The Canarian Camel: A Traditional Dromedary Population

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
2.1. Genetic Variability

{kind=link}
{kind=link}
{kind=link}
| Name | Primer sequences (5'-3') | No of alleles | Reference |
|---|---|---|---|
| VOLP03 | AGACGGTTGGGAAGGTGGTA | 15 | [7] |
| CGACAGCAAGGCACAGGA | |||
| VOLP08 | CCATTCACCCCATCTCTC | 4 | [7] |
| TCGCCAGTGACCTTATTTAGA | |||
| VOLP10 | CTTTCTCCTTTCCTCCCTACT | 8 | [7] |
| CGTCCACTTCCTTCATTTC | |||
| VOLP32 | GTGATCGGAATGGCTTGAAA | 2 | [7] |
| CAGCGAGCACCTGAAAGAA | |||
| YWLL08 | ATCAAGTTTGAGGTGCTTTCC | 21 | [8] |
| CCATGGCATTGTGTTGAAGAC | |||
| YWLL38 | GGCCTAAATCCTACTAGAC | 8 | [8] |
| CCTCTCACTCTTGTTCTCCTC | |||
| YWLL44 | CTCAACAATGCTAGACCTTGG | 8 | [8] |
| GAGAACACAGGCTGGTGAATA | |||
| CVRL01 | GAAGAGGTTGGGGCACTAC | 22 | [9] |
| CAGGCAGATATCCATTGAA | |||
| CVRL02 | TGTCACAAATGGCAAGAT | 4 | [9] |
| AGTGTACGTAGCAGCATTATTT | |||
| CVRL05 | CCTTGGACCTCCTTGCTCTG | 13 | [9] |
| GCCACTGGTCCCTGTCATT | |||
| CVRL06 | TTTTAAAAATTCTGACCAGGAGTCTG | 4 | [9] |
| CATAATAGCCAAAACATGGAAACAAC | |||
| CVRL07 | AATACCCTAGTTGAAGCTCTGTCCT | 20 | [9] |
| GAGTGCCTTTATAAATATGGGTCTG | |||
| LCA66 | GTGCAGCGTCCAAATAGTCA | 10 | [10] |
| CCAGCATCGTCCAGTATTCA |
| Origin | FIS | Heterozygosity | Allelic Richness | |
|---|---|---|---|---|
| Expected | Observed | |||
| Canarian | 0.01 | 0.593 | 0.586 | 6.0 |
| Arabian | 0.13* | 0.633 | 0.552 | 5.3 |
| Kenyan | 0.05* | 0.581 | 0.552 | 5.8 |
| Pakistani | -0.02 | 0.624 | 0.640 | 4.9 |
| Tindouf (Algeria) | 0.01 | 0.588 | 0.585 | 5.4 |
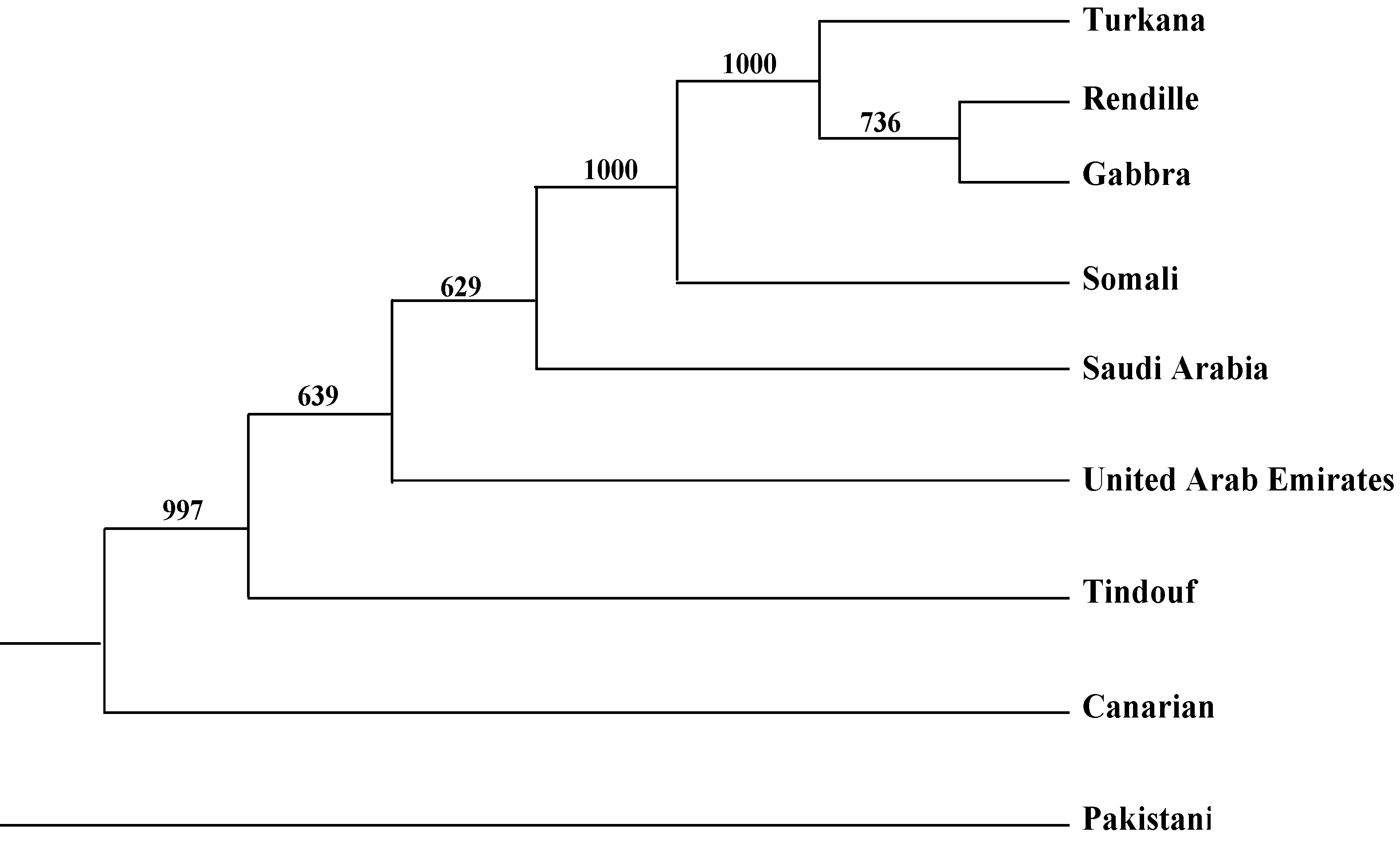
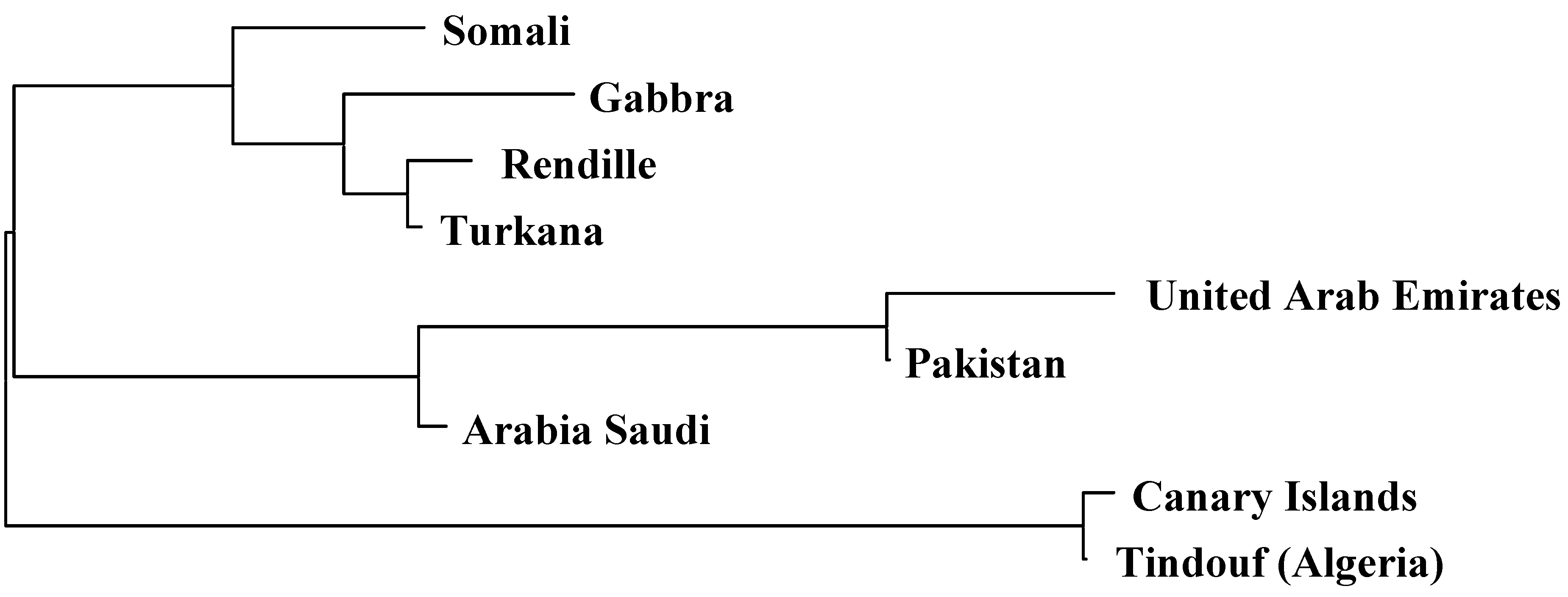
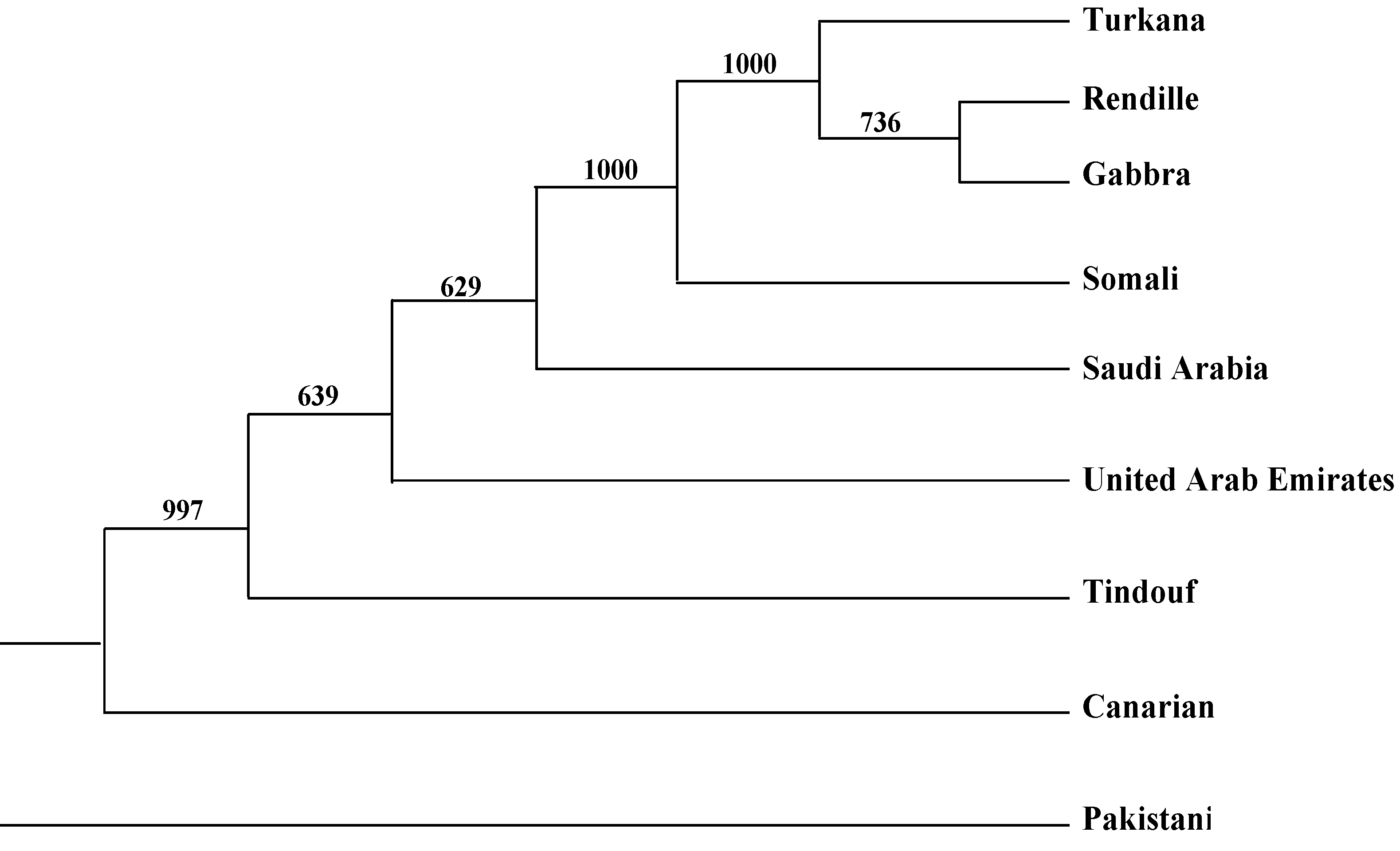
2.2. Genetic Distances and Clustering
| Somali | Rendille | Turkana | Gabbra | Pakistani | Saudi Arabia | United Arab Emirates | Tindouf (Algeria) | Average | |
|---|---|---|---|---|---|---|---|---|---|
| Canarian | 0.14 | 0.15 | 0.15 | 0.15 | 0.11 | 0.08 | 0.11 | 0.01 | 0.11 |
| Somali | 0.01 | 0.02 | 0.02 | 0.11 | 0.08 | 0.15 | 0.14 | 0.08 | |
| Rendille | 0.00 | 0.00 | 0.13 | 0.10 | 0.17 | 0.16 | 0.09 | ||
| Turkana | 0.00 | 0.11 | 0.09 | 0.16 | 0.16 | 0.09 | |||
| Gabbra | 0.13 | 0.09 | 0.17 | 0.16 | 0.09 | ||||
| Pakistani | 0.07 | 0.09 | 0.10 | 0.11 | |||||
| Saudi Arabia | 0.07 | 0.08 | 0.08 | ||||||
| United Arab Emirates | 0.12 | 0.13 | |||||||
| Tindouf (Algeria) | 0.12 |
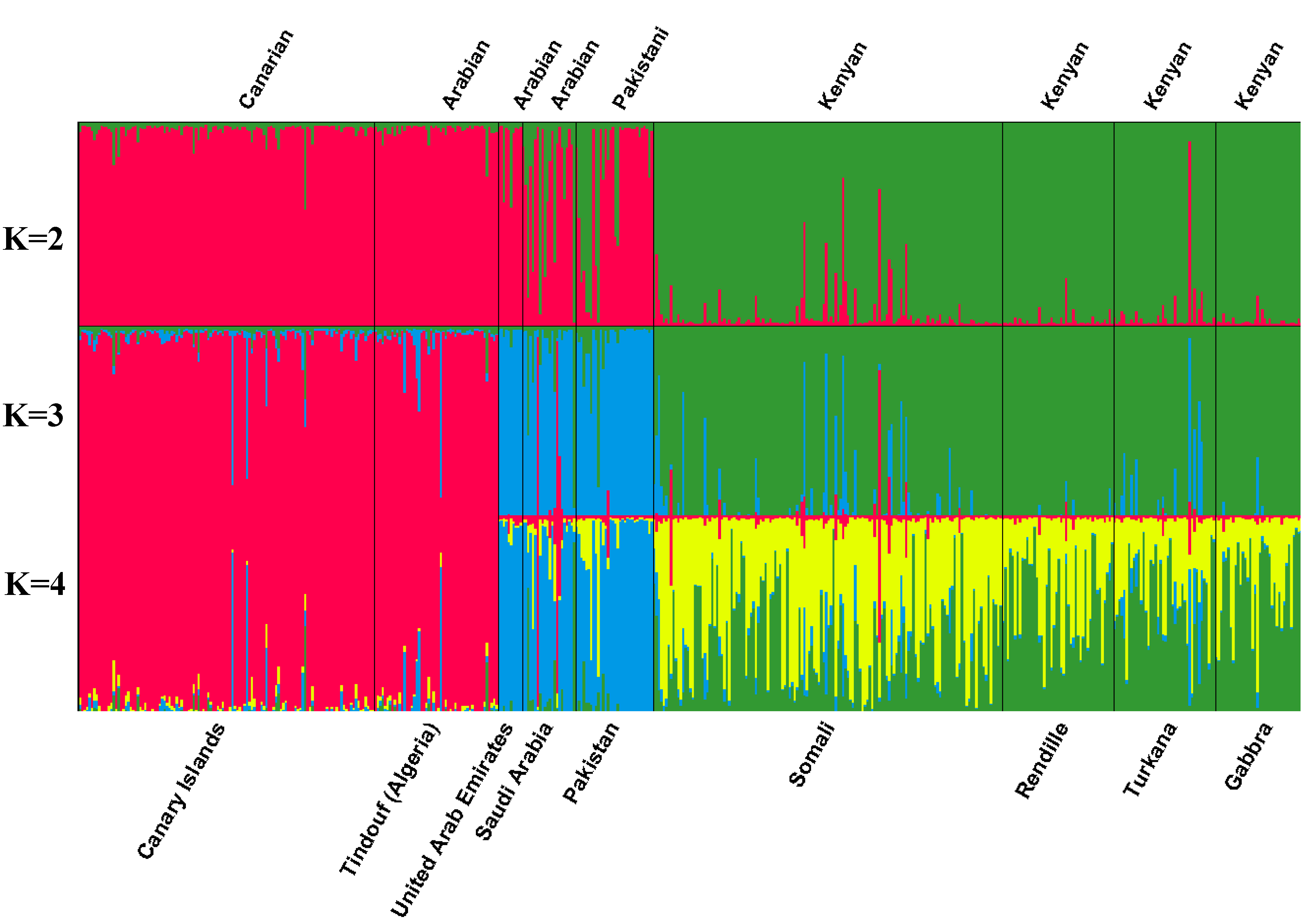
| Number of inferred clusters | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Two clusters (k=2) | Three clusters (k = 3) | Four clusters (k = 4) | |||||||||
| 1 | 2 | 1 | 2 | 3 | 1 | 2 | 3 | 4 | |||
| Canary Islands | 0.967 | 0.033 | 0.936 | 0.026 | 0.038 | 0.912 | 0.027 | 0.024 | 0.037 | ||
| Somali | 0.057 | 0.943 | 0.034 | 0.877 | 0.089 | 0.030 | 0.367 | 0.551 | 0.051 | ||
| Rendille | 0.024 | 0.976 | 0.018 | 0.962 | 0.020 | 0.017 | 0.586 | 0.381 | 0.015 | ||
| Turkana | 0.055 | 0.945 | 0.021 | 0.876 | 0.104 | 0.021 | 0.565 | 0.343 | 0.072 | ||
| Gabbra | 0.022 | 0.978 | 0.016 | 0.959 | 0.025 | 0.015 | 0.661 | 0.308 | 0.016 | ||
| Pakistan | 0.720 | 0.280 | 0.018 | 0.079 | 0.903 | 0.019 | 0.046 | 0.102 | 0.834 | ||
| Saudi Arabia | 0.674 | 0.326 | 0.133 | 0.124 | 0.742 | 0.136 | 0.122 | 0.085 | 0.656 | ||
| United Arab Emirates | 0.902 | 0.098 | 0.028 | 0.021 | 0.951 | 0.029 | 0.023 | 0.029 | 0.919 | ||
| Tindouf (Algeria) | 0.969 | 0.031 | 0.923 | 0.024 | 0.053 | 0.899 | 0.027 | 0.023 | 0.052 | ||
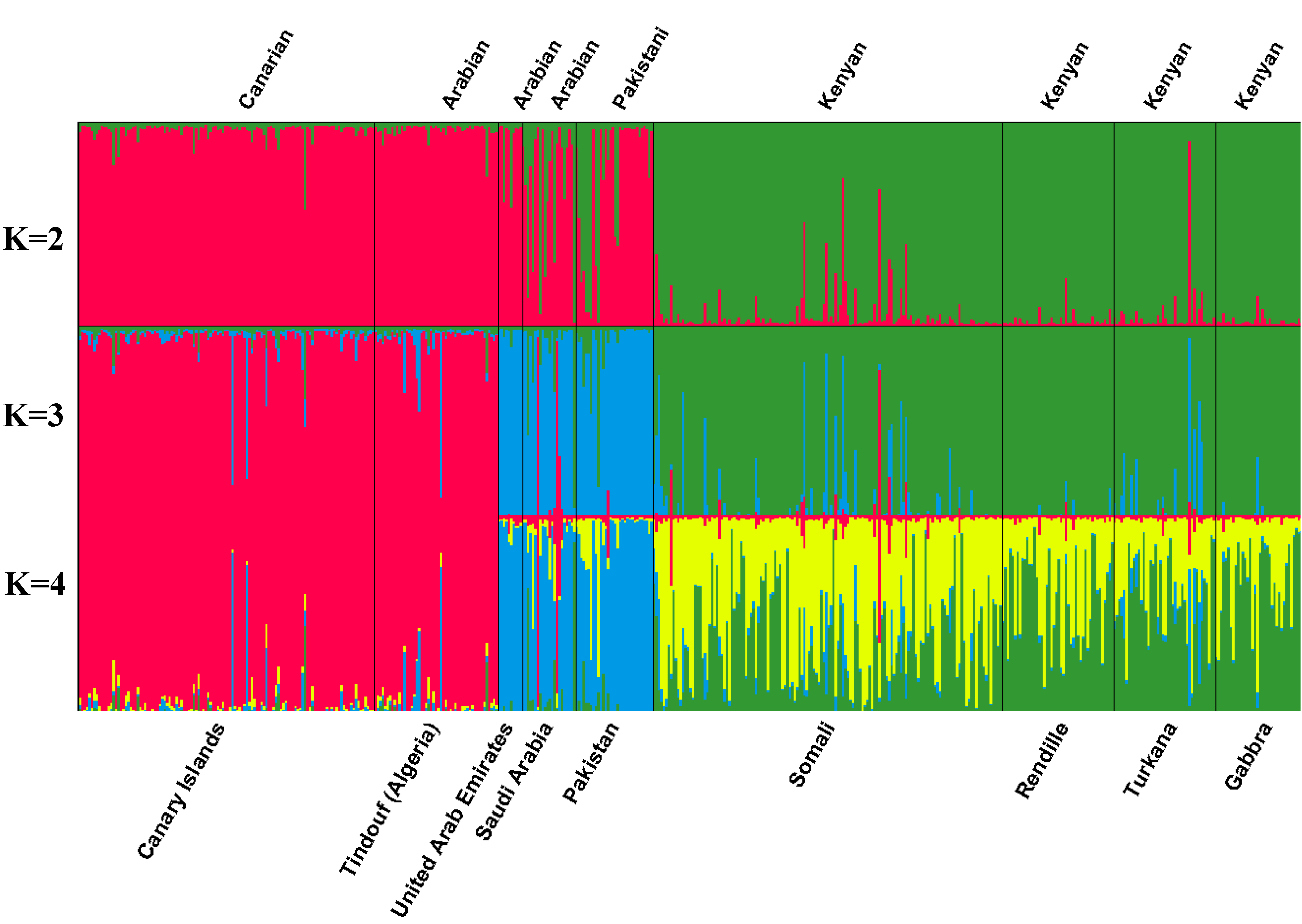
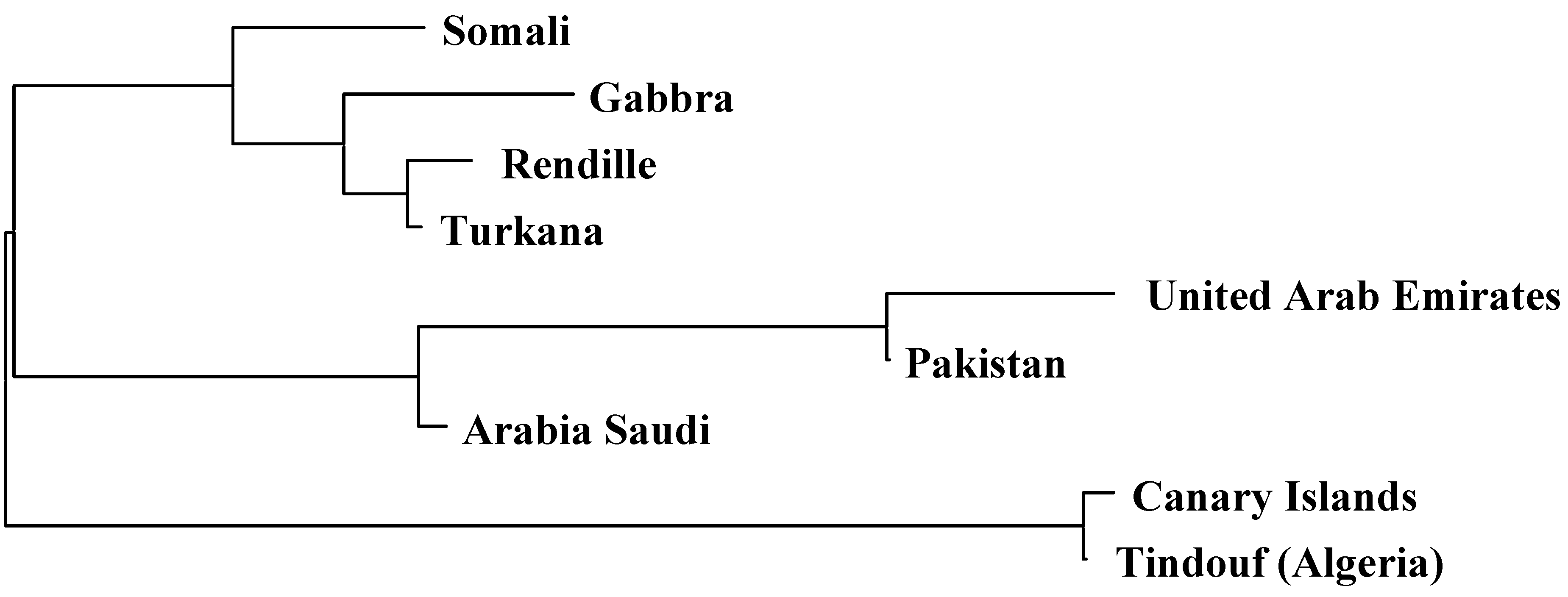
3. Experimental Section
3.1. Sample Collection
| Origin | Population | N |
|---|---|---|
| Canary Islands | Canarian | 122 |
| Arabian countries | Tindouf (Algeria) | 51 |
| Saudi Arabian | 22 | |
| United Arab Emirates | 10 | |
| Kenya | Somali | 144 |
| Rendille | 46 | |
| Turkana | 42 | |
| Gabbra | 36 | |
| Pakistan | Pakistani | 32 |
3.2. DNA Extraction, Microsatellite Markers and Genotyping
3.3. StatisticalAnalyses
4. Conclusions
Acknowledgements
References
- Morera, M. La tradición del camello en Canarias. Estudios Atlánticos, n37; Patronato de la Casa de Colón: Madrid-Las Palmas, Spain, 1991; pp. 167–204. [Google Scholar]
- Schulz, U. El Camello en Lanzarote; ADERLAN: Lanzarote, Spain, 2008. [Google Scholar]
- Phillipson, N.E. Camels in Australia. Proc. Royal Geog. Soc. Aust. 1899, 3, 83–92. [Google Scholar]
- Mburu, D.N.; Ochieng, J.W.; Kuria, S.G.; Jianlin, H.; Kaufmann, B.; Rege, J.E.O.; Hanotte, O. Genetic diversity and relationships of indigenous Kenyan camel (Camelus dromedarius) populations: implications for their classification. Anim. Genet. 2003, 34, 26–32. [Google Scholar]
- Baumung, R.; Simianer, H.; Hoffmann, I. Genetic diversity studies in farm animals—a survey. J. Anim. Breed. Genet. 2004, 121, 361–373. [Google Scholar] [CrossRef]
- Schulz, U.; Minguez, Y.; Checa, M.L.; Garcia-Atance, P.; Dunner, S.; Garcia, D.; Cañón, J. The Majorero camel (Camelus dromedarius) breed. Anim. Genet. Res. Inf. 2005, 36, 61–72. [Google Scholar] [CrossRef]
- Obreque, V.; Coogle, L.; Henney, P.J.; Bailey, E.; Mancilla, R.; Garcia-Huidobro, J.; Hinrichsen, P.; Cothran, E.G. Characterization of 10 polymorphic alpaca dinucleotide microsatellites. Anim. Genet. 1998, 29, 460–467. [Google Scholar]
- Lang, K.D.M.; Wang, Y.; Plante, Y. Fifteen polymorphic dinucleotide microsatellites in llamas and alpacas. Anim. Genet. 1996, 27, 285–294. [Google Scholar]
- Sasse, J.; Mariasegaram, M.; Jahabar Ali, M.K.; Pullenayegum, R.; Kinne, B.R.; Werney, U. Development of a microsatellite parentage and identity verification test for dromedary racing camels. In Presented at the 27th International Conference on Animal Genetics, St Paul/Minneapolis, MN, USA, July 2000.
- Penedo, C.; Caetano, A.; Cordova, K. Eight microsatellite markers for South American camelids. Anim. Genet. 1998, 30, 166–167. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP. (Phylogeny Inference Package) Version 3.69; Department of Genetics, University of Eashington: Seattle, WA, USA, 2009. Available online: http://evolution.gs.washington.edu/phylip.html (accessed on 10 February 2010).
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure from multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Cañón, J.; García, D.; García-Atance, M.A.; Obexer-Ruff, G.; Lenstra, J.A.; Ajmone-Marsan, P.; Dunner, S. the ECONOGENE Consortium Geographical partitioning of goat diversity in Europe and the Middle East. Anim. Genet. 2006, 37, 327–334. [Google Scholar] [CrossRef]
- Tamura, K.; Dudley, J.; Nei, M.; Kumar, S. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol. Biol. Evol. 2007, 24, 1596–1599. [Google Scholar] [CrossRef]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor: New York, NY, USA, 1989. [Google Scholar]
- Park, S.D.E. Trypanotolerance in West African Cattle and the Population Genetic Effects of Selection. Ph.D. Thesis, University of Dublin, Dublin, Ireland, 2001. [Google Scholar]
- Goudet, J. FSTAT, A Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9.3.2); University of Lausanne: Lausanne, Switzerland, 2001. Available online: http://www2.unil.ch/popgen/softwares/fstat.htm (accessed on 10 February 2010).
- Wright, S. The interpretation of population structure by F-statistics with special regard to systems of mating. Evolution 1965, 19, 395–420. [Google Scholar] [CrossRef]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. Genetix, logiciel sous Windows TM pour la génétique des populations, Laboratoire Génome, Populations, Interactions, CNRS UPR 9060; Université de Montpellier II: Montpellier, France, 2001. Available online: http://www.genetix.univ-montp2.fr/genetix/intro.htm (accessed on 10 February 2010).
- Rosenberg, N.A. Distruct: a Program for the Graphical Display of Structure Results; University of Michigan: MI, USA, 2002. Available online: http://rosenberglab.bioinformatics.med.umich.edu/ distruct.html (accessed on 10 February 2010).
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schulz, U.; Tupac-Yupanqui, I.; Martínez, A.; Méndez, S.; Delgado, J.V.; Gómez, M.; Dunner, S.; Cañón, J. The Canarian Camel: A Traditional Dromedary Population. Diversity 2010, 2, 561-571. https://doi.org/10.3390/d2040561
Schulz U, Tupac-Yupanqui I, Martínez A, Méndez S, Delgado JV, Gómez M, Dunner S, Cañón J. The Canarian Camel: A Traditional Dromedary Population. Diversity. 2010; 2(4):561-571. https://doi.org/10.3390/d2040561
Chicago/Turabian StyleSchulz, Ursula, Isabel Tupac-Yupanqui, Amparo Martínez, Susy Méndez, Juan Vicente Delgado, Mariano Gómez, Susana Dunner, and Javier Cañón. 2010. "The Canarian Camel: A Traditional Dromedary Population" Diversity 2, no. 4: 561-571. https://doi.org/10.3390/d2040561
APA StyleSchulz, U., Tupac-Yupanqui, I., Martínez, A., Méndez, S., Delgado, J. V., Gómez, M., Dunner, S., & Cañón, J. (2010). The Canarian Camel: A Traditional Dromedary Population. Diversity, 2(4), 561-571. https://doi.org/10.3390/d2040561