Molecular Analysis of Bacterial Community DNA in Sludge Undergoing Autothermal Thermophilic Aerobic Digestion (ATAD): Pitfalls and Improved Methodology to Enhance Diversity Recovery

Abstract
:1. Introduction
2. Experimental Section
2.1. Sampling, ATAD Location, Feed Characteristics and Sampling Procedure
2.2. DNA Extraction
2.2.1. Solvent-based method
2.2.2. Extraction of genomic DNA via the MoBIO commercial kit
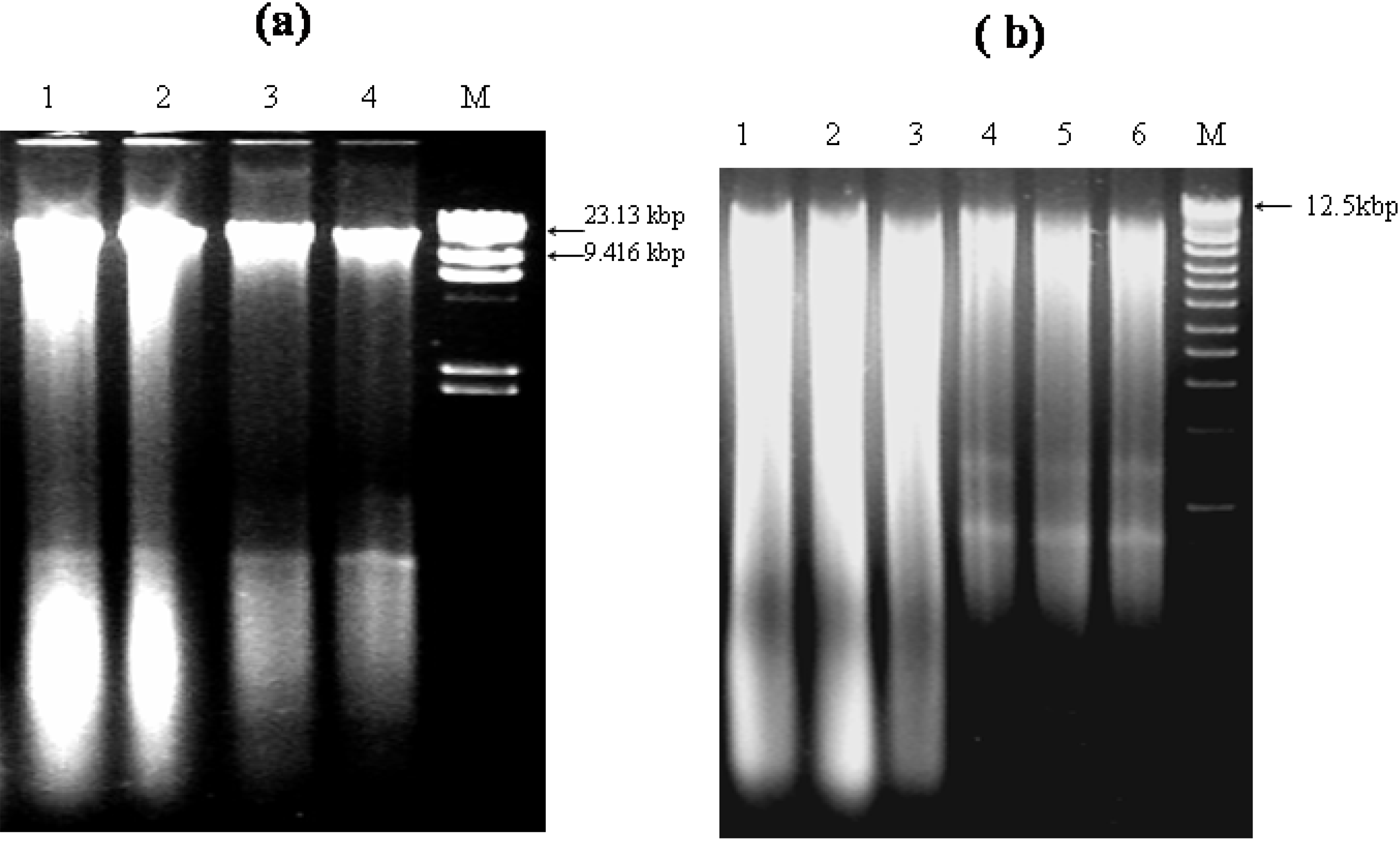
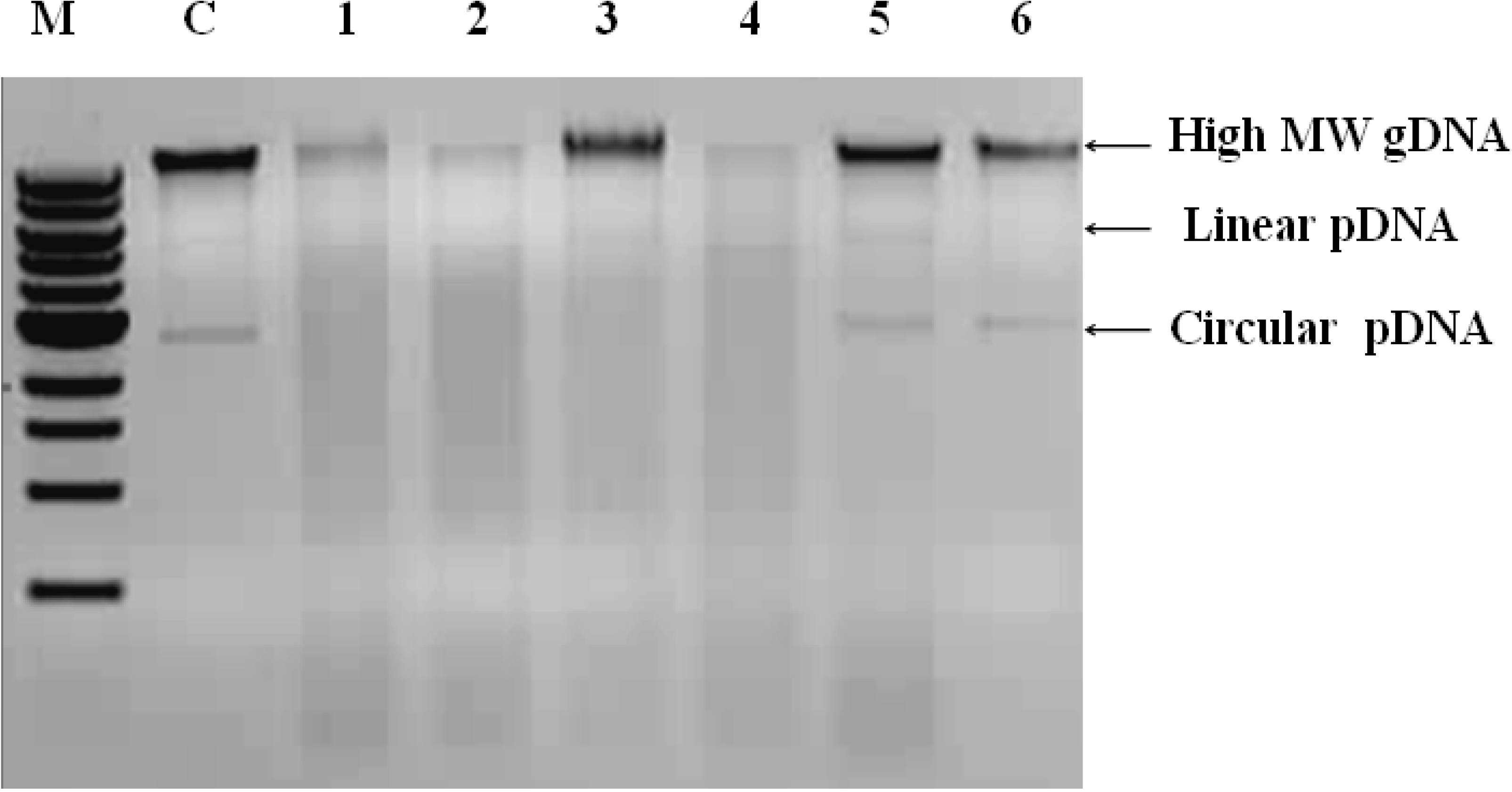
2.3. Analysis of Integrity and Size of Extracted Genomic DNA
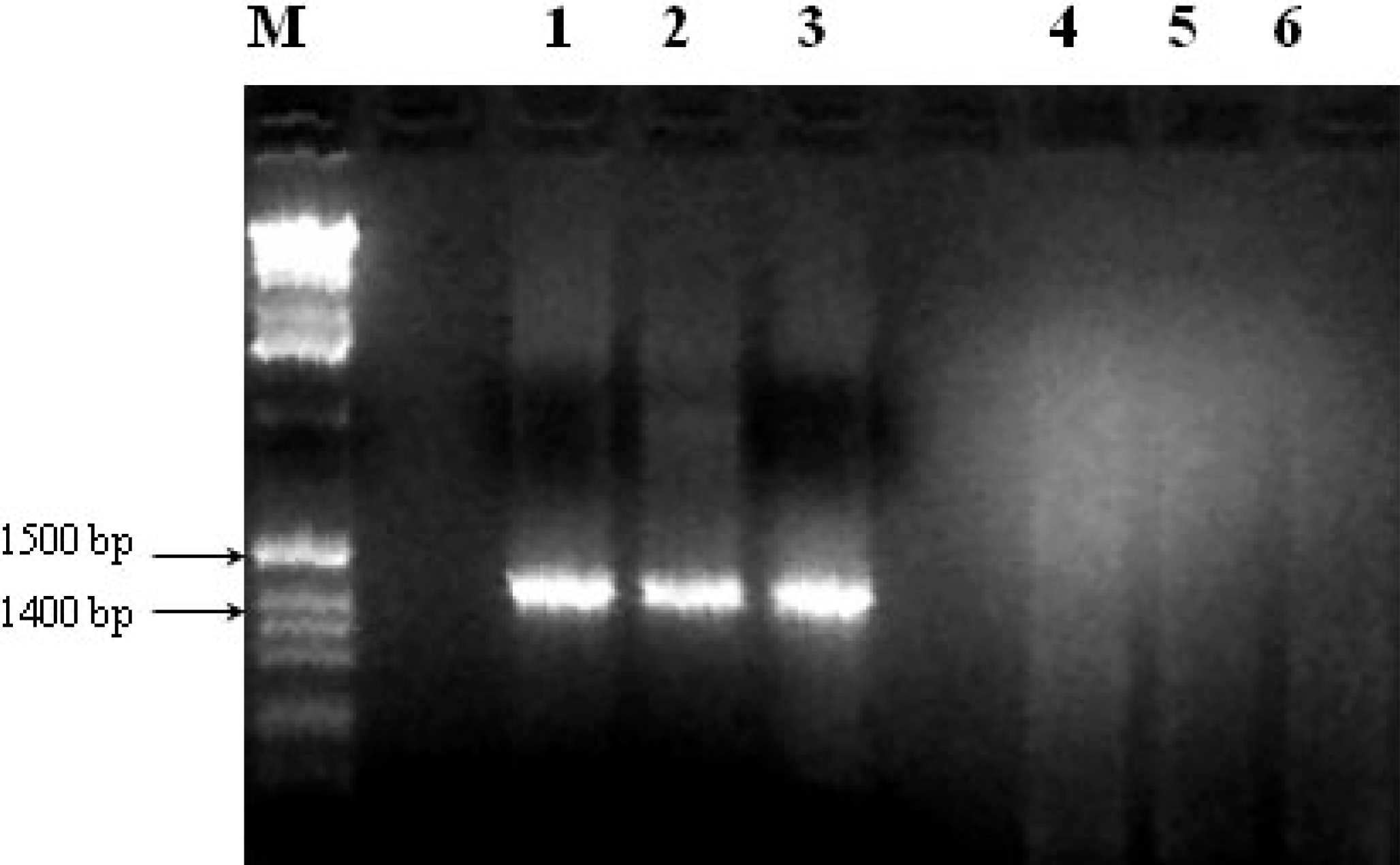
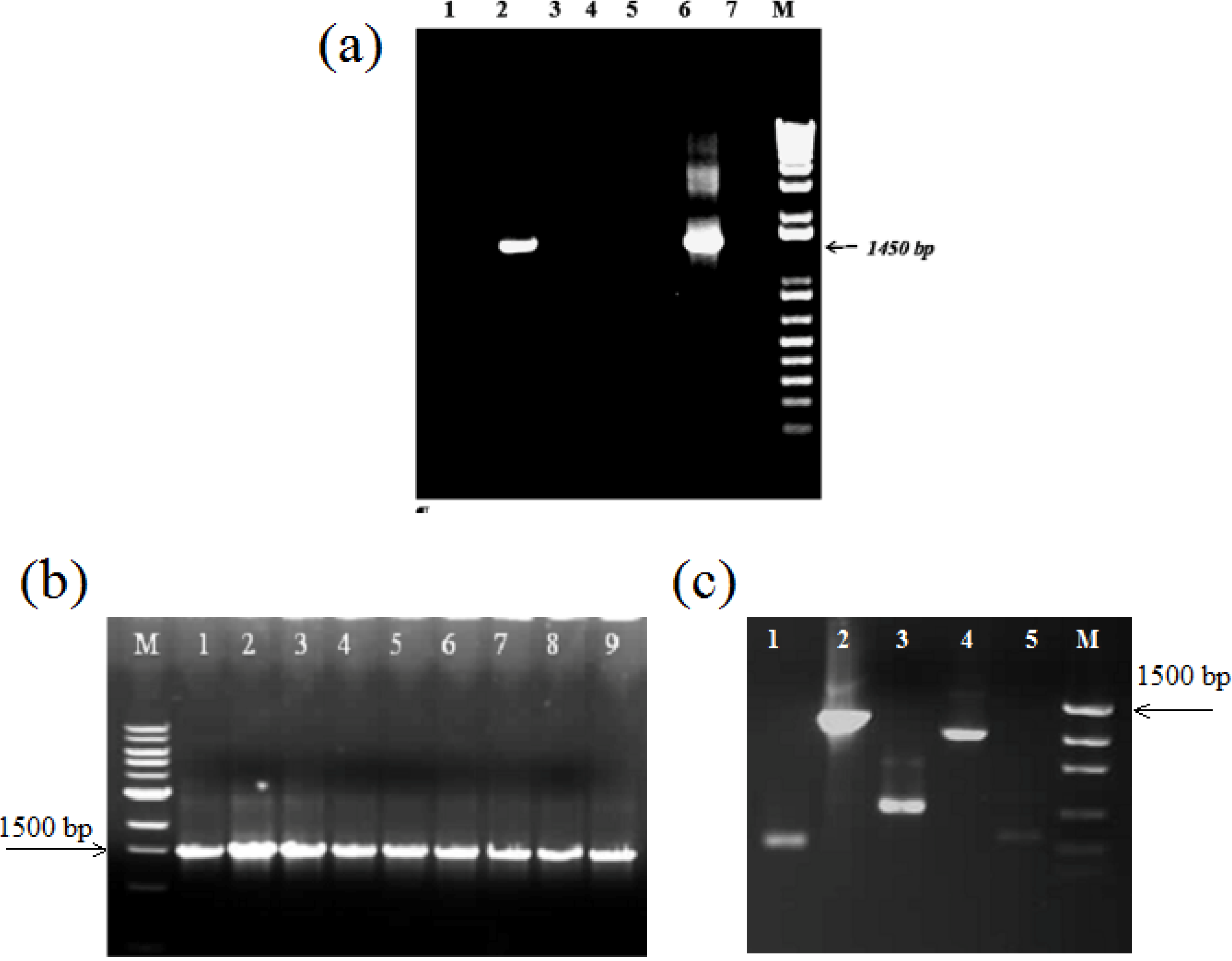
2.4. Amplification of Extracted DNA and Detection of Co-Extracted Contaminants
2.4.1. PCR conditions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer name | Nucleotide sequence (5’–3’) | Target sequences | Techniques | Reference |
|---|---|---|---|---|
| 27f | AGAGTTTGATCCTGGCTCAG | V1, 16S rRNA gene | PCR | [44] |
| 1492r | GGTTACCTTGTTACGACTT | V 9 , 16S rRNA gene | PCR | [44] |
| 518r | ATTACCGCGGCTGCTGG | V5, 16S rRNA gene | PCR, DGGE, | [45] |
| 338fGC a | ACTCCTACGGGAGGCAGCAG | V3, 16S rRNA gene | DGGE | [45] |
| 968f | AACGCGGAAGAACCTTAC | V6, 16S rRNA gene | PCR | [46] |
| 1401r | CGGTGTGTACAAGAAGACCC | V8 , 16S rRNA gene | PCR | [44] |
| 1698f | AACATCGGTTTGATCAAC | rpoB | PCR | [47] |
| 2041r | CGTTGCATGTTGGTACCCAT | rpoB | PCR | [47] |
| Int R391f | AACTAGGGCTGGGCTTATAACATGGCC | Integrase, R391 | PCR | [48,49] |
| Int R391r | AAAGATGGCAGCTTGCCGCAACCTC | Integrase, R391 | PCR | [48,49] |
| Kanf | tatcgattgtatgggaagcc | aph, R391 [46] | PCR, cloning, qRT-PCR | [This study] |
| Kanr | cagcgcatcaacaatattttca | aph, R391 [46] | PCR, cloning, qRT-PCR | [This study] |
| T7f | ATTTAGGTGACACTATAG | pGEM vector | sequencing | Promega, UK |
| SP6r | TAATACGACTCACTATAGGG | pGEM vector | sequencing | Promega, UK |
2.4.2. Addition of additives to improve PCR amplification and relieve inhibition of ATAD sludge derived components
2.5. The Effect of Inhibiting Co-Contaminants on the Activity of Taq-Polymerases of Different Origin
2.6. Development of an Internal Standard for the qRT-PCR Assay and Inhibition Assessment
2.7. Analysis of the Total Bacterial Community Profile and Predominant and Rare Species as a Function of Extraction Procedures
3. Results and Discussion
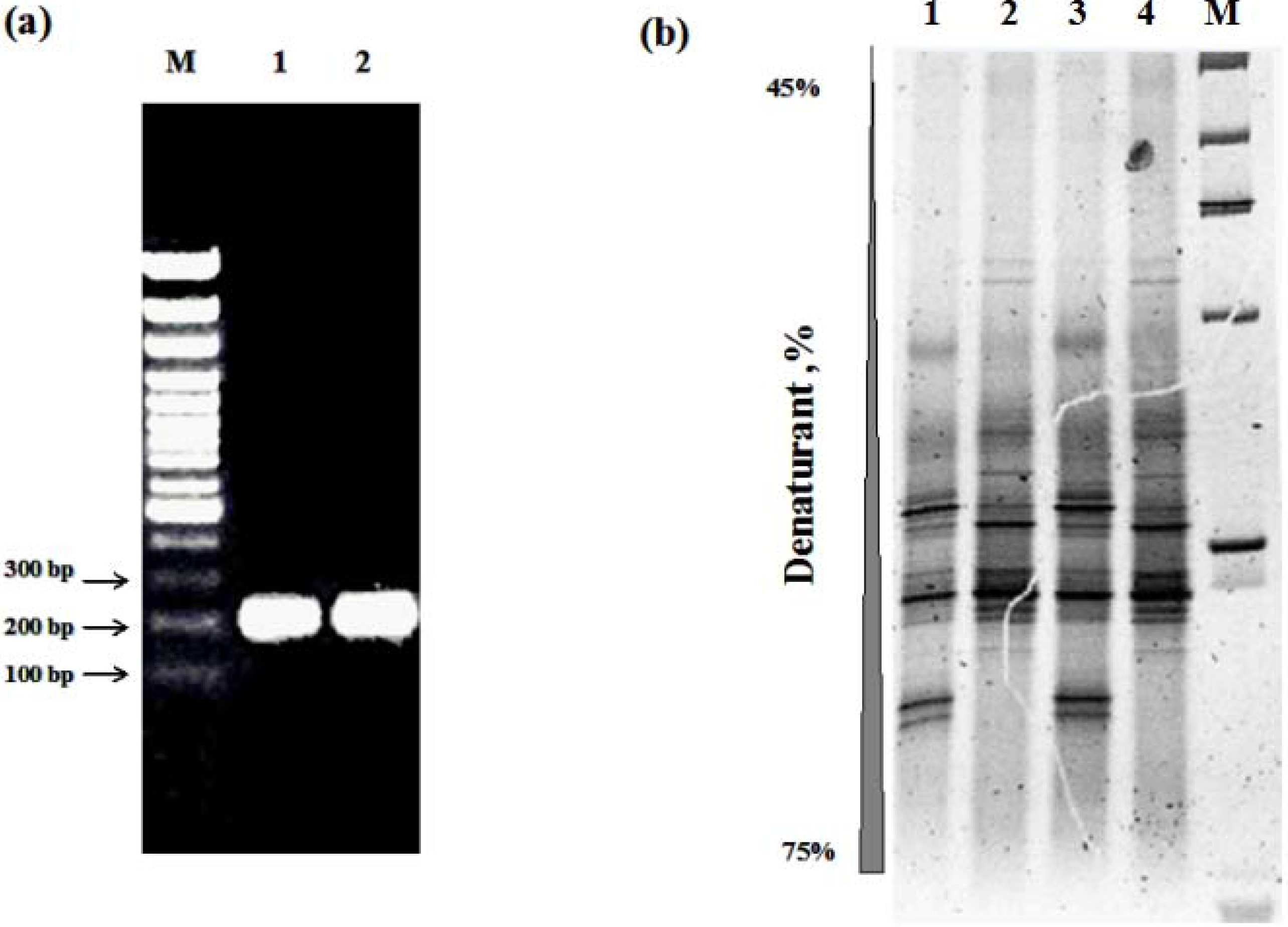
3.1. Integrity, Size and Stability of the Extracted Nucleic Acid
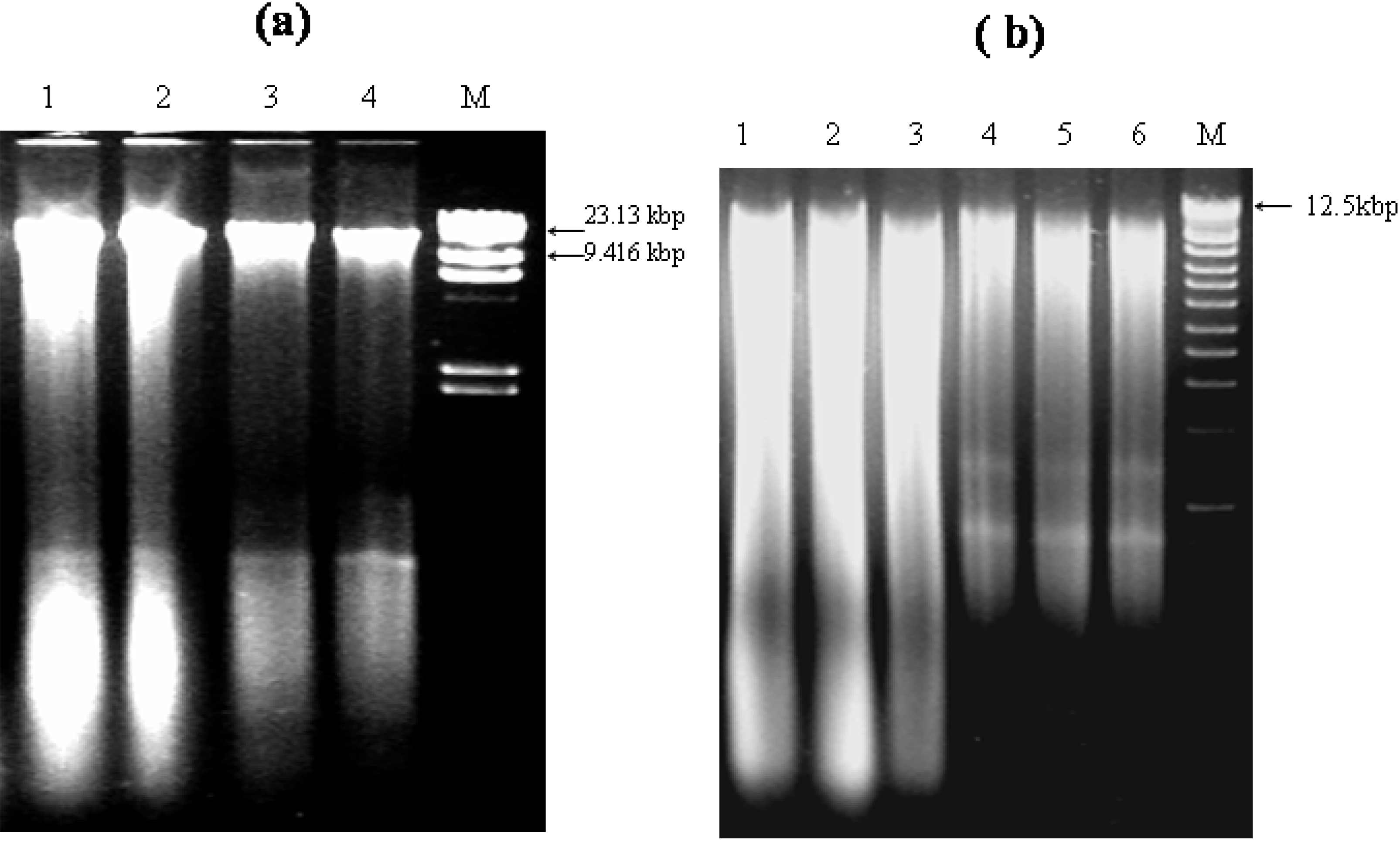
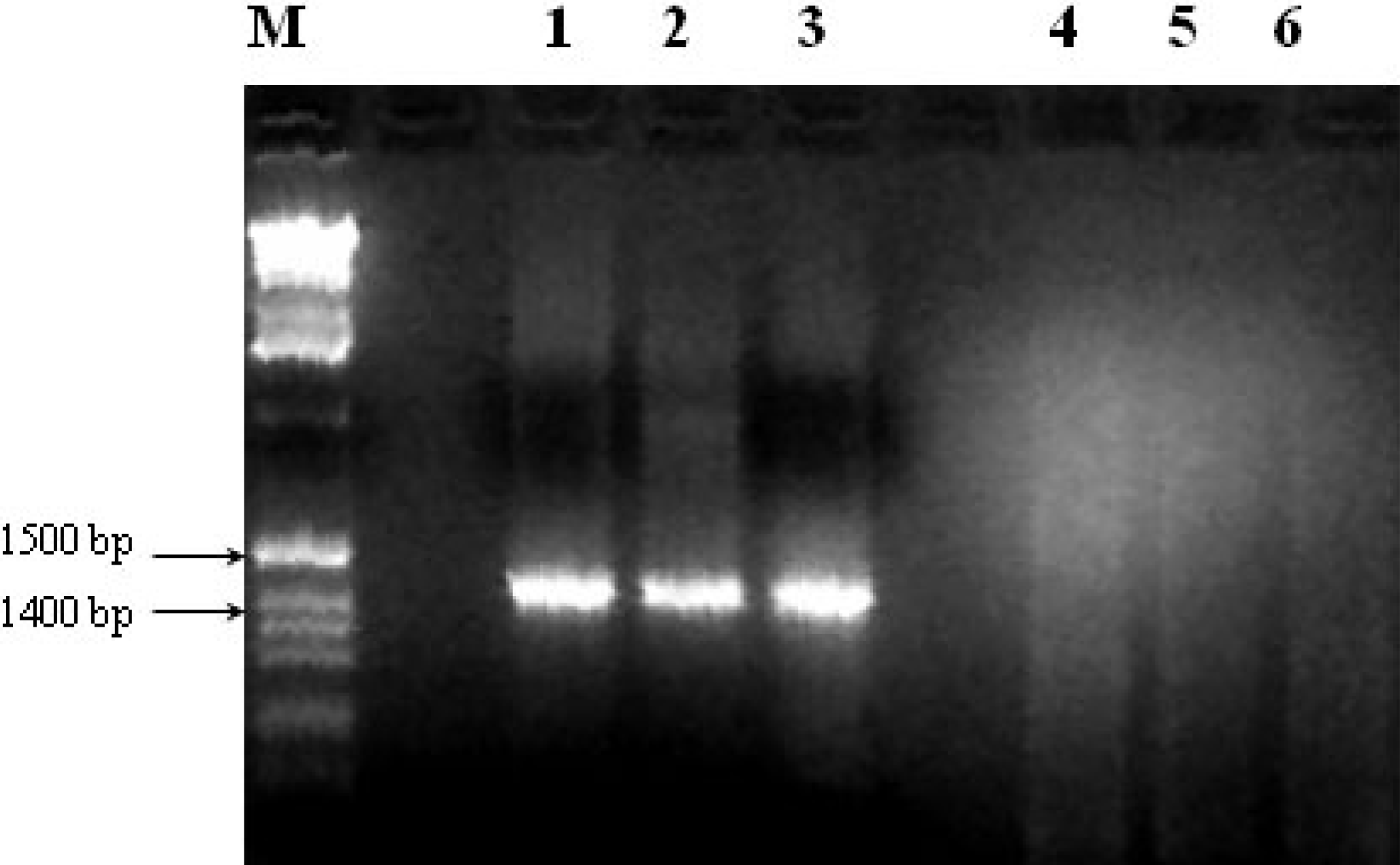
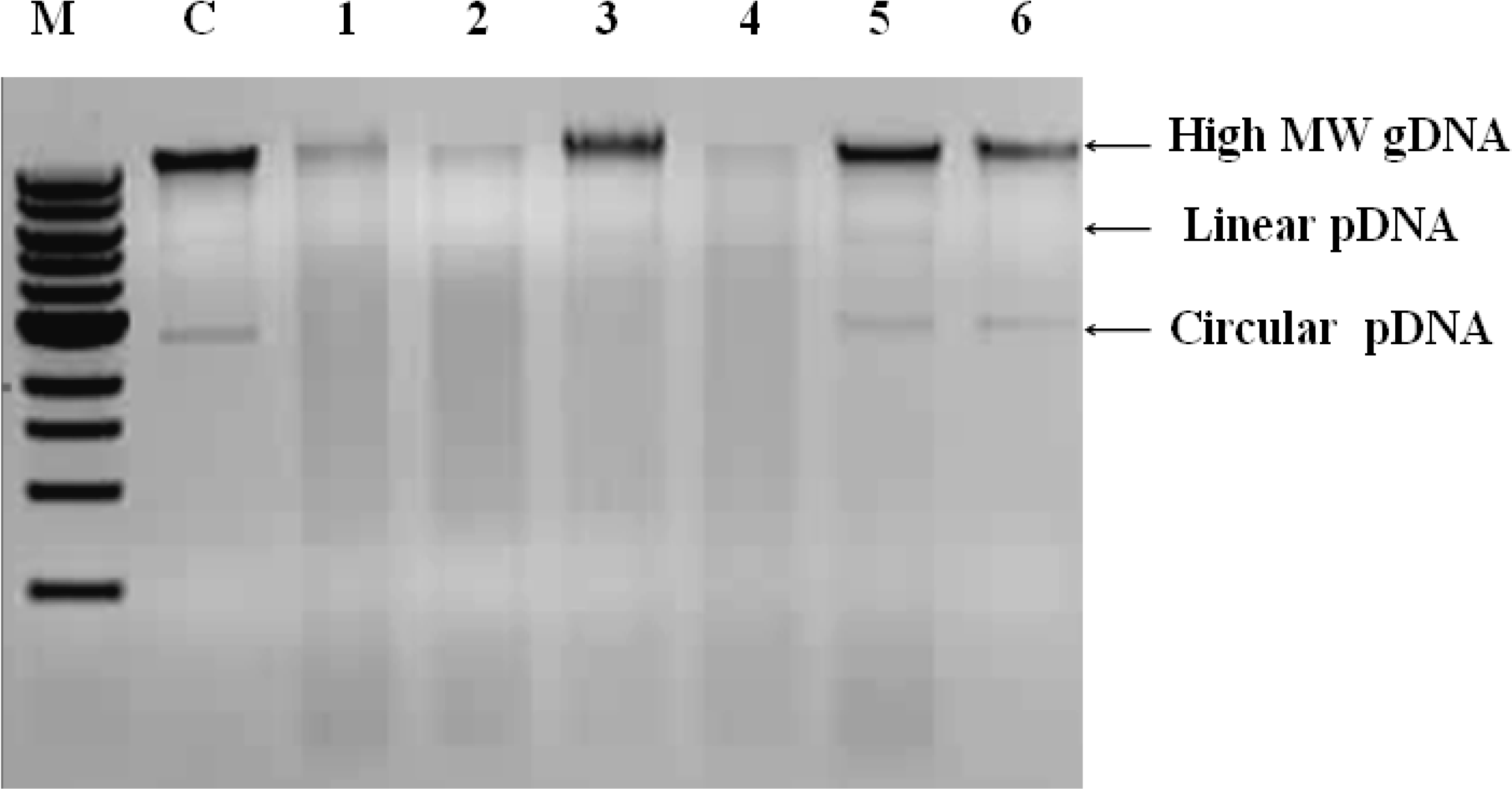
3.3. Other Strategies to Overcome Inhibition of PCR Amplification
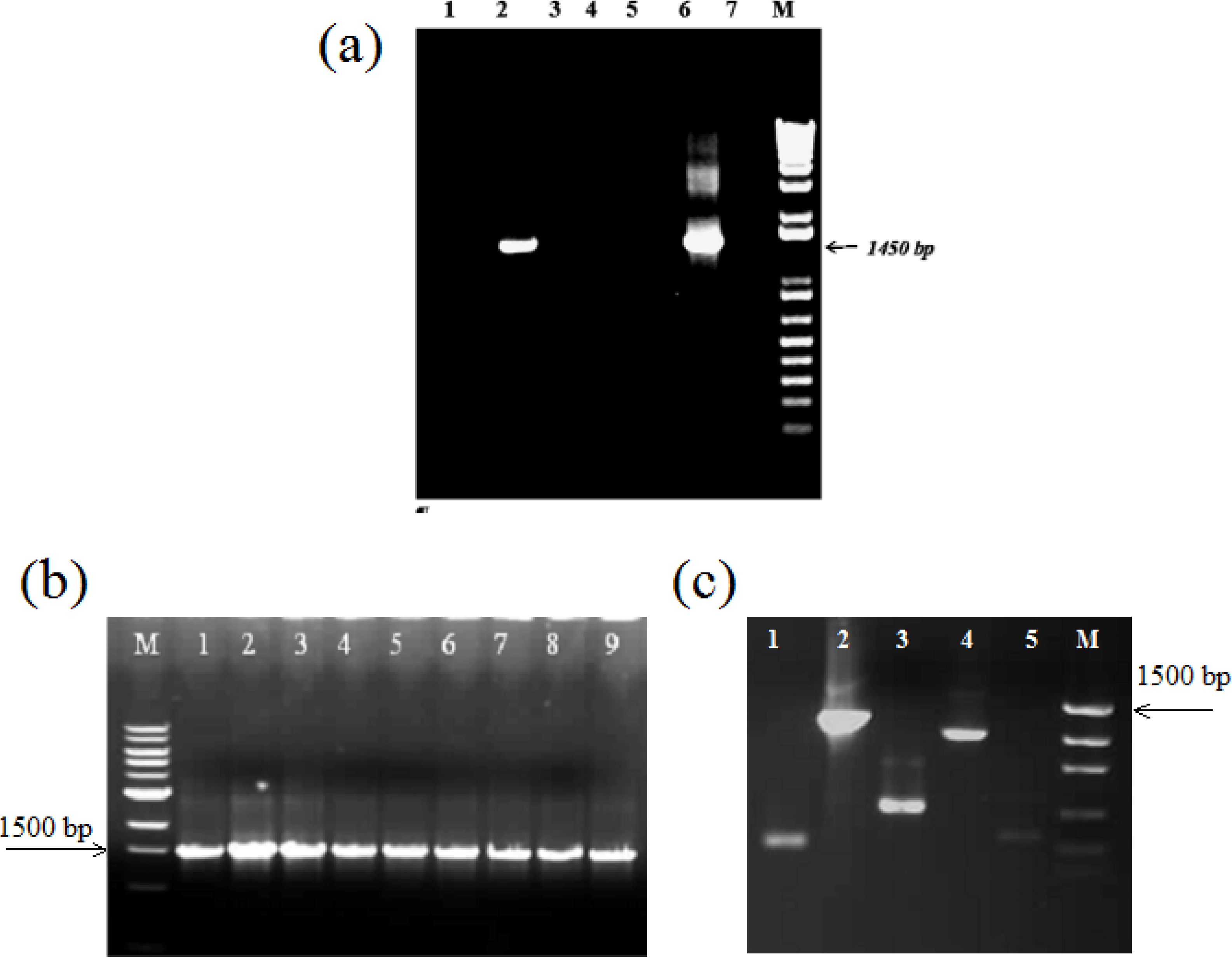
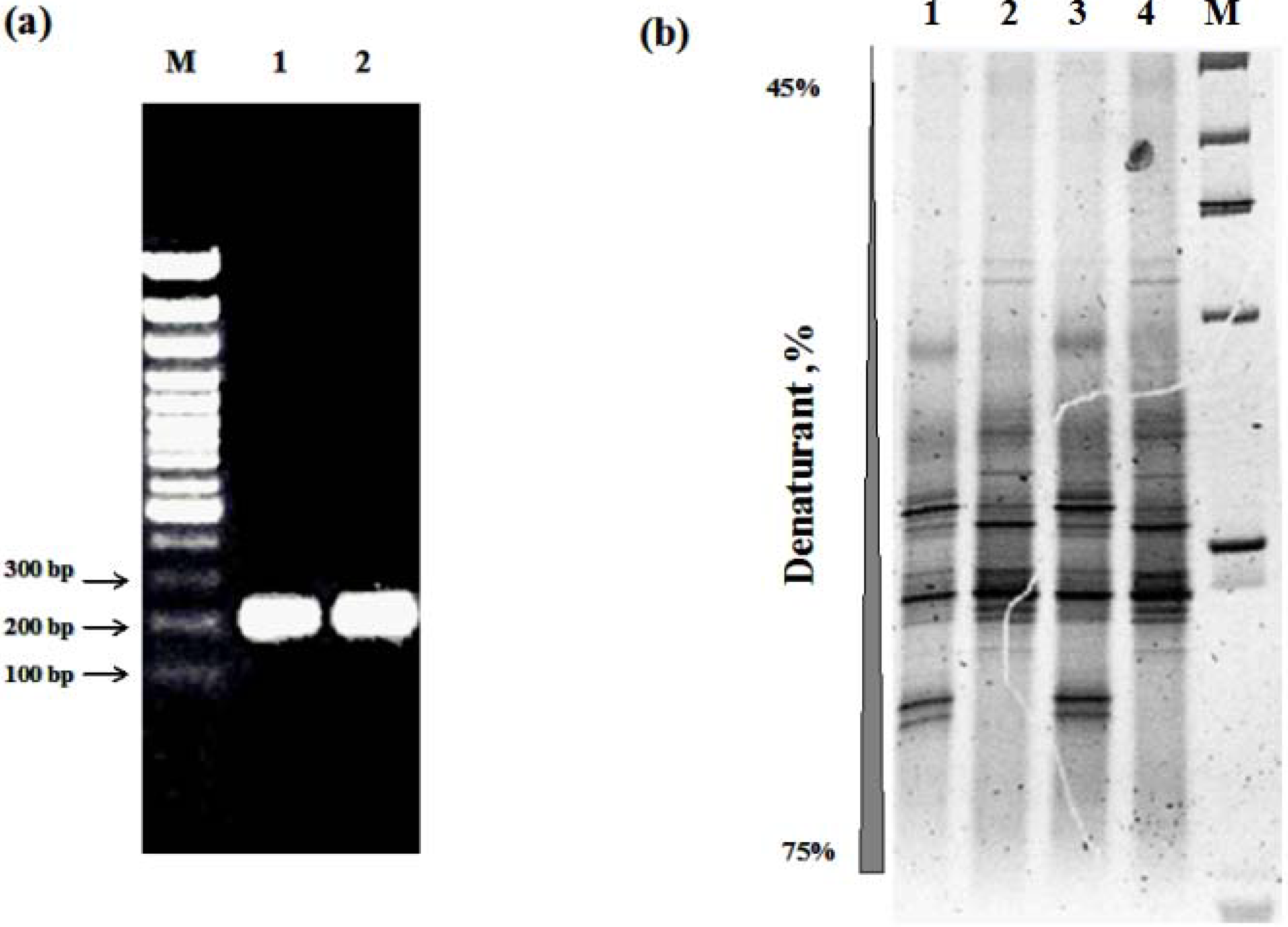
3.4. Capacity of DNA Extraction Methods to Reproduce a Total Community Profile
| OTU from ATAD library | Accesssion number | Closest affiliation | % | Closest affiliation to cultivated strains | % | % of total clones in the library | |
|---|---|---|---|---|---|---|---|
| Library type | |||||||
| MoBIO kit | Solvent- based method | ||||||
| CK5 | GU320654 | Uncultured bacterium, clone H1-814, EF174267 | 99 | Clostridium ultunense Z69293 | 94 | 17.5 | 8 |
| ER78 | GU325838 | Uncultured compost bacterium, clone 4B18, DQ346582 | 99 | Bacillus sp. MSP06G, AB084065 Bacillus thermocloaceae; DSM 5250, Z26939 | 98 98 | 0 | 14.5 |
| CK27 | GU320661 | Uncultured bacterium, clone SMQ30, AM930327 | 99 | Bacillus sp. MSP06G ; AB084065 Bacillus thermocloaceae; DSM 5250; Z26939 | 98 97.5 | 4 | 17.5 |
| CK28 | GU320662 | Uncultured bacterium clone F24; AM500822 | 99 | Bacillus sp. 50LAy-1, AB375754 Ureibacillus thermosphaericus; S7, AF403019 | 99 99 | 3 | 9.5 |
| ER32 | GU325832 | Symbiobacterium sp. KY38, AB361629 | 99 | Symbiobacterium thermophilum IAM 14863 DNA AP006840 | 97 | 1 | 30.5 |
| ER 9 | GU325829 | Uncultured compost bacterium clone 1B07, DQ346486 | 98 | Moorella glycerini SQL, GQ872425 | 97 | 0 | 2.5 |
Acknowledgements
References and Notes
- Nocker, A.; Burr, M.; Camper, A.K. Genotypic microbial community profiling: a critical technical review. Microb. Ecol. 2007, 54, 276–893. [Google Scholar] [CrossRef]
- Holben, W.E.; Harris, D. DNA-based monitoring of total bacterial community structure in environmental samples. Mol. Ecol. 1995, 4, 627–631. [Google Scholar] [CrossRef]
- Giraffa, G.; Neviani, E. DNA-based, culture-independent strategies for evaluating microbial communities in food-associated ecosystems. Int. J. Food Microbiol. 2001, 20, 19–34. [Google Scholar] [CrossRef]
- Ranjard, L.; Poly, F.; Nazaret, S. Monitoring complex bacterial communities using culture-independent molecular techniques: application to soil environment. Res. Microbiol. 2000, 151, 167–177. [Google Scholar] [CrossRef]
- Kawai, M.; Matsutera, E. 16S ribosomal DNA-based analysis of bacterial diversity in purified water used in pharmaceutical manufacturing processes by PCR and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 2000, 68, 699–704. [Google Scholar] [CrossRef]
- Zhou, X.; Bent, S.J.; Schneider, M.G.; Davis, C.C.; Islam, M.R.; Forney, L.J. Characterizationof vaginal microbial communities in adult healthy women using cultivation-independent methods. Microbiology 2004, 150, 2565–2573. [Google Scholar] [CrossRef]
- Amann, R.I.; Ludwig, W.; Schleifer, K.H. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 1995, 59, 143–146. [Google Scholar]
- Dahllöf, I. Molecular community analysis of microbial diversity. Curr. Opin. Microbiol. 2002, 13, 213–217. [Google Scholar]
- Colwell, R.; Grimes, J. Semantics and strategies. In Non-culturable Microorganisms in the Environment; Colwell, R., Grimes, J., Eds.; ASM: Washington. DC, USA, 2000; pp. 1–5. [Google Scholar]
- Kazuhiro, K. Nonculturable bacterial populations that control environmental processes. Biosci. Ind. 1999, 57, 731–736. [Google Scholar]
- Frostegard, A.; Courtois, S.; Ramisse, V.; Clerc, S.; Bernillon, D.; Le Gall, F.; Jeannin, P.; Nesme, X.; Simonet, P. Quantification of bias related to the extraction of DNA directly from soils. Appl. Environ. Microbiol. 1999, 65, 5409–5420. [Google Scholar]
- Wu, L.; Li, F.; Deng, C.; Xu, D.; Jiang, S; Xiong, Y. A method for obtaining DNA from compost. Appl. Biochem. Biotechnol. 2009, 84, 389–395. [Google Scholar]
- Al-Soud, W.A.; Radstrom, P. Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl. Environ. Microbiol. 1998, 64, 3748–3753. [Google Scholar]
- Weiss, A.; Jerome, V.; Freitag, R. Comparison of strategies for the isolation of PCR-compatible, genomic DNA from a municipal biogas plants. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2007, 15, 190–197. [Google Scholar] [CrossRef]
- Stach, J.E.; Bathe, S.; Clapp, J.P.; Burns, R.G. PCR-SSCP comparison of 16S rDNA sequence diversity in soil DNA obtained using different isolation and purification methods. FEMS Microbiol. Ecol. 2001, 36, 139–151. [Google Scholar]
- McIlroy, S.J.; Porter, K.; Seviour, R.J; Tillett, D. Extracting nucleic acids from activated sludge which reflect community population diversity. Antonie van Leeuwenhoek 2009, 96, 593–605. [Google Scholar] [CrossRef]
- McOrist, A.L.; Jackson, M.; Bird, A.R. A comparison of five methods for extraction of bacterial DNA from human faecal samples. J. Microbiol. Meth. 2002, 50, 131–139. [Google Scholar] [CrossRef]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of Complex Microbial populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar]
- Fischer, S.G.; Lerman, L.S. DNA fragments differing by single base-pair substitutions are separated in denaturing gradient gels: correspondence with melting theory. Proc. Natl. Acad. Sci. USA 1983, 80, 1579–1583. [Google Scholar] [CrossRef]
- Durtschi, J.D.; Voelkerding, K.V.; Wittwer, C.T.; Seipp, M.T. Multiplex amplicon genotyping by high-resolution melting. J. Biomol.Tech. 2009, 20, 160–164. [Google Scholar]
- Relman, A. Universal bacterial 16S rDNA amplification and sequencing. In Diagnostic Molecular Microbiology, Principles and Applications; Persing, D.H., Smith, T.F., Tenover, F.C., White, T.J., Eds.; ASM: Washington, DC, USA, 1993; pp. 489–495. [Google Scholar]
- Petti, C.A.; Polage, C.R.; Schreckenberger, P. The role of 16S rRNA gene sequencing in identification of microorganisms misidentified by conventional methods. J. Clin. Microbiol. 2005, 43, 6123–6125. [Google Scholar] [CrossRef]
- Alvarez, A.J.; Buttner, M.P.; Stetzenbach, L.D. PCR for bioaerosol monitoring: sensitivity and environmental interference. Appl. Environ. Microbiol. 1995, 61, 3639–3644. [Google Scholar]
- Lantz, P.G.; Abu Al-Soud, W.A.; Knutsson, R.; Hahn-Hägerdal, B.; Radstrom, P. Biotechnical use of polymerase chain reaction for microbiological analysis of biological samples. Biotechnol. Annu. Rev. 2000, 5, 87–130. [Google Scholar] [CrossRef]
- Heuer, H.; Smalla, K. Application of denaturing gradient gel electrophoresis and temperature gradient gel electrophoresis for studying soil microbial communities. In Modern Soil Microbiology; van Elsas, J.D., Trevors, J.T., Wellington, E.M.H., Eds.; Marcel Dekker: New York, NY, USA, 1997; pp. 353–373. [Google Scholar]
- Chandler, D.P.; Frederickson, J.K.; Brockman, J. Effect of PCR template concentration on the composition and distribution of total community 16S rDNA clone libraries. Mol. Ecol. 1997, 6, 475–482. [Google Scholar]
- Pomp, D.; Medrano, J.F. Organic solvents as facilitators of polymerase chain reaction. BioTechniques 1991, 10, 58–59. [Google Scholar]
- Sidhu, M.K.; Liao, M.J.; Rashidbaigi, A. Dimethyl sulfoxide improves RNA amplification. BioTechniques 1996, 21, 44–47. [Google Scholar]
- Nol, P.; Williamson, J.L.; Rocke, T.E.; Yuill, T.M. Detection of Clostridium botulinum type C cells in the gastrointestinal tracts of Mozambique Tilapia (Oreochromis mossambicus) by polymerase chain reaction. J. Wildl Dis. 2004, 40, 749–753. [Google Scholar] [CrossRef]
- Klammer, S.; Mondini, C.; Insam, H. Microbial community fingerprint of compost stored under different condition. Ann. Microbiol. 2005, 55, 299–305. [Google Scholar]
- Maarit-Niemi, R.; Heiskanen, I.; Wallenius, K.; Lindstrom, K. Extraction and purification of DNA in rhizosphere soil samples for PCR-DGGE analysis of bacterial consortia. J. Microbiol. Meth. 2001, 45, 155–165. [Google Scholar] [CrossRef]
- Bramucci, M.G.; Nagarajan, V. Industrial wastewater bioreactors: sources of novel microorganisms for biotechnology. Trends Biotechnol. 2000, 18, 501–505. [Google Scholar] [CrossRef]
- Sen, R; Chakrabarti, S. Biotechnology—applications to environmental remediation in resource exploitation. Curr. Sci. 2009, 97, 768–775. [Google Scholar]
- Layden, N.; Mavinic, D.; Kelly, H.; Moles, R.; Bartlett, J. Autothermal thermophilic aerobic digestion (ATAD)–Part II: Review of research and full-scale operating experiences. J. Environ. Eng. Sci. 2007, 6, 679–690. [Google Scholar] [CrossRef]
- Sonnleitner, B.; Fiechter, A. Bacterial diversity in thermophilic aerobic sewage sludge II. Types of organisms and their capacities. Eur. J. Appl. Microbiol. Biotechnol. 1983, 18, 174–180. [Google Scholar] [CrossRef]
- Sonnleitner, B.; Fiechter, A. Microbial flora studies in thermophilic aerobic sludge treatment. Conserv. Recycl. 1985, 8, 303–313. [Google Scholar] [CrossRef]
- Juteau, P.; Tremblay, D.; Villemur, R.; Bisaillon, J.G.; Beaudet, R. Analysis of the bacterial community inhabiting an aerobic thermophilic sequencing batch reactor (AT-SBR) treating swine waste. Appl. Microbiol. Biotechnol. 2004, 66, 115–122. [Google Scholar] [CrossRef]
- LaPara, T.M.; Nakatsu, C.H.; Pantea, L.; Alleman, J.E. Phylogenetic analysis of bacterial communities in mesophilic and thermophilic bioreactors treating pharmaceutical wastewater. Appl. Environ. Microbiol. 2000, 66, 3951–3959. [Google Scholar] [CrossRef]
- Piterina, A.V.; MacCausland, C.; Bartlett, J.; Pembroke, J.T. Microbial ecology of autothermal aerobic digestion (ATAD): diversity, dynamics and activity of bacterial communities involved in treatment of municipal wastewater. In Modern Multidisciplinary Applied Microbiology, Exploiting Microbes and Their Interactions; Mendez-Vilas, A., Ed.; Wiley-VCH: Weinheim, Germany, 2006; pp. 526–535. [Google Scholar]
- Piterina, A.V.; Barlett, J.; Pembroke, T.J. 13C-NMR assessment of the pattern of organic matter transformation during domestic wastewater treatment by Autothermal Aerobic Digestion (ATAD). Int. J. Environ. Res. Public Health 2009, 6, 2288–2306. [Google Scholar] [CrossRef]
- Miller, D.N.; Bryant, J.E.; Madsen, E.L.; Ghiorse, W.C. Evaluation and optimization of DNA extraction and purification procedures for soil and sediment samples. Appl. Environ. Microbiol. 1999, 65, 4715–4724. [Google Scholar]
- Martin-Laurent, F.; Philippot, L.; Hallet, S.; Chaussod, R.; Germon, J.C.; Soulas, G.; Catroux, G. DNA extraction from soils: old bias for new microbial diversity analysis method. Appl. Environ. Microbiol. 2001, 67, 2354–2359. [Google Scholar] [CrossRef]
- Sambrook, J. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Lane, D.J. 16S/23S rRNA Sequencing. In Nucleic Acid Techniques in Bacterial Systematics; Stackebrandt, E., Goodfellow, M., Eds.; John Wiley & Sons: New York, NY, USA, 1991; pp. 115–175. [Google Scholar]
- Muyzer, G.; Hottentrager, S.; Teske, A.; Wawer, C. Denaturing gradient gel electrophoresis of PCR-amplified 16S rDNA. A new molecular approach to analyse the genetic diversity of mixed microbial communities. In Molecular Microbial Ecology Manual; Akkermans, A., Ed.; Kluwer Academic Publ.: Norwell, MA, USA, 1996; p. 23. [Google Scholar]
- Nubel, U.; Engelen, B.; Felske, A.; Snaidr, J.; Wieshuber, A.; Amann, R.I.; Ludwig, W.; Backhaus, H. Sequence heterogeneities of genes encoding 16S rRNAs in Paenibacillus polymyxa detected by temperature gradient gel electrophoresis. J. Bacteriol. 1996, 178, 5636–5643. [Google Scholar]
- Dahllof, I.; Baillie, H.; Kjelleberg, S. rpoB-based microbial community analysis avoids limitations inherent in 16S rRNA gene intraspecies heterogeneity. Appl. Environ. Microbiol. 2000, 66, 3376–3380. [Google Scholar] [CrossRef]
- Böltner, D.; MacMahon, C.; Pembroke, J.T.; Strike, P.; Osborn, A.M. R391: a conjugative integrating mosaic comprised of phage, plasmid and transposon elements. J. Bacteriol. 2002, 184, 5158–5169. [Google Scholar] [CrossRef]
- McGrath, B.M; O’Halloran, J.A; Piterina, A.V.; Pembroke, J.T. Molecular tools to detect the IncJ elements: a family of integrating, antibiotic resistant mobile genetic elements. J. Microbiol. Methods 2006, 66, 32–42. [Google Scholar] [CrossRef]
- Wilson, I.G. Inhibition and facilitation of nucleic acid amplification. Appl. Environ. Microbiol. 1997, 63, 3741–3751. [Google Scholar]
- Demeke, T.; Adams, R.P. The effects of plant polysaccharides and buffer additives on PCR. Biotechniques 1992, 12, 332–334. [Google Scholar]
- Furrer, B.; Candrian, U.; Wieland, P.; Luthy, J. Improving PCR efficiency. Nature 1990, 26, 324. [Google Scholar]
- Kreader, C.A. Relief of amplification inhibition in PCR with bovine serum albumin or T4 gene 32 protein. Appl. Environ. Microbiol. 1996, 62, 1102–1106. [Google Scholar]
- Sarkar, G.; Kapelner, S.; Sommer, S.S. Formamide can dramatically improve the specificity of PCR. Nucl. Acids Res. 1990, 25, 7465. [Google Scholar] [CrossRef]
- Rossen, L.; Norskov, P.; Holmstrom, K.; Rasmussen, O.F. Inhibition of PCR by components of food samples, microbial diagnostic assays and DNA-extraction solutions. Int. J. Food Microbiol. 1992, 17, 37–45. [Google Scholar] [CrossRef]
- Akane, A.; Matsubara, K.; Nakamura, H.; Takahashi, S.; Kimura, K. Identification of the heme compound copurified with deoxyribonucleic acid (DNA) from bloodstains, a major inhibitor of polymerase chain reaction (PCR) amplification. J. Forensic Sci. 1994, 39, 362–372. [Google Scholar]
- Suarez, D. Molecular diagnostic techniques: can we identify influenza viruses, differentiate subtypes and determine pathogenicity potential of viruses by RT-PCR? Avian Dis. 1997, 47, 318–325. [Google Scholar]
- Sarkar, G.; Kapelner, S.; Sommer, S.S. Formamide can dramatically improve the specificity of PCR. Nucl. Acids Res. 1990, 18, 7465. [Google Scholar] [CrossRef]
- Abu Al-Soud, W.A.; Radstrom, P. Effects of amplification facilitators on diagnostic PCR in the presence of blood, feces, and meat. J. Clin. Microbiol. 2000, 38, 4463–4470. [Google Scholar]
- Chou, S.W. Optimizing polymerase chain reaction technology for clinical diagnosis. Clin. Chem. 1991, 37, 1893–1894. [Google Scholar]
- Kleiboeker, S.B. Applications of competitor RNA in diagnostic reverse transcription-PCR. J. Clin. Microbiol. 2003, 41, 2055–2061. [Google Scholar] [CrossRef]
- Sipos, R.; Székely, A.; Révész, S.; Márialigeti, K. Addressing PCR biases in environmental microbiology studies. Methods Mol. Bio 2010, 599, 37–58. [Google Scholar] [CrossRef]
- Don, R.H.; Cox, P.T.; Wainwright, B.J.; Baker, K.; Mattick, J.S. “Touchdown” PCR to circumvent spurious priming during gene amplification. Nucl. Acids Res. 1991, 19, 4008. [Google Scholar] [CrossRef]
- Hongoh, Y.; Yuzawa, H.; Ohkuma, M.; Kudo, T. Evaluation of primers and PCR conditions for the analysis of 16S rRNA genes from a natural environment. FEMS Microbiol. Lett. 2003, 25, 299–304. [Google Scholar]
- Massol-Deya, A.A.; Odelson, D.A.; Hickey, R.F.; Tiedje, J.M. Bacterial Community Fingerprinting of Amplified 16S and 16-23S Ribosomal DNA Gene Sequences and Restriction Endonuclease Analysis (ARDRA). In Molecular Microbial Ecology Manual; Akkermans, A.D.L., Ed.; Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; pp. 1–8. [Google Scholar]
- CLC bio’s Workbenches. ( http://www.clcbio.com).
- VecScreen. ( http://www.ncbi.nlm.nih.gov/VecScreen/VecScreen.html).
- Sequence manipulation suite. ( http://www.gobozzy.com/sequence-analysis-tools).
- Maidak, B.L; Cole, J.R.; Lilburn, T.G.; Parker, C.T., Jr; Saxman, P.R.; Farris, R.J.; Garrity, G.M.; Olsen, G.J.; Schmidt, T.M.; Tiedje, J.M. The RDP-II (Ribosomal Database Project). Nucl. Acids Res. 2001, 29, 173–174. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Wang, G.C.T.; Wang, Y. The frequency of chimeric molecules as a consequence of PCR co-amplification of 16S rRNA genes from different bacterial species. Microbiology 1996, 142, 1107–1114. [Google Scholar] [CrossRef]
- Kuske, C.R.; Banton, K.L.; Adorada, D.L.; Stark, P.C.; Jackson, P.J. Small-scale DNA sample preparation method for field PCR detection of microbial cells and spores in soil. Appl. Environ. Microbiol. 1998, 64, 2463–2472. [Google Scholar]
- DellAnno, A.; Fabiano, M.; Duineveld, G.C.A.; Kok, A.; Danovaro, R. Nucleic acid (DNA, RNA) quantification and RNA/DNA ratio determination in marine sediments: comparison of spectrophotometric, fluorometric and High Performance Liquid Chromatography methods and estimation of detrital DNA. Appl. Environ. Microbiol. 1998, 64, 3238–3245. [Google Scholar]
- Jackson, C.R.; Harper, J.P.; Willoughby, D.; Roden, E.E.; Churchill, P.F. A simple, efficient method for the separation of humic substances and DNA from environmental samples. Appl. Environ. Microbiol. 1997, 63, 4993–4995. [Google Scholar]
- Radstrom, P.; Knutsson, R.; Wolffs, P.; Lövenklev, M.; Löfström, C. Pre-PCR processing: Strategies to generate PCR-compatible samples. Mol. Biotechnol. 2004, 26, 133–146. [Google Scholar] [CrossRef]
- Abu Al-Soud, W.; Râdström, P. Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl. Environ. Microbiol. 1998, 64, 3748–3753. [Google Scholar]
- LaMontagne, M.G.; Michel, F.C., Jr.; Holden, P.A.; Reddy, C.A. Evaluation of extraction and purification methods for obtaining PCR-amplifiable DNA from compost for microbial community analysis. J. Microbiol. Meth. 2002, 49, 255–264. [Google Scholar] [CrossRef]
- Arbeli, Z.; Fuentes, C.L. Improved purification and PCR amplification of DNA from environmental samples. FEMS Microbiol. Lett. 2007, 272, 269–275. [Google Scholar] [CrossRef]
- Blanchard, M.M.; Tailon-Miller, P.; Nowotny, P.; Nowotny, V. PCR buffer optimization with a uniform temperature regimen to facicilate automation. PCR Meth. Applic. 1993, 2, 234–240. [Google Scholar]
- Innis, M.A.; Gelfand, D.H. Optimization of PCRs. In PCR protocols. A guide to methods and applications; Innis, M.A., Gelfand, D.H., Sninsky, J.J., White, T.J., Eds.; Academic Press Inc.: San Diego, CA, USA, 1990; p. 12. [Google Scholar]
- Loomis, W.D. Overcoming problems of phenolics and quinones in the isolation of plant enzymes and organelles. Meth. Enzymol. 1974, 31, 528–545. [Google Scholar] [CrossRef]
- Zhang, W.; Hu, G.Y.; Deisseroth, A. Improvement of PCR sequencing by formamide. Nucleic Acids Res. 1991, 19, 6649. [Google Scholar] [CrossRef]
- Loeffelholz, M.; Deng, H. PCR and Its Variations. In Advanced techniques in Diagnostic Microbiology; Tang, Y.W., Stratton, C.W., Eds.; Springer: New York, NY, USA, 2006; Volume 1, pp. 166–183. [Google Scholar]
- Wiedbrauk, D.L.; Werner, J.C.; Drevon, A.M. Inhibition of PCR by aqueous and vitreous fluids. J. Clin. Microbiol. 1995, 33, 2643–2646. [Google Scholar]
- Wilson, I.G.; Cooper, J.E.; Gilmour, A. Some factors inhibiting amplification of the Staphylococcus aureus enterotoxin C1 (sec1) by PCR. Int. J. Food Microbiol. 1994, 22, 55–62. [Google Scholar]
- Gibson, J.R.; Sutherland, K.; Owen, R.J. Inhibition of DNAse activity in PFGE analysis of DNA from Campylobacter jejuni. Lett. Appl. Microbiol. 1994, 19, 357–358. [Google Scholar] [CrossRef]
- Singer, G.A.; Hickey, D.A. Thermophilic prokaryotes have characteristic patterns of codon usage, amino acid composition and nucleotide content. Gene 2003, 23, 39–47. [Google Scholar] [CrossRef]
- Klappenbach, J.A.; Saxman, P.R.; Cole, J.R.; Schmidt, T.M. rrndb: the Ribosomal RNA Operon Copy Number Database. Nucleic Acids Res. 2001, 29, 181–184. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Piterina, A.V.; Bartlett, J.; Pembroke, J.T. Molecular Analysis of Bacterial Community DNA in Sludge Undergoing Autothermal Thermophilic Aerobic Digestion (ATAD): Pitfalls and Improved Methodology to Enhance Diversity Recovery. Diversity 2010, 2, 505-526. https://doi.org/10.3390/d2040505
Piterina AV, Bartlett J, Pembroke JT. Molecular Analysis of Bacterial Community DNA in Sludge Undergoing Autothermal Thermophilic Aerobic Digestion (ATAD): Pitfalls and Improved Methodology to Enhance Diversity Recovery. Diversity. 2010; 2(4):505-526. https://doi.org/10.3390/d2040505
Chicago/Turabian StylePiterina, Anna V., John Bartlett, and J. Tony Pembroke. 2010. "Molecular Analysis of Bacterial Community DNA in Sludge Undergoing Autothermal Thermophilic Aerobic Digestion (ATAD): Pitfalls and Improved Methodology to Enhance Diversity Recovery" Diversity 2, no. 4: 505-526. https://doi.org/10.3390/d2040505
APA StylePiterina, A. V., Bartlett, J., & Pembroke, J. T. (2010). Molecular Analysis of Bacterial Community DNA in Sludge Undergoing Autothermal Thermophilic Aerobic Digestion (ATAD): Pitfalls and Improved Methodology to Enhance Diversity Recovery. Diversity, 2(4), 505-526. https://doi.org/10.3390/d2040505