
Different Ribotypes of Akashiwo sanguinea Harbor Distinct Bacterial Communities in Their Phycospheres


 , and
, and 
Abstract
1. Introduction
2. Materials and Methods
2.1. Algal Cultures of Akashiwo sanguinea
2.2. Sample Collection and DNA Extraction
2.3. Amplicon Sequencing of 28 and 16S rRNA Genes
2.4. Sequencing Data Processing and Bioinformatics Analysis
2.5. Functional Predictions of the Presented Bacterial Communities
2.6. Phylogenetic Analysis for the LSU rRNA Gene Sequences
3. Results
3.1. General Descriptions of Pyrosequencing the Eukaryotic 28S rRNA and the Prokaryotic 16S rRNA Gene Amplicons
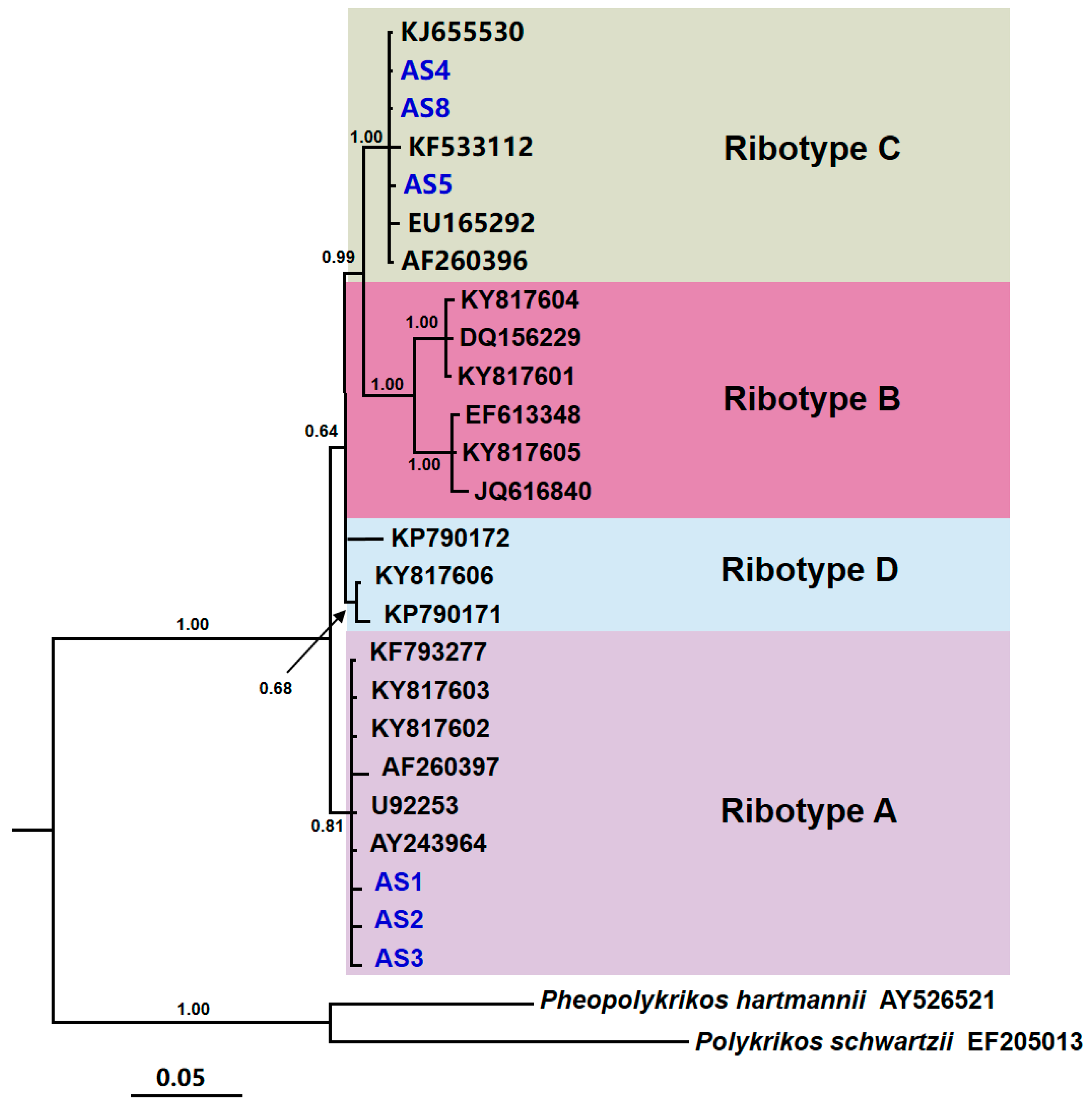
3.2. Phylogenetic Analysis
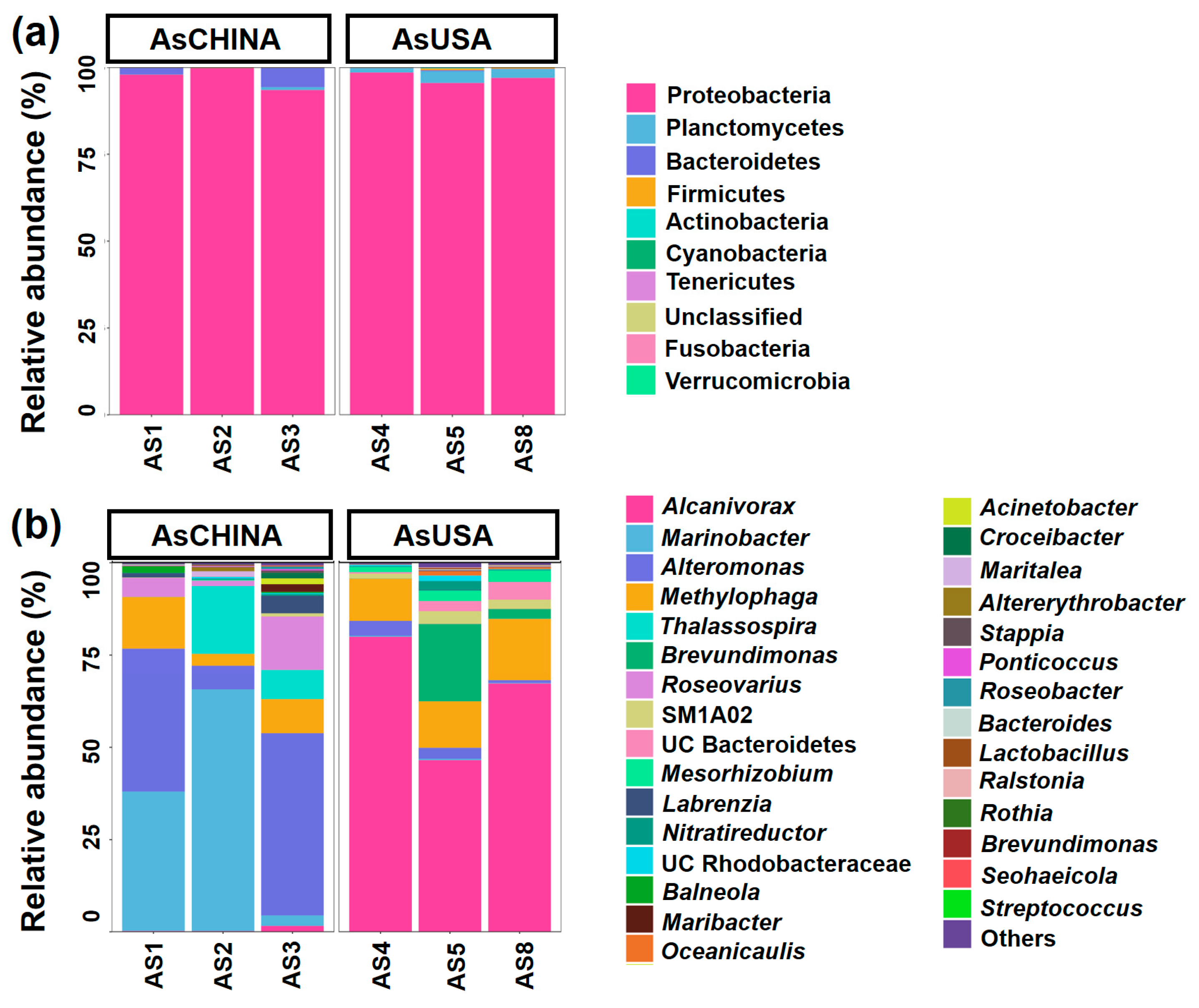
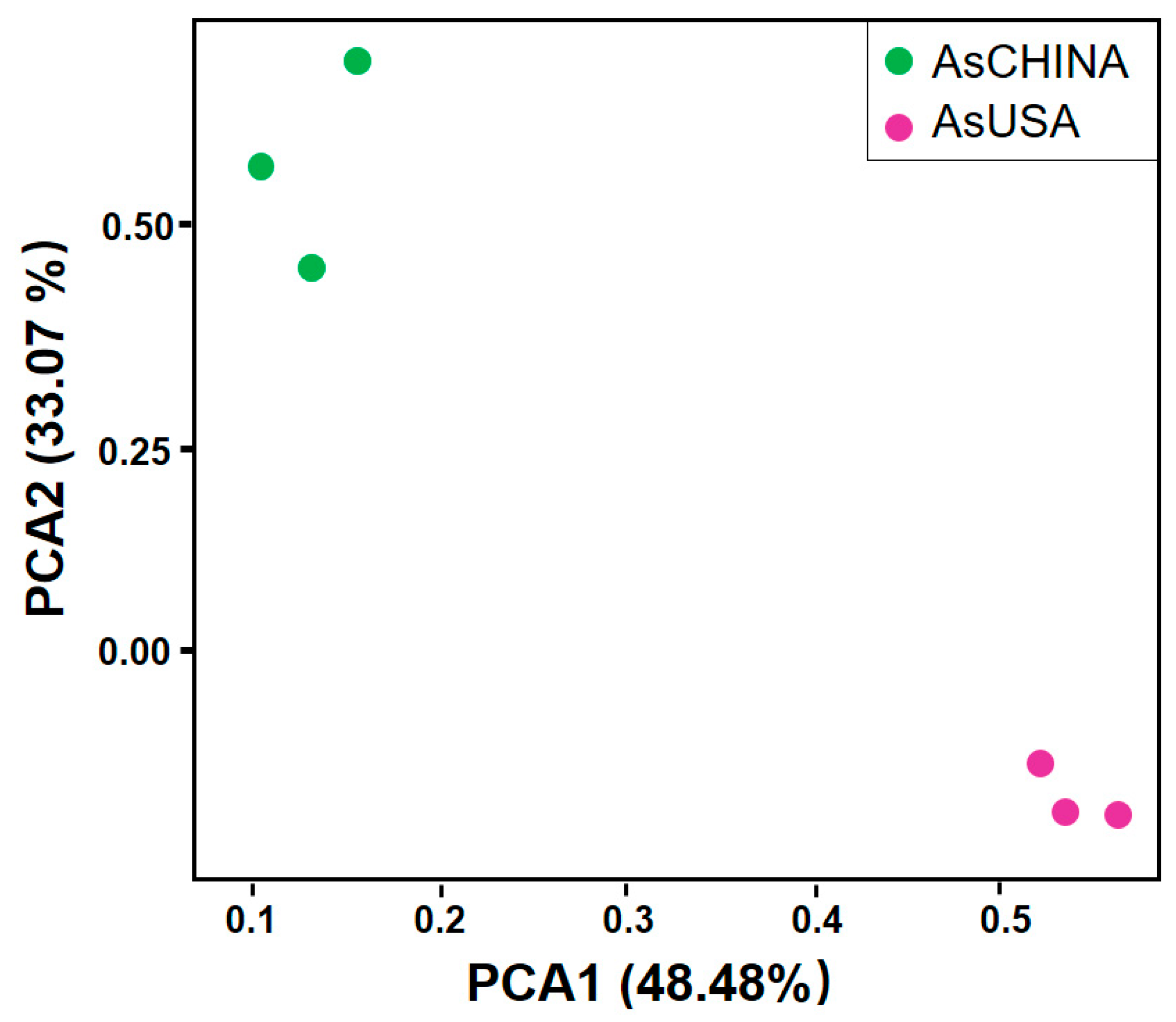
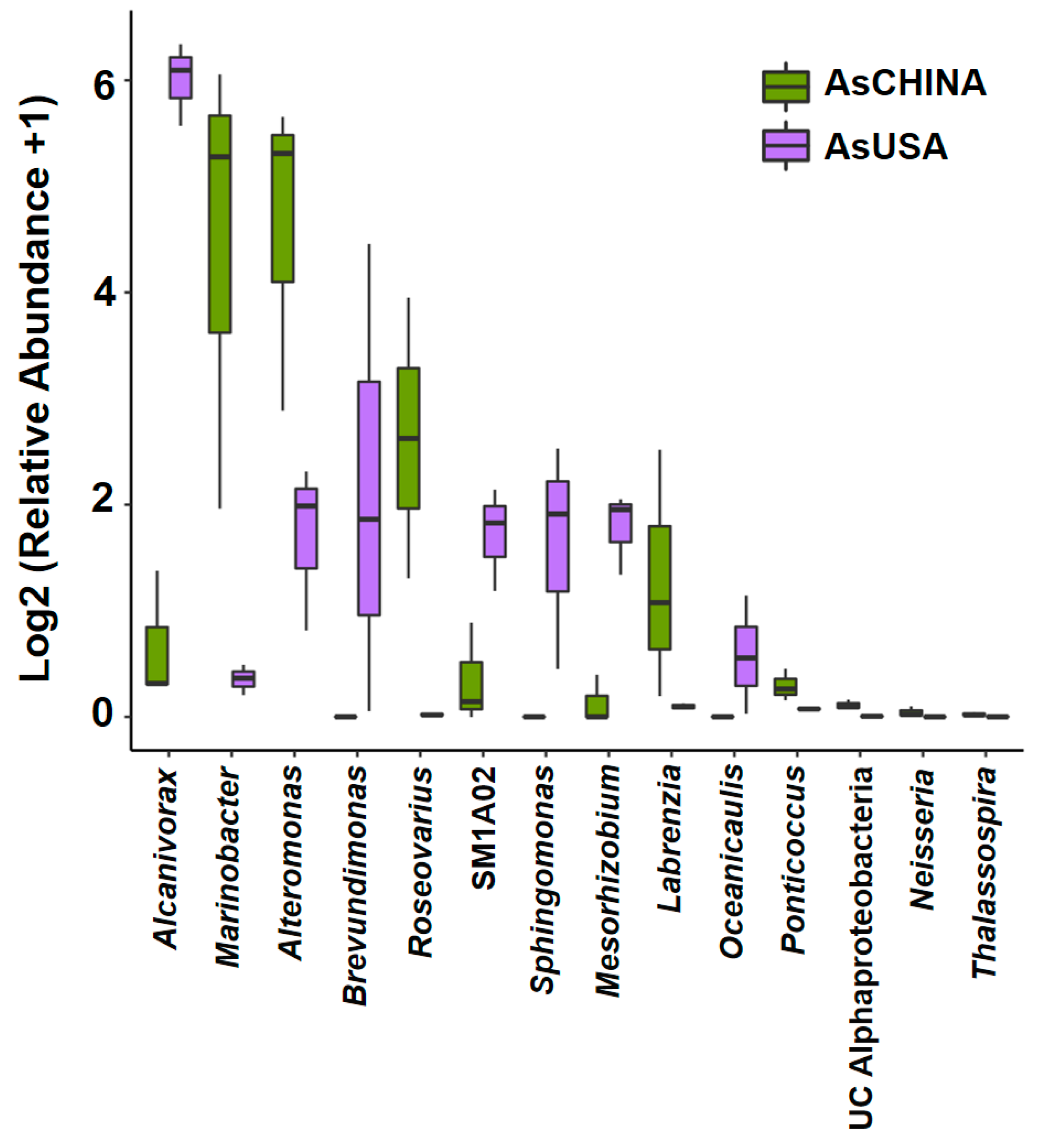
3.3. Comparative Analysis of ASV Diversity and Structural Composition of Bacterial Communities Between AsCHINA and AsUSA Groups
3.4. Comparative Analysis of Predicted Function of Bacterial Communities Between AsCHINA and AsUSA Groups
4. Discussion
4.1. Highly Diverse Bacterial Communities Associated with Laboratory-Cultured A. sanguinea Strains from Different Geographic Origins
4.2. Ribotype A and Ribotype C Harbor Distinct Bacterial Communities Characterized by Different Metabolic Capabilities in Nitrogen Acquisition
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| HAB | Harmful algal bloom |
| ASVs | Amplicon sequence variants |
| PCA | Principal component analysis |
| BI | Bayesian inference |
References
- Daugbjerg, N.; Hansen, G.; Larsen, J.; Moestrup, O. Phylogeny of some of the major genera of dinoflagellates based on ultrastructure and partial LSU rDNA sequence data, including the erection of three new genera of unarmoured dinoflagellates. Phycologia 2000, 39, 302–317. [Google Scholar] [CrossRef]
- Steidinger, K.A.; Tangen, K. Dinoflagellates. In Identifying Marine Diatoms and Dinoflagellates; Tomas, C.R., Ed.; Academic Press: New York, NY, USA, 1996; pp. 387–598. [Google Scholar]
- Tang, Y.Z.; Gobler, C.J. Sexual resting cyst production by the dinoflagellate Akashiwo sanguinea: A potential mechanism contributing to the ubiquitous distribution of a harmful alga. J. Phycol. 2015, 51, 298–309. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Kang, W.; Hui, L. Akashiwo sanguinea blooms in Chinese waters in 1998–2017. Mar. Pollut. Bull. 2019, 149, 110652. [Google Scholar] [CrossRef]
- Li, X.D.; Su, L.; Li, X.J.; Li, J.; Xu, Y.J.; Chang, L.R.; Yu, R.C.; Yang, D.Z.; Pang, S.J. Comprehensive analysis of large-scale Saccharina japonica damage in the principal farming area of Rongcheng in Shandong Province from 2021 to 2022. J. Agric. Sci. Technol. 2023, 25, 206–222. [Google Scholar]
- Geng, H.X.; Kong, F.Z.; Wang, J.X.; Zhang, Q.C.; Li, F.; Hong, X.; Song, M.J.; Lian, Z.R.; Cai, Y.L.; Yu, R.C. An unusual winter bloom of dinoflagellates with notable damage to kelp cultivation around Shandong peninsula, China. Mar. Environ. Res. 2024, 201, 106687. [Google Scholar] [CrossRef]
- Horner, R.A.; Garrison, D.L.; Plumley, F.G. Harmful algal blooms and red tide problems on the US west coast. Limnol. Oceanogr. 1997, 42, 1076–1088. [Google Scholar] [CrossRef]
- Matsubara, T.; Nagasoe, S.; Yamasaki, Y.; Shikata, T.; Shimasaki, Y.; Oshima, Y.; Honjo, T. Effects of temperature, salinity, and irradiance on the growth of the dinoflagellate Akashiwo sanguinea. J. Exp. Mar. Biol. Ecol. 2007, 342, 226–230. [Google Scholar] [CrossRef]
- Jessup, D.A.; Miller, M.A.; Ryan, J.P.; Nevins, H.M.; Kerkering, H.A.; Mekebri, A.; Crane, D.B.; Johnson, T.A.; Kudela, R.M. Mass stranding of marine birds caused by a surfactant-producing red tide. PLoS ONE 2009, 4, e4550. [Google Scholar] [CrossRef]
- Xu, N.; Wang, M.; Tang, Y.; Zhang, Q.; Duan, S.; Gobler, C.J. Acute toxicity of the cosmopolitan bloom-forming dinoflagellate Akashiwo sanguinea to finfish, shellfish, and zooplankton. Aquat. Microb. Ecol. 2017, 80, 209–222. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Wang, K.; Hu, Z.X.; Tang, Y.Z. Abundant species diversity and essential functions of bacterial communities associated with dinoflagellates as revealed from metabarcoding sequencing for laboratory-raised clonal cultures. Int. J. Environ. Res. Public Health 2022, 19, 4446. [Google Scholar] [CrossRef]
- Yang, X.; Liu, Z.; Zhang, Y.; Shi, X.; Wu, Z. Dinoflagellate–Bacteria Interactions: Physiology, Ecology, and Evolution. Biology 2024, 13, 579. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Chen, G.F.; Ying, K.Z.; Jin, H.; Song, J.T.; Cai, Z.H. Phycosphere microbial succession patterns and assembly mechanisms in a marine dinoflagellate bloom. Appl. Environ. Microbiol. 2019, 85, e00349-19. [Google Scholar] [CrossRef] [PubMed]
- Patin, N.V.; Brown, E.; Chebli, G.; Garfield, C.; Kubanek, J.; Stewart, F.J. Microbial and chemical dynamics of a toxic dinoflagellate bloom. Peer J. 2020, 8, e9493. [Google Scholar] [CrossRef] [PubMed]
- Hu, J.M.; Camerón, H.; Rilling, J.I.; Campos, M.; Ruiz-Gil, T.; Gonzalez, M.A.; Gajardo, G.; Vergara, K.; Guzmán, L.; Espinoza-González, O.; et al. Differentiation of microbial communities in coastal seawater before and during an Akashiwo sanguinea (Dinophyceae) bloom in the urban area of Antofagasta city (northern Chile). Harmful Algae 2025, 142, 102782. [Google Scholar] [CrossRef]
- Li, S.F.; Chen, M.C.; Chen, Y.F.; Tong, J.; Wang, L.Y.; Xu, Y.; Hu, Z.L.; Chen, H. Epibiotic bacterial community composition in red-tide dinoflagellate Akashiwo sanguinea culture under various growth conditions. FEMS Microbiol. Ecol. 2019, 95, fiz057. [Google Scholar] [CrossRef]
- Yang, C.Y.; Li, Y.; Zhou, Y.Y.; Zheng, W.; Tian, Y.; Zheng, T.L. Bacterial community dynamics during a bloom caused by Akashiwo sanguinea in the Xiamen sea area, China. Harmful Algae 2012, 20, 132–141. [Google Scholar] [CrossRef]
- Yang, C.Y.; Li, Y.; Zhou, B.; Zhou, Y.Y.; Zheng, W.; Tian, Y.; Van Nostrand, J.D.; Wu, L.Y.; He, Z.L.; Zhou, J.Z.; et al. Illumina sequencing-based analysis of free-living bacterial community dynamics during an Akashiwo sanguinea bloom in Xiamen sea, China. Sci. Rep. 2015, 5, 8476. [Google Scholar]
- Yang, C.Y.; Li, Y.; Zhou, Y.Y.; Lei, X.Q.; Zheng, W.; Tian, Y.; Van Nostrand, J.D.; He, Z.L.; Wu, L.Y.; Zhou, J.Z.; et al. A comprehensive insight into functional profiles of free-living microbial community responses to a toxic Akashiwo sanguinea bloom. Sci. Rep. 2016, 6, 34645. [Google Scholar] [CrossRef]
- Chen, H.R.; Jiang, J.J.; Jiang, F.J.; Li, S.F.; Hu, Z.L. Temporal variability of free-living microbial culturability and community composition after an Akashiwo sanguinea bloom in Shenzhen, China. Ecotoxicology 2021, 30, 975–985. [Google Scholar] [CrossRef]
- Kang, J.; Park, J.S.; Jung, S.W.; Kim, H.J.; Joo, H.M.; Kang, D.; Seo, H.; Kim, S.; Jang, M.C.; Lee, K.W.; et al. Zooming on dynamics of marine microbial communities in the phycosphere of Akashiwo sanguinea (Dinophyta) blooms. Mol. Ecol. 2021, 30, 207–221. [Google Scholar] [CrossRef]
- Cirri, E.; Pohnert, G. Algae-bacteria interactions that balance the planktonic microbiome. New Phytol. 2019, 223, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Bickford, D.; Lohman, D.J.; Sodhi, N.S.; Ng, P.K.L.; Meier, R.; Winker, K.; Ingram, K.K.; Das, I. Cryptic species as a window on diversity and conservation. Trends Ecol. Evol. 2007, 22, 148–155. [Google Scholar] [CrossRef] [PubMed]
- Montresor, M.; Sgrosso, S.; Procaccini, G.; Kooistra, W.H. Intraspecific diversity in Scrippsiella trochoidea (Dinopbyceae): Evidence for cryptic species. Phycologia 2003, 42, 56–70. [Google Scholar] [CrossRef]
- Kim, E.; Wilcox, L.; Graham, L.; Graham, J. Genetically distinct populations of the dinoflagellate Peridinium limbatum in neighboring Northern Wisconsin lakes. Microb. Ecol. 2004, 48, 521–527. [Google Scholar] [CrossRef]
- Lilly, E.L.; Halanych, K.M.; Anderson, D.M. Phylogeny, biogeography, and species boundaries within the Alexandrium minutum group. Harmful Algae 2005, 4, 1004–1020. [Google Scholar] [CrossRef]
- Lowe, C.D.; Montagnes, D.J.; Martin, L.E.; Watts, P.C. High genetic diversity and fine-scale spatial structure in the marine flagellate Oxyrrhis marina (Dinophyceae) uncovered by microsatellite loci. PLoS ONE 2010, 5, e15557. [Google Scholar] [CrossRef]
- Genovesi, B.; Shin-Grzebyk, M.S.; Grzebyk, D.; Laabir, M.; Gagnaire, P.A.; Vaquer, A.; Pastoureaud, A.; Lasserre, B.; Collos, Y.; Berrebi, P. Assessment of cryptic species diversity within blooms and cyst bank of the Alexandrium tamarense complex (Dinophyceae) in a Mediterranean lagoon facilitated by semimultiplex PCR. J. Plankton Res. 2011, 33, 405–414. [Google Scholar] [CrossRef]
- Nagahama, Y.; Murray, S.; Tomaru, A.; Fukuyo, Y. Species boundaries in the toxic dinoflagellate Prorocentrum lima (Dinophyceae, Prorocentrales), based on morphological and phylogenetic characters. J. Phycol. 2011, 47, 178–189. [Google Scholar] [CrossRef]
- Anglès, S.; Reñé, A.; Garcés, E.; Lugliè, A.; Sechi, N.; Camp, J.; Satta, C.T. Morphological and molecular characterization of Bysmatrum subsalsum (Dinophyceae) from the western Mediterranean Sea reveals the existence of cryptic species. J. Phycol. 2017, 53, 833–847. [Google Scholar] [CrossRef]
- Wang, N.; Mertens, K.N.; Krock, B.; Luo, Z.H.; Derrien, A.; Pospelova, V.; Liang, Y.B.; Bilien, G.; Smith, K.F.; De Schepper, S.; et al. Cryptic speciation in Protoceratium reticulatum (Dinophyceae): Evidence from morphological, molecular and ecophysiological data. Harmful Algae 2019, 88, 101610. [Google Scholar] [CrossRef]
- Rissler, L.J.; Apodaca, J.J. Adding more ecology into species delimitation: Ecological niche models and phylogeography help define cryptic species in the black salamander (Aneides flavipunctatus). Syst. Biol. 2007, 56, 924–942. [Google Scholar] [CrossRef] [PubMed]
- De Meester, N.; Derycke, S.; Bonte, D.; Moens, T. Salinity effects on the coexistence of cryptic species: A case study on marine nematodes. Mar. Biol. 2011, 158, 2717–2726. [Google Scholar] [CrossRef]
- Luo, Z.H.; Yang, W.D.; Leaw, C.P.; Pospelova, V.; Bilien, G.; Liow, G.R.; Lim, P.T.; Gu, H.F. Cryptic diversity within the harmful dinoflagellate Akashiwo sanguinea in coastal Chinese waters is related to differentiated ecological niches. Harmful Algae 2017, 66, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Reñé, A.; Camp, J.; Garcés, E. Diversity and phylogeny of Gymnodiniales (Dinophyceae) from the NW Mediterranean Sea revealed by a morphological and molecular approach. Protist 2015, 166, 234–263. [Google Scholar] [CrossRef]
- Guillard, R.R.L. Culture of Phytoplankton for Feeding Marine Invertebrates. In Culture of Marine Invertebrate Animals; Smith, W.L., Chanley, M.H., Eds.; Plenum Press: New York, NY, USA, 1975; pp. 29–60. [Google Scholar]
- Deng, Y.Y.; Hu, Z.X.; Zhan, Z.F.; Ma, Z.P.; Tang, Y.Z. Differential expressions of an Hsp70 gene in the dinoflagellate Akashiwo sanguinea in response to temperature stress and transition of life cycle and its implications. Harmful Algae 2015, 50, 57–64. [Google Scholar] [CrossRef]
- Chai, Z.Y.; He, Z.L.; Deng, Y.Y.; Yang, Y.F.; Tang, Y.Z. Cultivation of seaweed Gracilaria lemaneiformis enhanced biodiversity in a eukaryotic plankton community as revealed via metagenomic analyses. Mol. Ecol. 2018, 27, 1081–1093. [Google Scholar] [CrossRef]
- Logue, J.B.; Stedmon, C.A.; Kellerman, A.M.; Nielsen, N.J.; Andersson, A.F.; Laudon, H.; Lindström, E.S.; Kritzberg, E.S. Experimental insights into the importance of aquatic bacterial community composition to the degradation of dissolved organic matter. ISME J. 2016, 10, 533–545. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. Peer J. 2016, 4, e2584. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef]
- Douglas, G.M.; Maffei, V.J.; Zaneveld, J.R.; Yurgel, S.N.; Brown, J.R.; Taylor, C.M.; Huttenhower, C.; Langille, M.G.I. PICRUSt2 for prediction of metagenome functions. Nat. Biotechnol. 2020, 38, 685–688. [Google Scholar] [CrossRef] [PubMed]
- Langille, M.G.; Zaneveld, J.; Caporaso, J.G.; McDonald, D.; Knights, D.; Reyes, J.A.; Clemente, J.C.; Burkepile, D.E.; Thurber, R.L.V.; Knight, R.; et al. Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat. Biotechnol. 2013, 31, 814–821. [Google Scholar] [CrossRef] [PubMed]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.I.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Hall, T.A. BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp. Ser. 1999, 41, 95–98. [Google Scholar]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef]
- Behringer, G.; Ochsenkühn, M.A.; Fei, C.; Fanning, J.; Koester, J.A.; Amin, S.A. Bacterial communities of diatoms display strong conservation across strains and time. Front. Microbiol. 2018, 9, 659. [Google Scholar] [CrossRef]
- Lawson, C.A.; Raina, J.B.; Kahlke, T.; Seymour, J.R.; Suggett, D.J. Defining the core microbiome of the symbiotic dinoflagellate, Symbiodinium. Environ. Microbiol. Rep. 2018, 10, 7–11. [Google Scholar] [CrossRef]
- Maire, J.; Girvan, S.K.; Barkla, S.E.; Perez-Gonzalez, A.; Suggett, D.J.; Blackall, L.L.; van Oppen, M.J.H. Intracellular bacteria are common and taxonomically diverse in cultured and in hospite algal endosymbionts of coral reefs. ISME J. 2021, 15, 2028–2042. [Google Scholar] [CrossRef]
- Deng, Y.Y.; Wang, K.; Hu, Z.X.; Hu, Q.; Tang, Y.Z. Identification and implications of a core bacterial microbiome in 19 clonal cultures laboratory-reared for months to years of the cosmopolitan dinoflagellate Karlodinium veneficum. Front. Microbiol. 2022, 13, 967610. [Google Scholar] [CrossRef]
- Mendes, R.; Garbeva, P.; Raaijmakers, J.M. The rhizosphere microbiome: Significance of plant beneficial, plant pathogenic, and human pathogenic microorganisms. Fems Microbiol. Rev. 2013, 37, 634–663. [Google Scholar] [CrossRef] [PubMed]
- Krohn-Molt, I.; Alawi, M.; Förstner, K.U.; Wiegandt, A.; Burkhardt, L.; Indenbirken, D.; Thiess, M.; Grundhoff, A.; Kehr, J.; Tholey, A.; et al. Insights into microalga and bacteria interactions of selected phycosphere biofilms using metagenomic, transcriptomic, and proteomic approaches. Front. Microbiol. 2017, 8, 1941. [Google Scholar] [CrossRef] [PubMed]
- Buchan, A.; LeCleir, G.R.; Gulvik, C.A.; González, J.M. Master recyclers: Features and functions of bacteria associated with phytoplankton blooms. Nat. Rev. Microbiol. 2014, 12, 686–698. [Google Scholar] [CrossRef] [PubMed]
- Seymour, J.R.; Amin, S.A.; Raina, J.B.; Stocker, R. Zooming in on the phycosphere: The ecological interface for phytoplankton-bacteria relationships. Nat. Microbiol. 2017, 2, 17065. [Google Scholar] [CrossRef]
- Kumar, V.; Gera, R. Isolation of a multi-trait plant growth promoting Brevundimonas sp. and its effect on the growth of Bt-cotton. 3 Biotech 2014, 4, 97–101. [Google Scholar] [CrossRef]
- Cavicchioli, R.; Fegatella, F.; Ostrowski, M.; Eguchi, M.; Gottschal, J. Sphingomonads from marine environments. J. Ind. Microbiol. Biotechnol. 1999, 23, 268–272. [Google Scholar] [CrossRef]
- Xie, C.H.; Yokota, A. Sphingomonas azotifigens sp. nov., a nitrogen-fixing bacterium isolated from the roots of Oryza sativa. Int. J. Syst. Evol. Microbiol. 2006, 56, 889–893. [Google Scholar] [CrossRef]
- Laranjo, M.; Alexandre, A.; Oliveira, S. Legume growth-promoting rhizobia: An overview on the Mesorhizobium genus. Microbiol. Res. 2014, 169, 2–17. [Google Scholar] [CrossRef]
- Summers, M.M.; Katz, S.; Allen, E.E.; Rouse, G.W. Association of rhizobia with a marine polychaete. Environ. Microbiol. Rep. 2013, 5, 492–498. [Google Scholar] [CrossRef]
- Eady, R.R.; Postgate, J.R. Nitrogenase. Nature 1974, 249, 805–810. [Google Scholar] [CrossRef]
- Kim, J.; Rees, D.C. Nitrogenase and biological nitrogen fixation. Biochemistry 1994, 33, 389–397. [Google Scholar] [CrossRef] [PubMed]
- Zehr, J.P.; Capone, D.G. Changing perspectives in marine nitrogen fixation. Science 2020, 368, eaay9514. [Google Scholar] [CrossRef] [PubMed]
- Fiore, C.L.; Jarett, J.K.; Olson, N.D.; Lesser, M.P. Nitrogen fixation and nitrogen transformations in marine symbioses. Trends Microbiol. 2010, 18, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Sohm, J.A.; Webb, E.A.; Capone, D.G. Emerging patterns of marine nitrogen fixation. Nat. Rev. Microbiol. 2011, 9, 499–508. [Google Scholar] [CrossRef]
- Lamas, A.; Leon-Miranda, E.; Tejada-Jimenez, M. Microalgal and nitrogen-fixing bacterial consortia: From interaction to biotechnological potential. Plants-Basel. 2023, 12, 2476. [Google Scholar] [CrossRef]
- Jeong, H.J.; Park, J.Y.; Nho, J.H.; Park, M.O.; Ha, J.H.; Seong, K.A.; Jeng, C.; Seong, C.N.; Lee, K.Y.; Yih, W.H. Feeding by red-tide dinoflagellates on the cyanobacterium Synechococcus. Aquat. Microb. Ecol. 2005, 41, 131–143. [Google Scholar] [CrossRef]
- Hansen, P.J. The role of photosynthesis and food uptake for the growth of marine mixotrophic dinoflagellates. J. Eukaryot. Microbiol. 2011, 58, 203–214. [Google Scholar] [CrossRef]
- Nakayama, T.; Nomura, M.; Takano, Y.; Tanifuji, G.; Shiba, K.; Inaba, K.; Inagaki, Y.; Kawata, M. Single-cell genomics unveiled a cryptic cyanobacterial lineage with a worldwide distribution hidden by a dinoflagellate host. Proc. Natl. Acad. Sci. USA 2019, 116, 15973–15978. [Google Scholar] [CrossRef]
- Wu, Z.; Yang, X.; Lin, S.; Lee, W.H.; Lam, P.K. A Rhizobium bacterium and its population dynamics under different culture conditions of its associated toxic dinoflagellate Gambierdiscus balechii. Mar. Life Sci. Tech. 2021, 3, 542–551. [Google Scholar] [CrossRef]
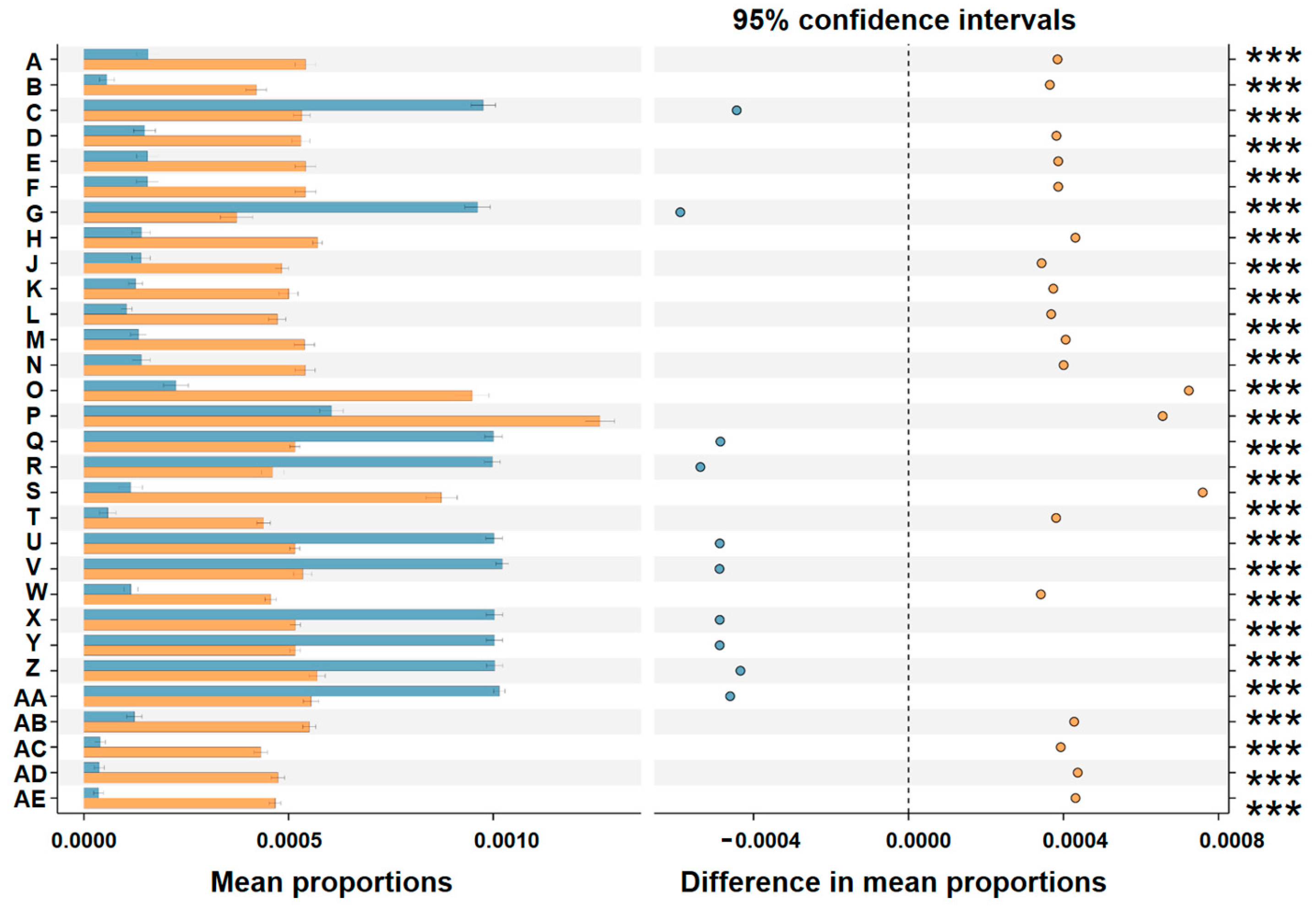

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Grouping | Sample ID | Strain Number | Origin/Isolation Date |
|---|---|---|---|
| AsCHINA | AS1 | ASND1 | Ningde, Fujian province, China, 2016 |
| AsCHINA | AS2 | ASND2 | Ningde, Fujian Province, China, 2016 |
| AsCHINA | AS3 | CCMA256 1 | Xiamen, Fujian province, China, 2011 |
| AsUSA | AS4 | ASNP6 | Northport Bay, New York, USA, 2011 |
| AsUSA | AS5 | ASNP2 | Northport Bay, New York, USA, 2011 |
| AsUSA | AS8 | ASNP2G | Northport Bay, New York, USA, 2011 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zou, H.; Li, F.; Lu, J.; Hu, Z.; Shang, L.; Tang, Y.Z.; Deng, Y. Different Ribotypes of Akashiwo sanguinea Harbor Distinct Bacterial Communities in Their Phycospheres. Diversity 2025, 17, 400. https://doi.org/10.3390/d17060400
Zou H, Li F, Lu J, Hu Z, Shang L, Tang YZ, Deng Y. Different Ribotypes of Akashiwo sanguinea Harbor Distinct Bacterial Communities in Their Phycospheres. Diversity. 2025; 17(6):400. https://doi.org/10.3390/d17060400
Chicago/Turabian StyleZou, Hanying, Fengting Li, Jiaqi Lu, Zhangxi Hu, Lixia Shang, Ying Zhong Tang, and Yunyan Deng. 2025. "Different Ribotypes of Akashiwo sanguinea Harbor Distinct Bacterial Communities in Their Phycospheres" Diversity 17, no. 6: 400. https://doi.org/10.3390/d17060400
APA StyleZou, H., Li, F., Lu, J., Hu, Z., Shang, L., Tang, Y. Z., & Deng, Y. (2025). Different Ribotypes of Akashiwo sanguinea Harbor Distinct Bacterial Communities in Their Phycospheres. Diversity, 17(6), 400. https://doi.org/10.3390/d17060400