, S.; Koljalg, U.; Zarre, S.; Tedersoo, L. Regional and local patterns of ectomycorrhizal fungal diversity and community structure along an altitudinal gradient in the Hyrcanian forests of northern Iran. New Phytol. 2012, 193, 465–473. [Google Scholar] [CrossRef]
- Green, J.L.; Holmes, A.J.; Westoby, M.; Oliver, I.; Briscoe, D.; Dangerfield, M.; Gillings, M.; Beattie, A.J. Spatial scaling of microbial eukaryote diversity. Nature 2004, 432, 747–750. [Google Scholar] [CrossRef]
- Lekberg, Y.; Koide, R.T.; Rohr, J.R.; Aldrich-Wolfe, L.; Morton, J.B. Role of niche restrictions and dispersal in the composition of arbuscular mycorrhizal fugal communities. J. Ecol. 2007, 95, 95–105. [Google Scholar] [CrossRef]
- Dennis, P.G.; Rushton, S.P.; Newsham, K.K.; Lauducina, V.A.; Ord, V.J.; Daniell, T.J.; O’Donnell, A.G.; Hopkins, D.W. Soil fungal community composition does not alter along a latitudinal gradient through the maritime and sub-Antarctic. Fungal Ecol. 2012, 5, 403–408. [Google Scholar] [CrossRef]
- Rice, W.C.; Gowda, P.H. Influence of geographical location, crop type and crop residue cover on bacterial and fungal community structures. Geoderma 2011, 160, 271–280. [Google Scholar] [CrossRef]
- Bao, Y.; Morimoto, S.; Takada-Hoshino, Y.; Okada, H.; Oba, H.; Takemoto, S.; Niwa, S.; Ohigashi, K.; Suzuki, C.; Nagaoka, K.; et al. Combined analyses of bacterial, fungal and nematode communities in andosolic agricultural Soils in Japan. Microbes Environ. 2012, 27, 72–79. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar]
- Brock, P.M.; Doring, H.; Bidartondo, M.I. How to know unknown fungi: The role of a herbarium. New Phytol. 2009, 181, 719–724. [Google Scholar] [CrossRef]
- Nagy, L.G.; Petkovits, T.; Kovács, G.M.; Voigt, K.; Csaba, V.l.; Papp, T. Where is the unseen fungal diversity hidden? A study of Mortierella reveals a large contribution of reference collections to the identification of fungal environmental sequences. New Phytol. 2011, 191, 789–794. [Google Scholar] [CrossRef]
- Hawksworth, D.L. The magnitude of fungal diversity: the 1.5 million species estimate revisited. Mycol. Res. 2001, 105, 1422–1432. [Google Scholar] [CrossRef]
- O’Brien, H.E.; Parrent, J.L.; Jackson, J.A.; Moncalvo, J.M.; Vilgalys, R. Fungal community analysis by large-scale sequencing of environmental samples. Appl. Environ. Microbiol. 2005, 71, 5544–5550. [Google Scholar] [CrossRef]
- Hibbett, D.S.; Ohman, A.; Glotzer, D.; Nuhn, M.; Kirk, P.; Nilsson, N.R.H. Progress in molecular and morphological taxon discovery in Fungi and options for formal classification of environmental sequences. Fung. Biol. Rev. 2011, 25, 38–47. [Google Scholar] [CrossRef]
- Schadt, C.W.; Martin, A.P.; Lipson, D.A.; Schmidt, S.K. Seasonal dynamics of previously unknown fungal lineages in tundra soils. Science 2003, 301, 1359–1361. [Google Scholar] [CrossRef]
- Porter, T.M.; Schadt, C.W.; Rizvi, L.; Martin, A.P.; Schmidt, S.K.; Scott-Denton, L.; Vilgalys, R.; Moncalvo, J.M. Widespread occurrence and phylogenetic placement of a soil clone group adds a prominent new branch to the fungal tree of life. Mol. Phylogenet. Evol. 2008, 46, 635–644. [Google Scholar] [CrossRef]
- Schoch, C.L.; Seifert, K.A.; Huhndorf, S.; Robert, V.; Spouge, J.L.; Levesque, C.A.; Chen, W. Nuclear ribosomal internal transcribed spacer (ITS) region as a universal DNA barcode marker for Fungi. Proc. Natl. Acad. Sci. USA 2012, 16, 6241–6246. [Google Scholar]
- Domsch, K.H.; Gams, W.; Anderson, T.-H. Compendium of Soil Fungi, 2nd ed; IHW-Verlag and Verlagsbuchhandlung: Eching, Germany, 2007. [Google Scholar]
- Fulthorpe, R.R.; Roesch, L.F.W.; Riva, A.; Triplett, E.W. Distantly sampled soils carry few species in common. ISME J. 2008, 2, 901–910. [Google Scholar] [CrossRef]
- Fierer, N.; Bradford, M.A.; Jackson, R.B. Toward an ecological classification of soil bacteria. Ecology 2007, 88, 1354–1364. [Google Scholar] [CrossRef]
- Hirsch, P.; Bernhard, M.; Cohen, S.; Ensign, J.; Jannasch, H.; Koch, A.; Marshall, K.; Matin, A.; Poindexter, J.; Rittenberg, S.; et al. Life under conditions of low nutrient concentrations: group report. In Strategies of Microbial Life in Extreme Environments; Shilo, M., Ed.; Dahlem Konferenzen Life Sciences Research Report: Weinheim, Germany, 1979; pp. 323–339. [Google Scholar]
- Gottschal, J. Some reflections on microbial competitive ness among heterotrophic bacteria. Anton. Leeuw. 1985, 51, 473–494. [Google Scholar] [CrossRef]
- Wainwright, M. Oligotrophic growth of fungi-stress or natural state? In Stress Tolerance of Fungi; Jennings, D.H., Ed.; Marcel Dekker: New York, NY, USA, 1993; pp. 127–144. [Google Scholar]
- Tribe, H.T.; Mabadeje, S.A. Growth of moulds on media prepared without organic nutrients. Trans. Br. Mycol. Soc. 1972, 58, 127–137. [Google Scholar] [CrossRef]
- Wainwright, M. Metabolic diversity of fungi in relation to growth and mineral cycling in soil-a review. Trans. Br. Mycol. Soc. 1988, 90, 159–170. [Google Scholar] [CrossRef]
- Parkinson, S.M.; Wainwrigh, M.; Killham, K. Observations on oligotrophic growth of fungi on silica gel. Mycol. Res. 1989, 93, 529–534. [Google Scholar] [CrossRef]
- Bergero, R.; Girlanda, M.; Varese, G.C.; Intili, D.; Luppi, A.M. Psychrooligotrophic fungi from Arctic soils of Franz Joseph Land. Polar Biol. 1998, 21, 361–368. [Google Scholar]
- Cooke, R.C.; Rayner, A.D.M. Ecology of Saprotrophic Fungi; Addison-Wesley Educational Publishers Inc.: London & New York, UK & USA, 1984. [Google Scholar]
- Murat, C.; Vizzini, A.; Bonfante, P.; Mello, A. Morphological and molecular typing of the below-ground fungal community in a natural Tuber magnatum truffle-ground. FEMS Microbiol. Lett. 2005, 245, 307–313. [Google Scholar] [CrossRef]
- Zampieri, E.; Mello, A.; Bonfante, P.; Murat, C. PCR primers specific for the genus Tuber reveal the presence of several truffle species in a truffle-ground. FEMS Microbiol. Lett. 2009, 297, 67–72. [Google Scholar] [CrossRef]
- Zampieri, E.; Murat, C.; Cagnasso, M.; Bonfante, P.; Mello, A. Soil analysis reveals the presence of an extended mycelial network in a Tuber magnatum truffle-ground. FEMS Microbiol. Ecol. 2010, 71, 43–49. [Google Scholar]
- Lagomarsino, A.; Benedetti, A.; Marinari, S.; Pompili, L.; Moscatelli, M.C.; Roggero, P.P.; Lai, R.; Ledda, L.; Grego, S. Soil organic C variability and microbial functions in a Mediterranean agro-forest ecosystem. Biol. Fertil. Soils 2011, 47, 283–291. [Google Scholar] [CrossRef]
- Bagella, S.; Caria, M.C. Vegetation series: a tool for the assessment of grassland ecosystem services in Mediterranean large-scale grazing systems. Fitosociologia 2011, 48, 47–54. [Google Scholar]
- Girlanda, M.; Favero-Longo, S.E.; Lazzari, A.; Segreto, R.; Perotto, S.; Siniscalco, C. Indigenous microfungi and plants reduce soil nonylphenol contamination and stimulate resident microfungal communities. Appl. Microbiol. Biot. 2009, 82, 359–370. [Google Scholar]
- Luis, P.; Walther, G.; Kellner, K.; Martin, F.; Buscot, F. Diversity of laccase genes from Basidiomycetes in a forest soil. Soil Biol. Biochem. 2004, 36, 1025–1036. [Google Scholar]
- Toju, H.; Tanabe, A.S.; Yamamoto, S.; Sato, H. High-coverage ITS primers for the DNA-based identification of Ascomycetes and Basidiomycetes in environmental samples. Plos One 2012, 7, e4086. [Google Scholar]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Gonzalez Pena, A.; Goodrich, J.K.; Gordon, J.I.; et al. QIIME allows analysis of high-throughput community sequencing data. Nature Methods 2010, 7, 335–336. [Google Scholar]
- Holland, S.M. Analytic rarefaction. Available online: http://www.uga.edu/strata/software/ (accessed on 18 February, 2013).
- Watanabe, M.; Yonezawa, T.; Lee, K.; Kumagai, S.; Sugita-Konishi, Y.; Goto, K.; Hara-Kudo, Y. Molecular phylogeny of the higher and lower taxonomy of the Fusarium genus and differences in the evolutionary histories of multiple genes. BMC Evol. Biol. 2011, 11, 322. [Google Scholar]
- Porter, T.M.; Golding, G.B. Factors that affect large subunit ribosomal DNA amplicon sequencing studies of fungal communities: Classification method, primer choice, and error. Plos One 2012, 7, e35749. [Google Scholar]
- Pukkila, P.J.; Skrzynia, C. Frequent changes in the number of reiterated ribosomal-RNA genes throughout the life-cycle of the basidiomycete Coprinus cinereus. Genetics 1993, 133, 203–211. [Google Scholar]
- Rooney, A.P.; Ward, T.J. Evolution of a large ribosomal RNA multigene family in filamentous fungi: birth and death of a concerted evolution paradigm. Proc. Natl. Acad. Sci. USA 2005, 102, 5084–5089. [Google Scholar]
- Klappenbach, J.A.; Dunbar, J.M.; Schmidt, T.M. RRNA operon copy number reflects ecological strategies of bacteria. Appl. Environ. Microbiol. 2000, 66, 1328–1333. [Google Scholar]
- Amend, A.S.; Seifert, K.A.; Bruns, T.D. Quantifying microbial communities with 454 pyrosequencing: does read abundance count? Mol. Ecol. 2010, 19, 5555–5565. [Google Scholar]
- Medinger, R.; Nolte, V.; Pandey, R.V.; Jost, S.; Ottenwalder, B.; Schlotterer, C.; Boenigk, J. Diversity in a hidden world: potential and limitation of next-generation sequencing for surveys of molecular diversity of eukaryotic microorganisms. Mol. Ecol. 2010, 19 (Suppl. 1), 32–40. [Google Scholar]
- Prosser, J.I. Replicate or lie. Environ. Microbiol. 2010, 12, 1806–1810. [Google Scholar]
- Van de Peer, Y.; Bonnet, E. ZT: A software tool for simple and partial Mantel tests. J. Stat. Soft. 2002, 7, 1–12. [Google Scholar]
- XLSTAT. Available online: http://www.xlstat.com/ (accessed on 18 February, 2013).
- Damon, C.; Lehembre, F.; Oger-Desfeux, C.; Luis, P.; Ranger, J.; Fraissinet-Tachet, L.; Marmeisse, R. Metatranscriptomics reveals the diversity of genes expressed by eukaryotes in forest soils. Plos One 2012, 7, e28967. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Orgiazzi, A.; Bianciotto, V.; Bonfante, P.; Daghino, S.; Ghignone, S.; Lazzari, A.; Lumini, E.; Mello, A.; Napoli, C.; Perotto, S.; et al. 454 Pyrosequencing Analysis of Fungal Assemblages from Geographically Distant, Disparate Soils Reveals Spatial Patterning and a Core Mycobiome. Diversity 2013, 5, 73-98. https://doi.org/10.3390/d5010073
Orgiazzi A, Bianciotto V, Bonfante P, Daghino S, Ghignone S, Lazzari A, Lumini E, Mello A, Napoli C, Perotto S, et al. 454 Pyrosequencing Analysis of Fungal Assemblages from Geographically Distant, Disparate Soils Reveals Spatial Patterning and a Core Mycobiome. Diversity. 2013; 5(1):73-98. https://doi.org/10.3390/d5010073
Chicago/Turabian StyleOrgiazzi, Alberto, Valeria Bianciotto, Paola Bonfante, Stefania Daghino, Stefano Ghignone, Alexandra Lazzari, Erica Lumini, Antonietta Mello, Chiara Napoli, Silvia Perotto, and et al. 2013. "454 Pyrosequencing Analysis of Fungal Assemblages from Geographically Distant, Disparate Soils Reveals Spatial Patterning and a Core Mycobiome" Diversity 5, no. 1: 73-98. https://doi.org/10.3390/d5010073
APA StyleOrgiazzi, A., Bianciotto, V., Bonfante, P., Daghino, S., Ghignone, S., Lazzari, A., Lumini, E., Mello, A., Napoli, C., Perotto, S., Vizzini, A., Bagella, S., Murat, C., & Girlanda, M. (2013). 454 Pyrosequencing Analysis of Fungal Assemblages from Geographically Distant, Disparate Soils Reveals Spatial Patterning and a Core Mycobiome. Diversity, 5(1), 73-98. https://doi.org/10.3390/d5010073