cTBP: A Successful Intron Length Polymorphism (ILP)-Based Genotyping Method Targeted to Well Defined Experimental Needs


Abstract
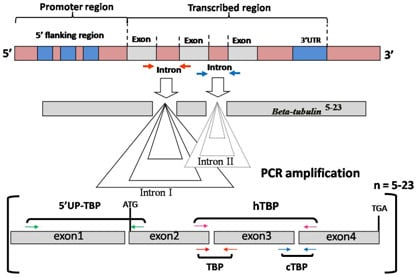
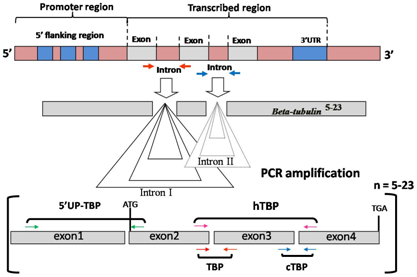
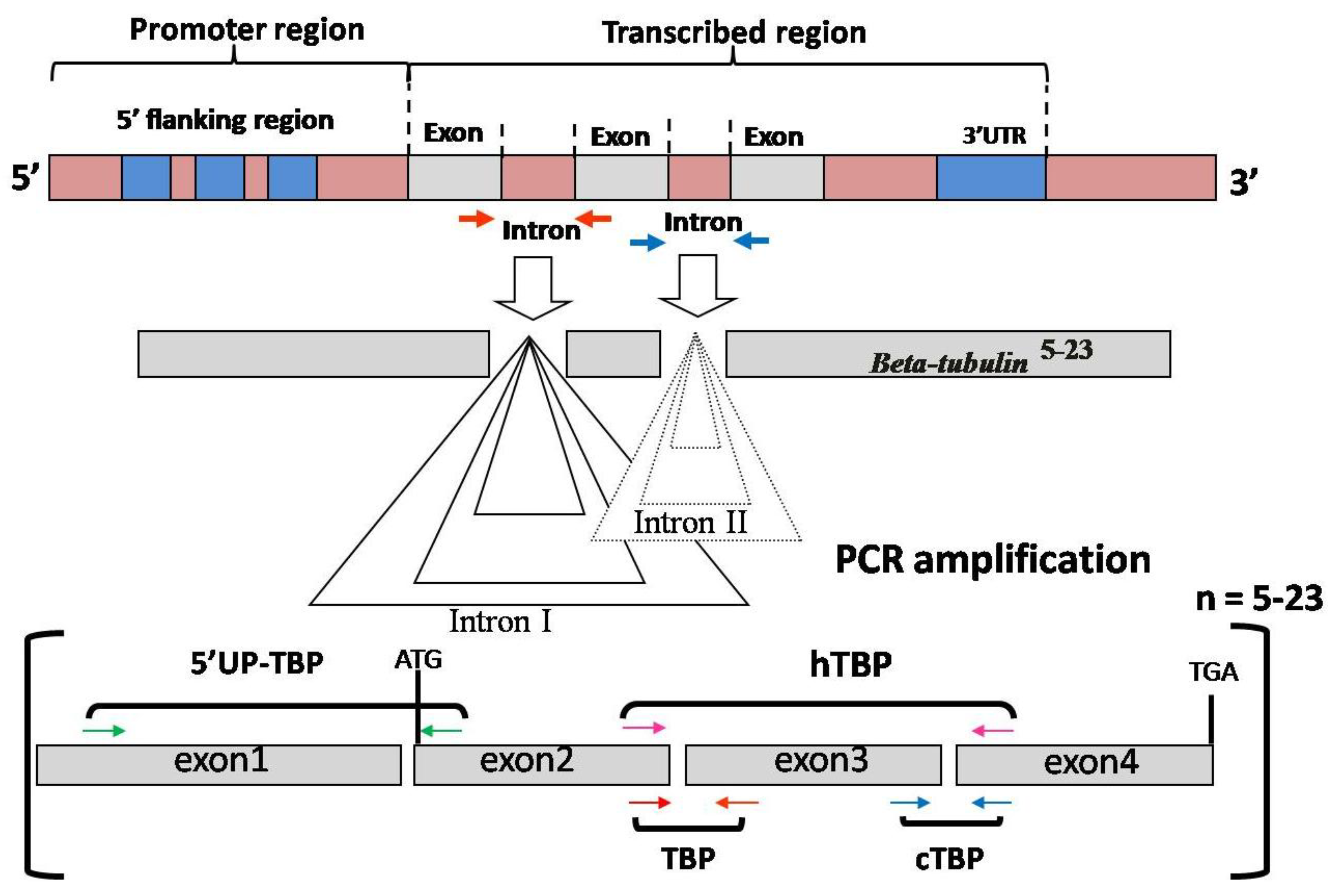
:
1. Introduction
2. Experimental Section
2.1. Plant Material

{kind=link}
{kind=link}
{kind=link}
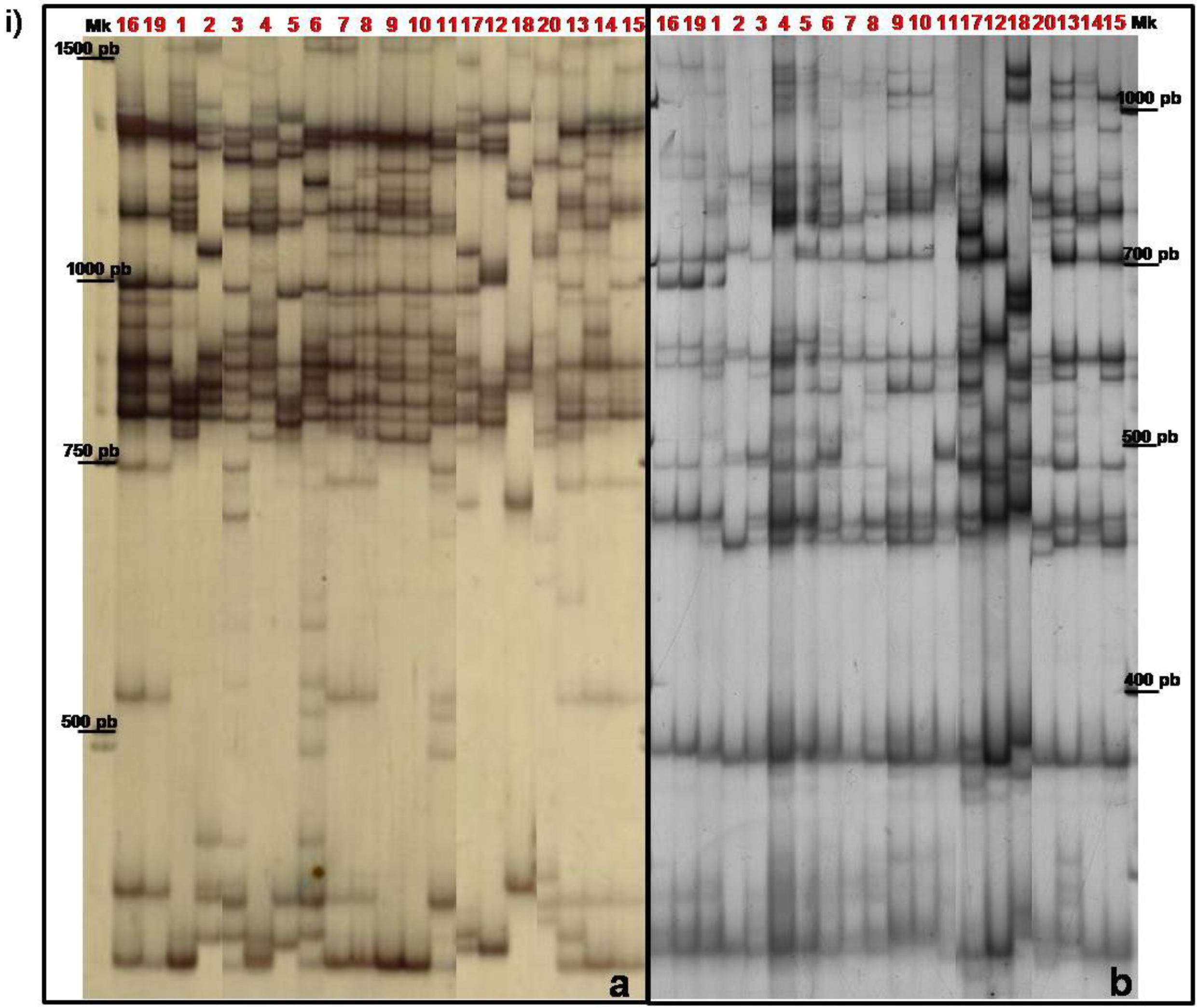
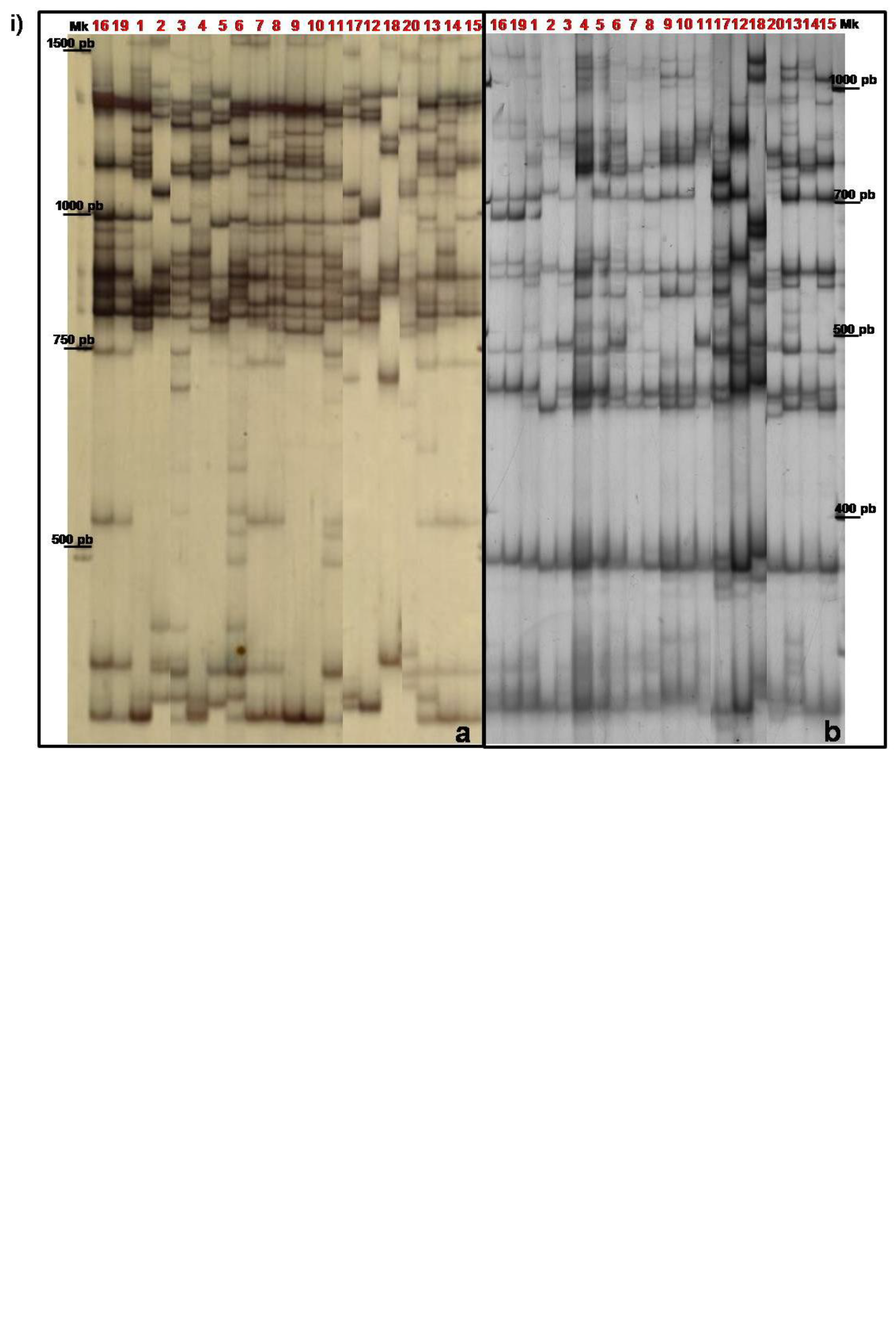{kind=link}
{kind=link}
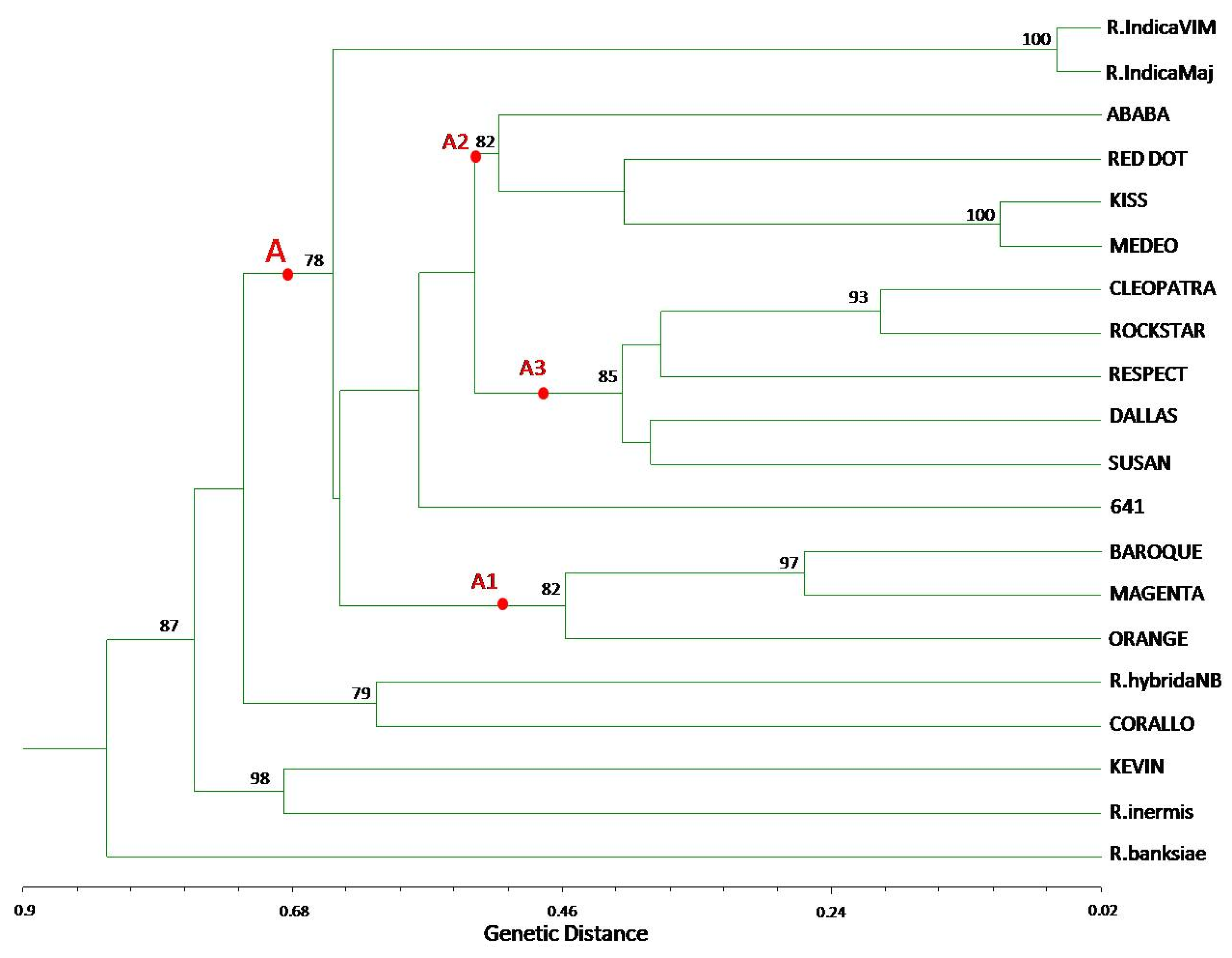
{kind=link}
{kind=link}
{kind=link}
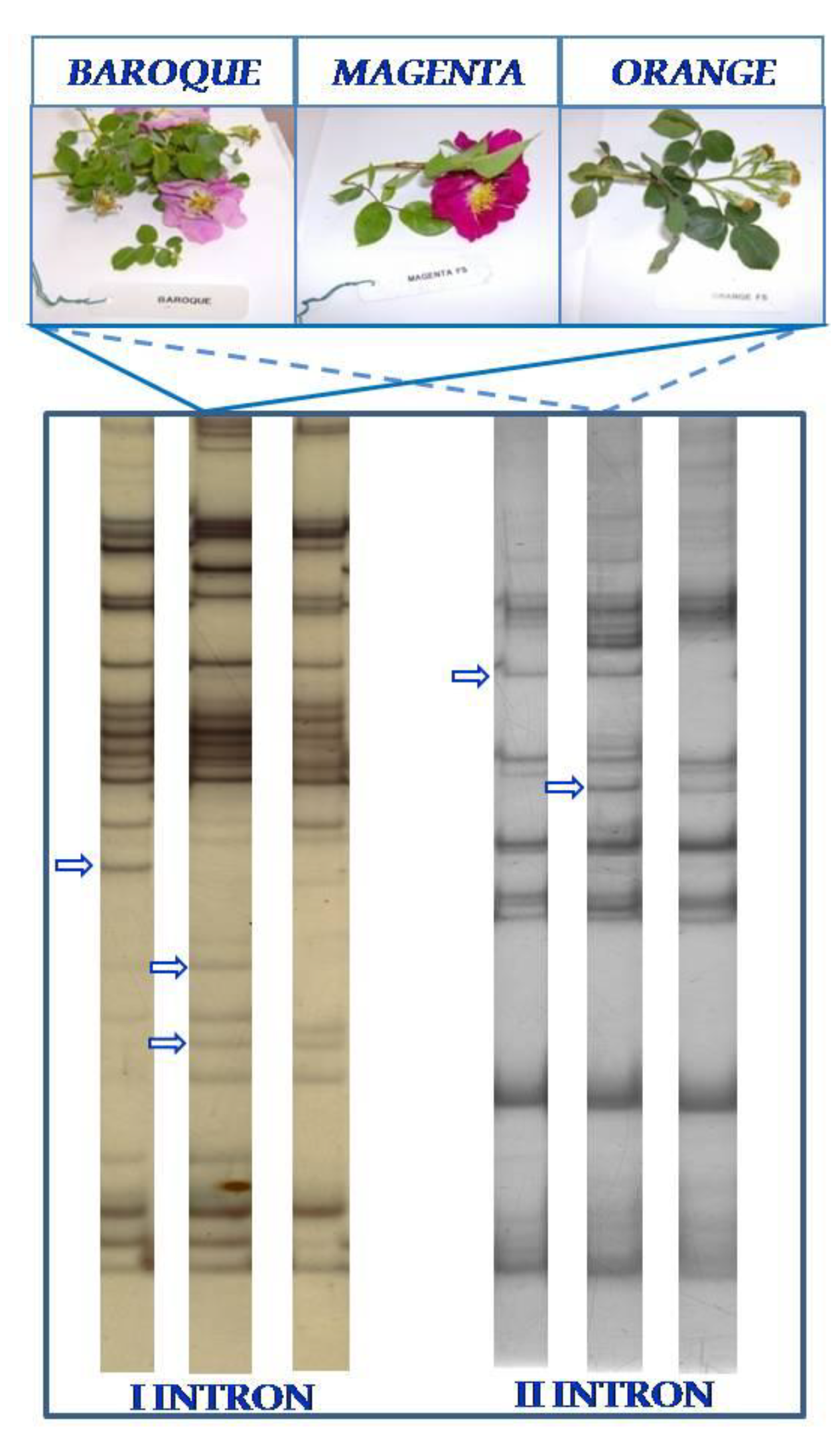{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample N° | Name | Sample N° | Name |
|---|---|---|---|
| 1 | ‘ABABA’ | 24 | Festuca violacea Gaudin |
| 2 | ‘KEVIN’ | 25 | Deschampsia cespitosa (L.) P. Beauv |
| 3 | ‘BAROQUE’ | 26 | Bromus hordaceus L. |
| 4 | ‘RED DOT’ | 27 | Poa annua L. |
| 5 | ‘641’ | 28 | Trifolium hybridum L. |
| 6 | ‘MAGENTA’ | 29 | Trifolium repens L. |
| 7 | ‘CLEOPATRA’ | 30 | Trifolium pretense L. |
| 8 | ‘RESPECT’ | 31 | Arrhenatherum elatius (L.) P. Beauv |
| 9 | ‘KISS’ | 32 | Holcus lanatus L. |
| 10 | ‘MEDEO’ | 33 | Phalaris arundinacea L. |
| 11 | ‘ORANGE’ | 34 | Arrenatherum spp |
| 12 | ‘CORALLO H’ | 35 | Dactilis glomerata L. |
| 13 | ‘DALLAS’ | 36 | Poa trivialis L |
| 14 | ‘ROCKSTAR’ | 37 | Crepis spp. |
| 15 | ‘SUSAN’ | 38 | Rumex acetosa L. |
| 16 | Rosa indica Thory. ‘Vera Indica Major®’ | 39 | Lotus spp. |
| 17 | Rosa hybrida L. ‘Natal Briar’ | 40 | Veronica persica Poiret |
| 18 | Rosa banksiae | 41 | Trifolium spp. |
| 19 | Rosa indica Thory ‘Major’ | 42 | Trifolium spp. |
| 20 | Rosa inermis Mill. | 43 | Trifolium spp. |
| 21 | Nardus stricta L. | 44 | Trifolium pratense ssp nivale (Sieber) |
| 22 | Trifolium alpinum L | 45 | Plantago lanceolata L. |
| 23 | Phleum pratense L. |
2.2. DNA Extraction and PCR Conditions
2.3. Data Analysis
3. Results and Discussion
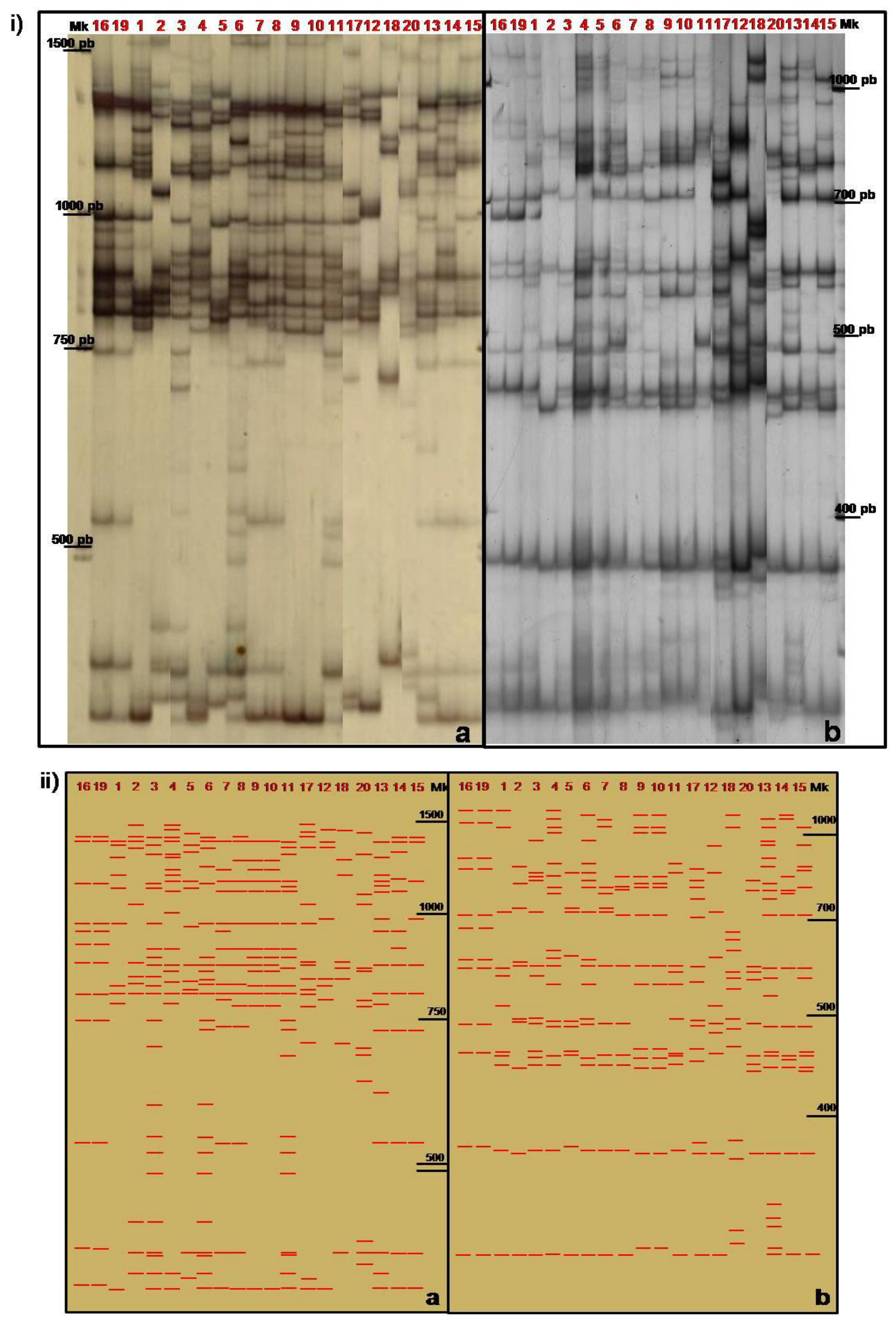
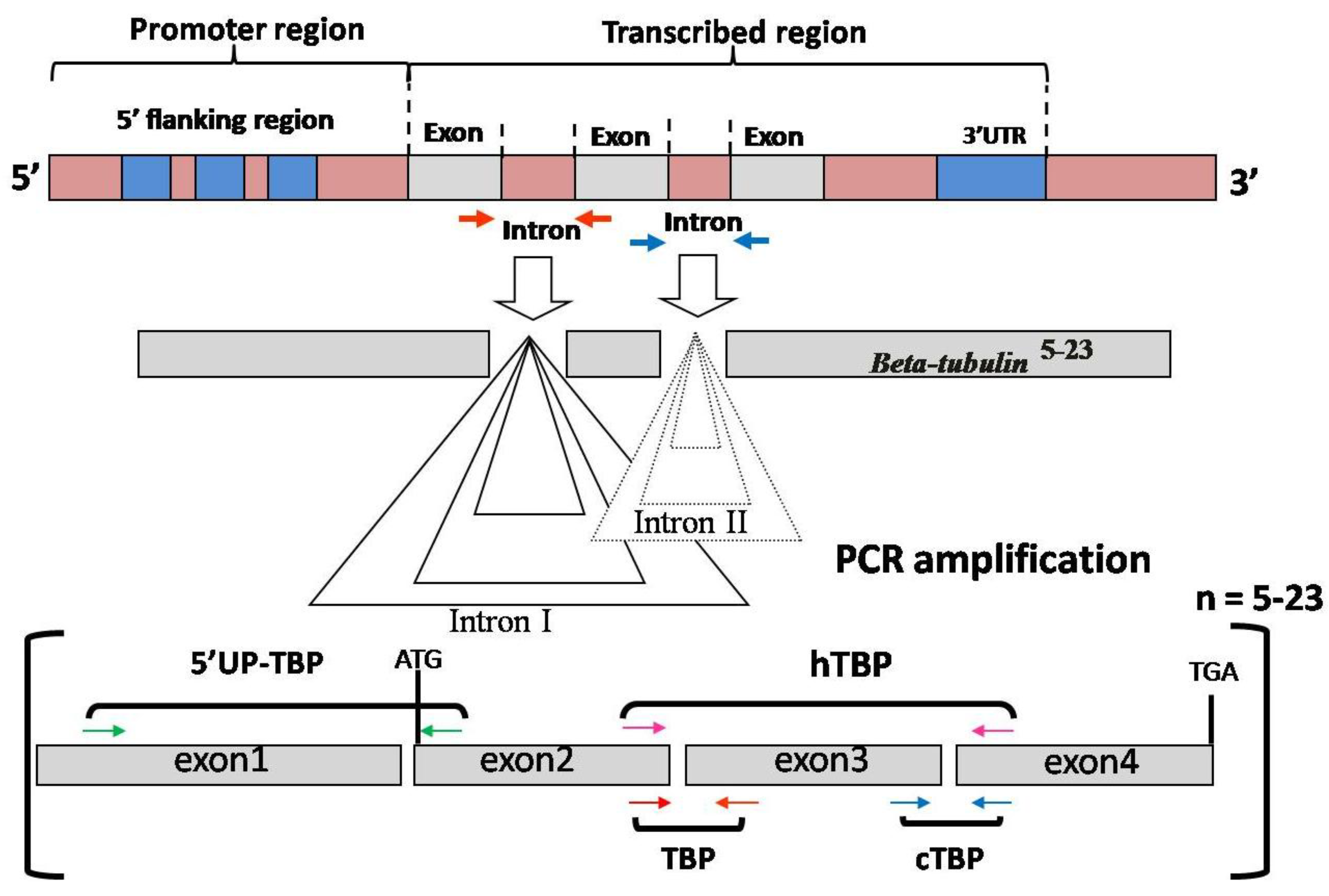
3.1. Intron Length Polymorphism Detected by the TBP/cTBP Method
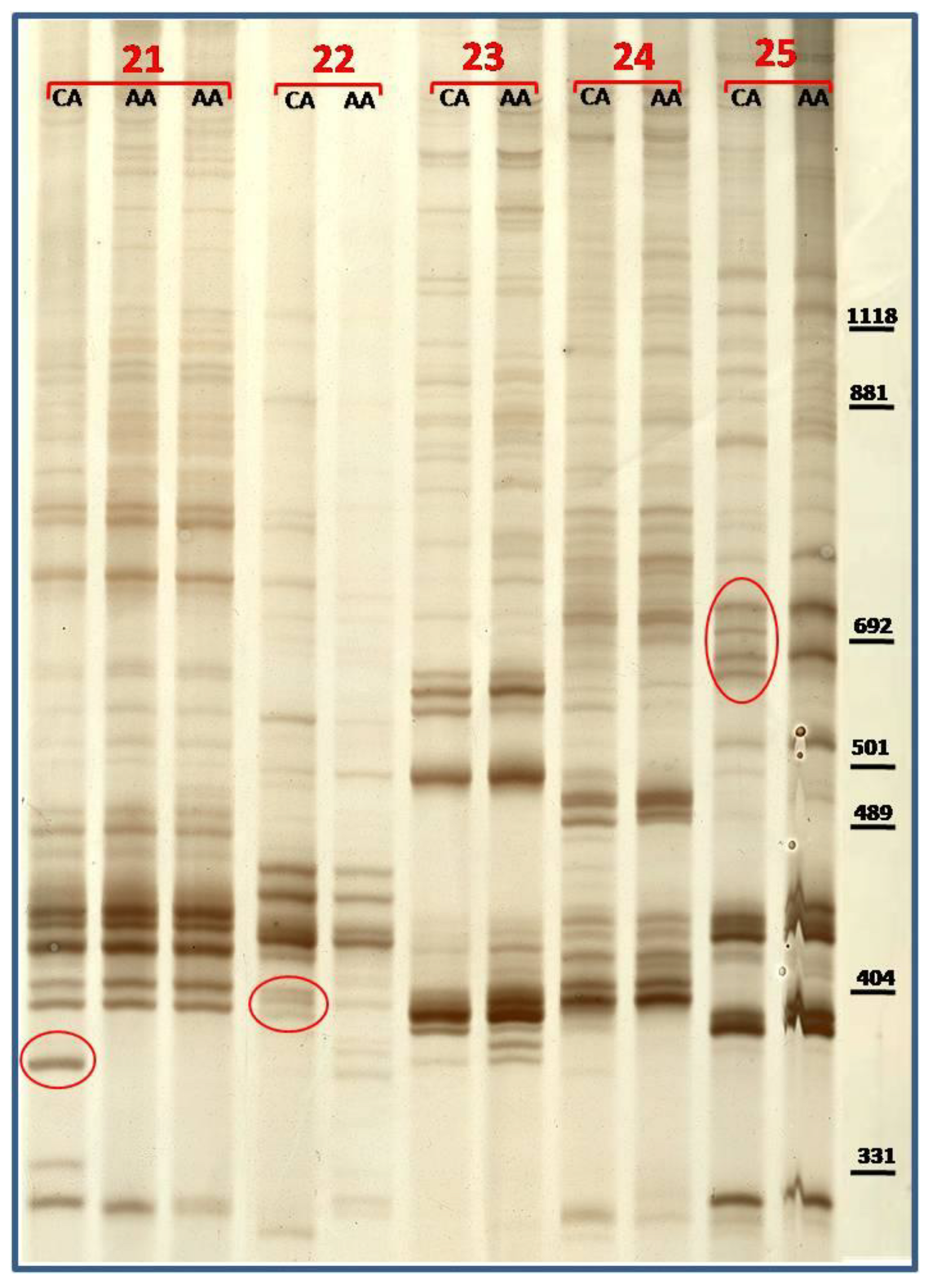
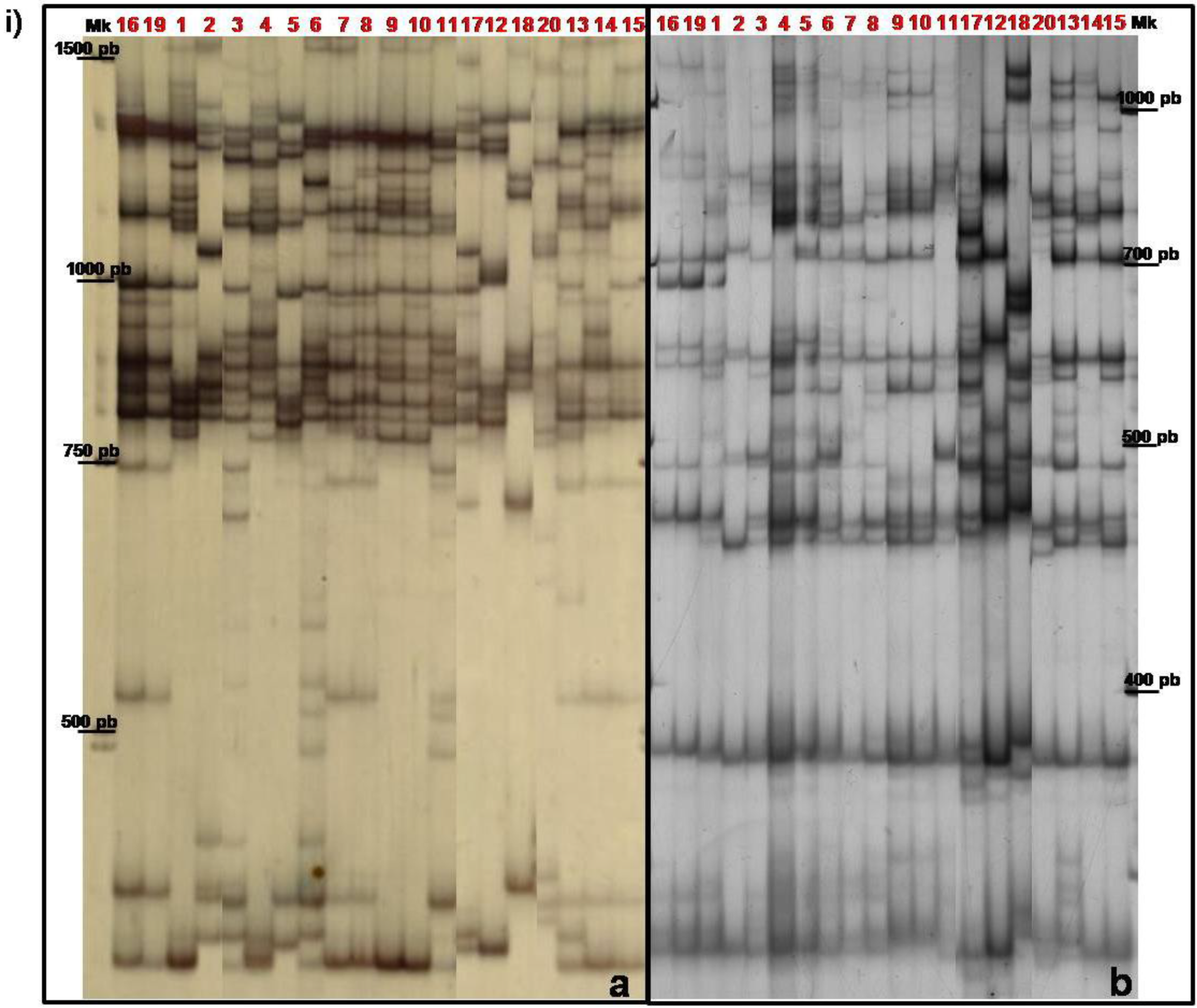
3.2. TBP/cTBP Analysis on Rosa spp.

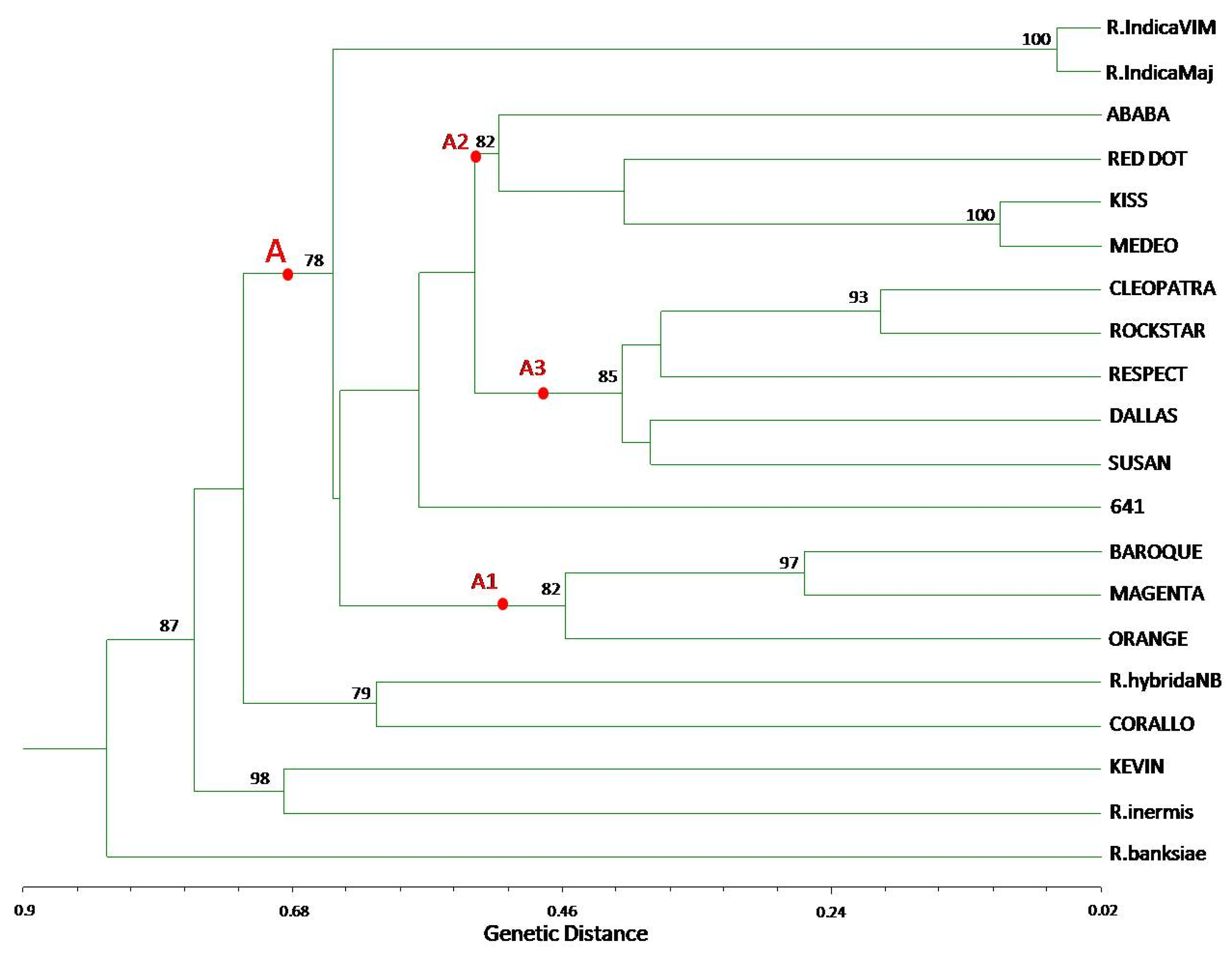
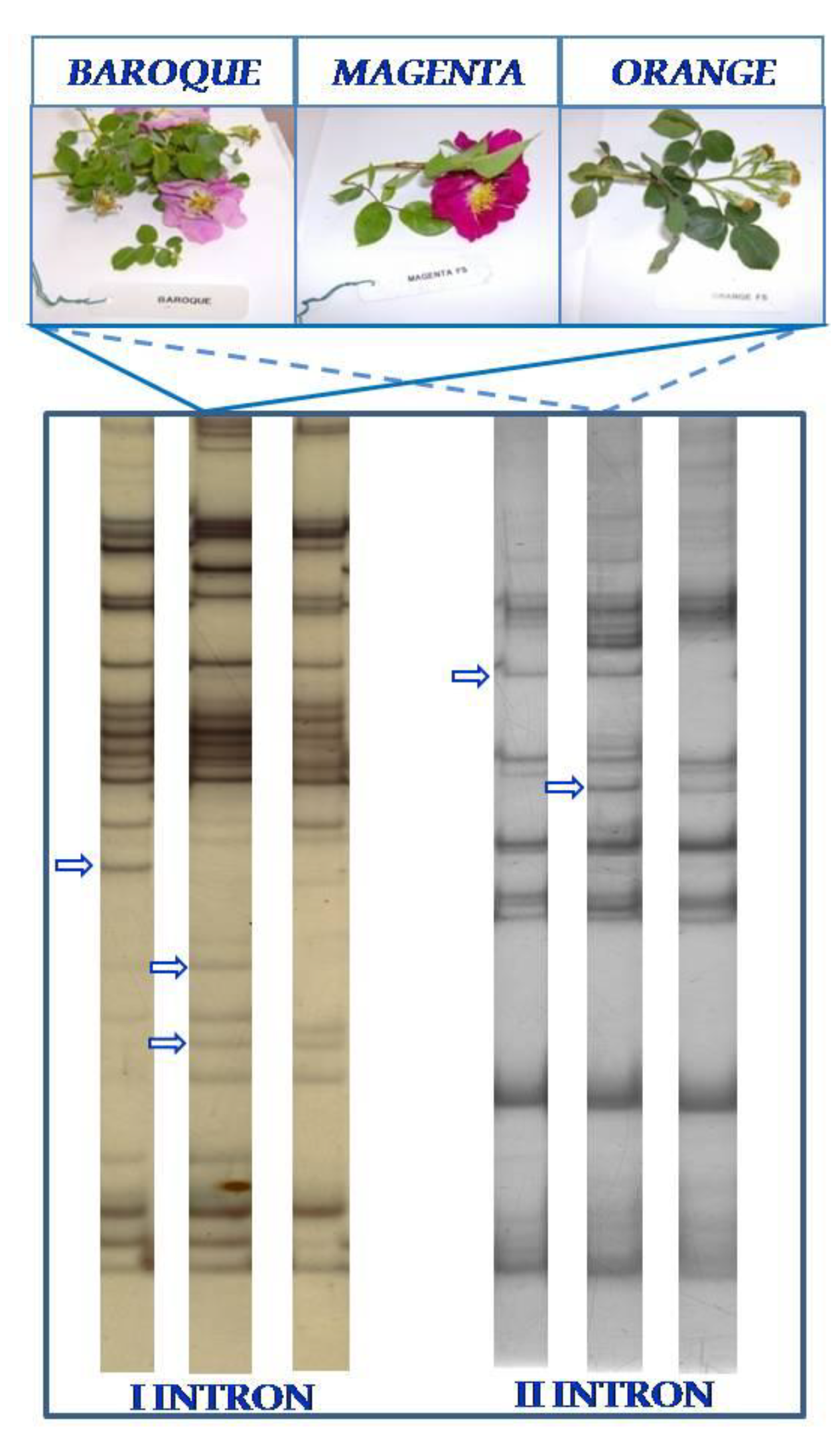
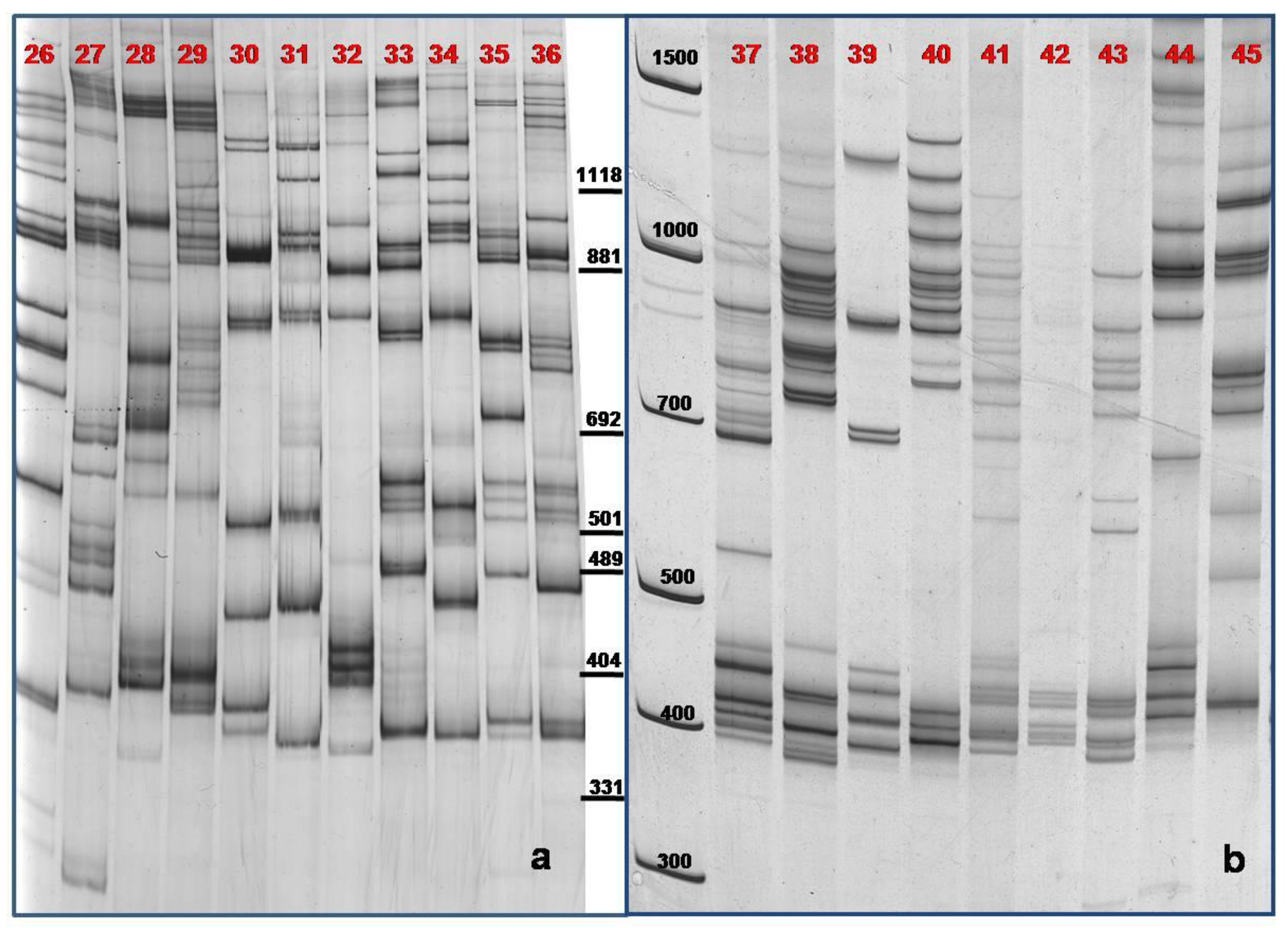
3.3. TBP/cTBP on Pasture and Garden Plants
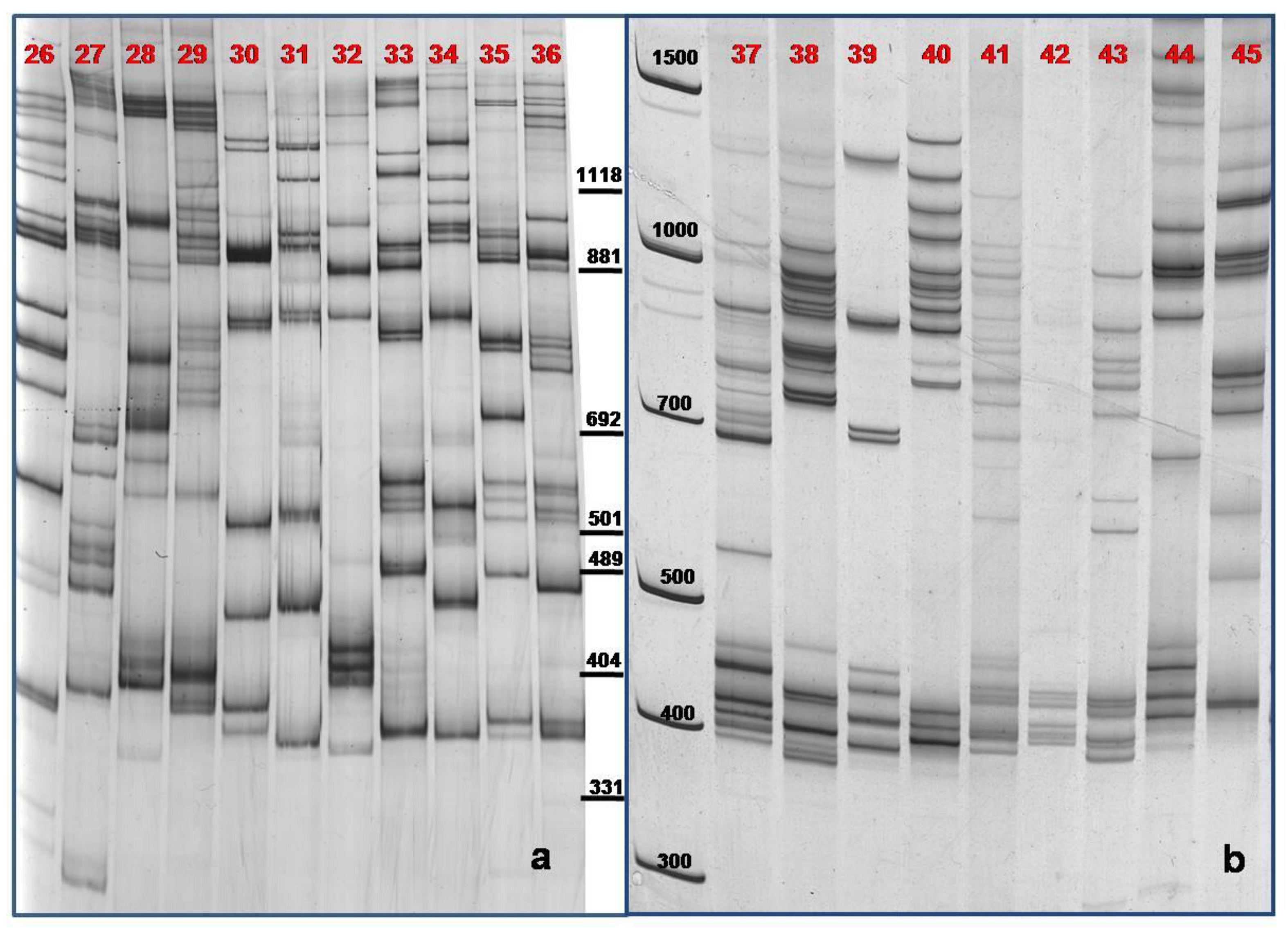

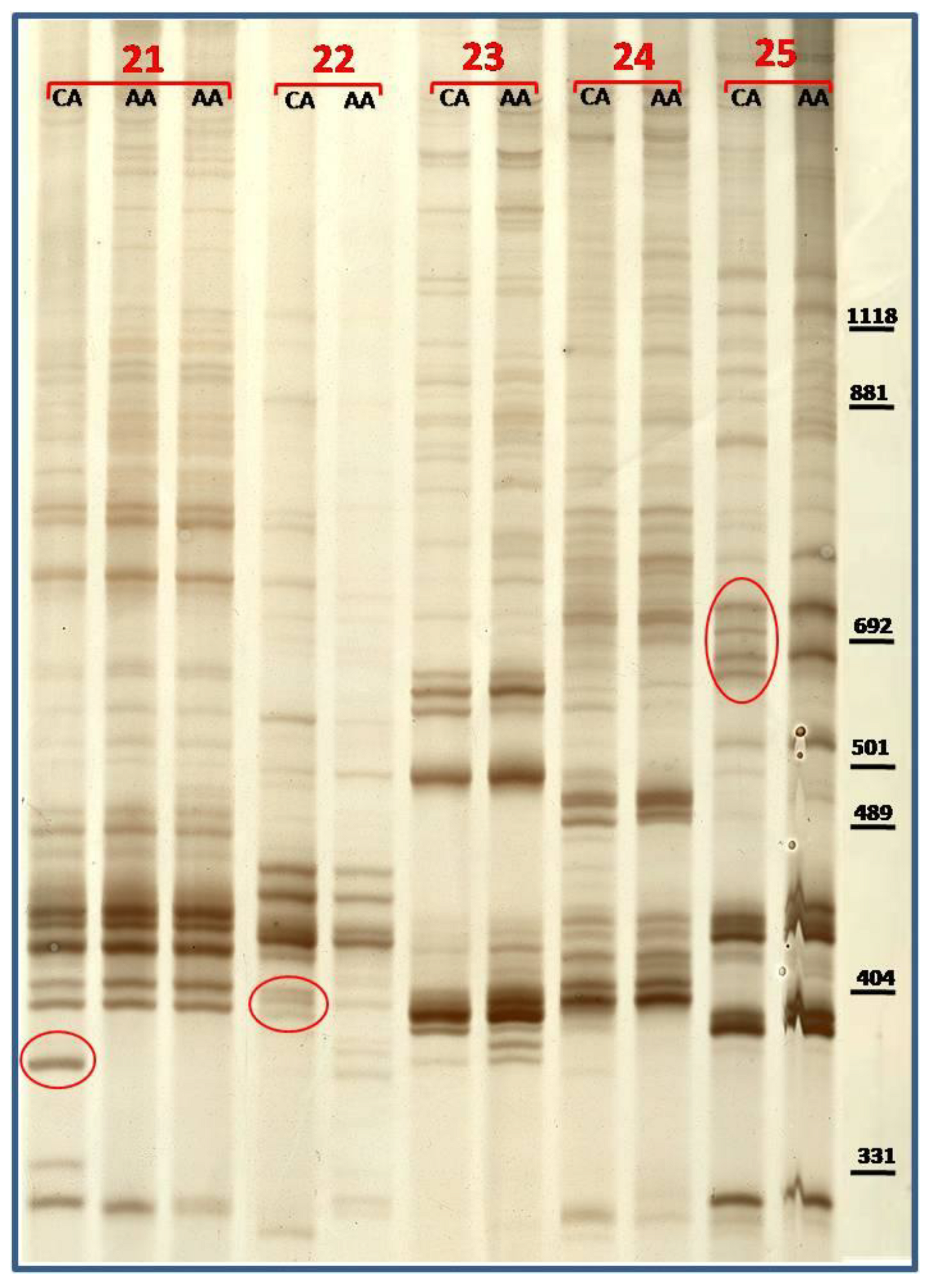
4. Conclusions
Acknowledgements
References
- Orgel, L.E.; Crick, F.H. Selfish DNA: the ultimate parasite. Nature 1980, 284, 604–607. [Google Scholar] [CrossRef]
- Cavalier-Smith, T. Selfish DNA and the origin of introns. Nature 1985, 315, 283–284. [Google Scholar] [CrossRef]
- Le Hir, H.; Nott, A.; Moore, M.J. How intron influence and enhance eukaryotic gene expression. Trends Biomedical Sci. 2003, 28, 215–220. [Google Scholar] [CrossRef]
- Morello, L.; Breviario, D. Plant Spliceosomal Introns: not only cut and paste. Curr. Genomics 2008, 9, 227–238. [Google Scholar] [CrossRef]
- Mascarenhas, D.; Mettler, I.J.; Pierce, D.A.; Lowe, H.W. Intron-mediated enhancement of heterologous gene expression in maize. Plant. Mol. Biol. 1990, 15, 913–920. [Google Scholar] [CrossRef]
- Gianì, S.; Altana, A.; Campanoni, P.; Morello, L.; Breviario, D. In transgenic rice, α- and β-tubulin regulatory sequences control GUS amount and distribution through intron mediated enhancement and intron dependent spatial expression. Transgenic Res. 2009, 18, 151–162. [Google Scholar] [CrossRef]
- Li, S.C.; Tang, P.; Lin, W.C. Intronic microRNA: discovery and biological implications. DNA Cell. Biol. 2007, 26, 195–207. [Google Scholar] [CrossRef]
- Ruby, J.G.; Jan, C.H.; Bartel, D.P. Intronic microRNA precursors that bypass Drosha processing. Nature 2007, 448, 83–87. [Google Scholar] [CrossRef]
- Chanfreau, G.C.; Legrain, R.P.; Jacquier, A. Processing of a dicistronic small nucleolar RNA precursor by the RNA endonuclease Rnt1. EMBO J. 1998, 17, 3726–3737. [Google Scholar] [CrossRef]
- Brown, J.W.S.; Marshall, D.F.; Echeverria, M. Intronic noncoding RNAs and splicing. Trends Plant Sci. 2008, 13, 335–342. [Google Scholar] [CrossRef]
- Fedorov, A.; Merican, A.F.; Gilbert, W. Large-scale comparison of intron positions among animal, plants and fungal genes. Proc Natl. Acad. Sci. USA 2002, 99, 16128–16133. [Google Scholar] [CrossRef]
- Wendel, J.F.; Cronn, R.C.; Alvarez, I.; Liu, B.; Small, R.L.; Senchina, D.S. Intron size and genome size in plants. Mol. Biol. Evol. 2002, 19, 2346–2352. [Google Scholar] [CrossRef]
- Roy, S.W.; Gilbert, W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nature Rev. Genet. 2006, 7, 211–221. [Google Scholar]
- Rodriguez-Trelles, F.; Tarrio, R.; Ayala, F.J. Origins and evolutionof spliceosomal introns. Annu. Rev. Genet. 2006, 40, 47–76. [Google Scholar] [CrossRef]
- Jeffares, D.C.; Mourier, T.; Penny, D. The biology of intron gain and loss. Trends Genet. 2006, 22, 16–22. [Google Scholar] [CrossRef]
- Gingeras, T.R. Origin of phenotypes: genes and transcripts. Genome Res. 2007, 17, 682–690. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Zhu, J.; Wu, W. Genome-wide investigation of Intron-Length Polymorphisms and their potential as molecular markers in rice (Oryza sativa L.). DNA Res. 2005, 12, 417–427. [Google Scholar] [CrossRef]
- Breviario, D. Plant tubulin genes: regulatory and evolutionary aspects. In Plant Microtubules, 2nd ed.; Nick, P., Ed.; Springer-Verlag: Berlin, Heidelberg, Germany, 2008; pp. 207–232. [Google Scholar]
- Bardini, M.; Lee, D.; Donini, P.; Mariani, A.; Gianì, S.; Toschi, M.; Lowe, C.; Breviario, B. TBP, a new tool for testing genetic diversity in plant species based on functionally relevant sequences. Genome 2004, 247, 281–291. [Google Scholar]
- Inada, D.C.; Bashir, A.; Lee, C.; Thomas, B.C.; Ko, C.; Goff, S.A. Conserved noncoding sequences in the grasses. Genome Res. 2003, 13, 2030–2041. [Google Scholar] [CrossRef]
- Thomas, B.C.; Rapaka, L.; Lyons, E.; Pedersen, B.; Freeling, M. Arabidopsis intragenomic conserved noncoding sequence. Proc. Natl. Acad. Sci. USA 2007, 104, 3348–3353. [Google Scholar] [CrossRef]
- Li, X.; Tan, L.; Wang, L.; Hu, S.; Sun, C. Isolation and characterization of conserved non-coding sequences among rice (Oryza sativa L.) paralogous regions. Mol. Genet. Genomics 2009, 281, 11–18. [Google Scholar] [CrossRef]
- Ferreira, A.O.; Cardoso, H.G.; Macedo, E.S.; Breviario, D.; Arnholdt-Schmitt, B. Intron polymorphism pattern in AOX1b of wild St John’s wort (Hipericum perforatum) allows discrimination between individual plants. Physiol. Plantarum 2009, 137, 520–531. [Google Scholar] [CrossRef]
- Perumal, B.S.; Sakharkar, K.R.; Tak Kwong Chow, V.; Kangueane, P.; Sakharkar, M.K. Intron position conservation across eukaryotic lineages in tubulin genes. Front. Biosci. 2005, 10, 2412–2419. [Google Scholar] [CrossRef]
- Dhonukshe, P.; Mathur, J.; Hulskamp, M.; Gadella, T.W., Jr. Microtubule plus-ends reveal essential links between intracellular polarization and localized modulation of endocytosis during division-plate establishment in plant cells. BMC Biol 2005, 14, 3–11. [Google Scholar]
- Oakley, R.V.; Wang, Y.S.; Ramakrishna, W.; Harding, S.A.; Tsai, C.J. Differential expansion and expression of α- and β-tubulin gene families in Populus. Plant. Physiol. 2007, 145, 961–973. [Google Scholar] [CrossRef]
- Bierne, N.; Lenhert, S.A.; Bedier, E. Screening for intron-length polymorphism in penacid shrimps using exon-primed intron-crossing (EPIC)-PCR. Mol. Ecol. 2000, 9, 233–235. [Google Scholar] [CrossRef]
- Breviario, D.; Baird, W.V.; Sangoi, S.; Hilu, K.; Blumetti, P.; Gianì, S. High polymorphism and resolution in targeted fingerprinting with combined beta-tubulin introns. Mol. Breed. 2007, 20, 249–259. [Google Scholar] [CrossRef]
- Bassam, B.J.; Anollés, G.C.; Gresshoff, P.M. Fast and sensitive silver staining of DNA in polyacrylamide gels. Anal. Biochem. 1991, 196, 80–83. [Google Scholar]
- Van de Peer, Y.; De Wachter, R. Treecon for Windows: a software package for the construction and drawing of evolutionary trees for the Microsoft Windows environment. Comput. Appl. Biosci. 1994, 10, 569–570. [Google Scholar]
- Nei, M.; Li, W.H. Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. 1979, 76, 5269–5273. [Google Scholar] [CrossRef]
- Sneath, P.H.A; Sokal, R.R. Numerical Taxonomy; Freeman, W.H. Co.: San Francisco, CA, USA, 1973; p. 573. [Google Scholar]
- Hillis, D.M.; Bull, J.J. An empirical test of bootstrapping as a method for assessing confidence in phylogenetic analysis. Syst. Biol. 1993, 42, 182–192. [Google Scholar] [CrossRef]
- Liaud, M.F.; Brinkmann, H.; Cerff, R. The β-tubulin gene family of pea: primary structure genomic organization and intron-dependent evolution of genes. Plant Mol. Biol. 1992, 18, 639–651. [Google Scholar] [CrossRef]
- Casazza, A.P.; Gavazzi, F.; Mastromauro, F.; Gianì, S.; Breviario, D. Certifying the feed to guarantee the quality of traditional food: an easy way to trace plant species in complex mixtures. Food Chem. 2010. in review, manuscript ID FOODCHEM-D-09-02959. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Braglia, L.; Manca, A.; Mastromauro, F.; Breviario, D. cTBP: A Successful Intron Length Polymorphism (ILP)-Based Genotyping Method Targeted to Well Defined Experimental Needs. Diversity 2010, 2, 572-585. https://doi.org/10.3390/d2040572
Braglia L, Manca A, Mastromauro F, Breviario D. cTBP: A Successful Intron Length Polymorphism (ILP)-Based Genotyping Method Targeted to Well Defined Experimental Needs. Diversity. 2010; 2(4):572-585. https://doi.org/10.3390/d2040572
Chicago/Turabian StyleBraglia, Luca, Antonella Manca, Francesco Mastromauro, and Diego Breviario. 2010. "cTBP: A Successful Intron Length Polymorphism (ILP)-Based Genotyping Method Targeted to Well Defined Experimental Needs" Diversity 2, no. 4: 572-585. https://doi.org/10.3390/d2040572
APA StyleBraglia, L., Manca, A., Mastromauro, F., & Breviario, D. (2010). cTBP: A Successful Intron Length Polymorphism (ILP)-Based Genotyping Method Targeted to Well Defined Experimental Needs. Diversity, 2(4), 572-585. https://doi.org/10.3390/d2040572