
Comparing the Utility of Capture–Mark–Recapture, Telemetry, and Genetic Data in Assessing Population-Level Dispersal

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Literature Search
2.2. Eligibility Criteria
2.3. Data Extraction
2.4. Statistical Analysis
3. Results
3.1. Taxonomic Coverage of Dispersal Research
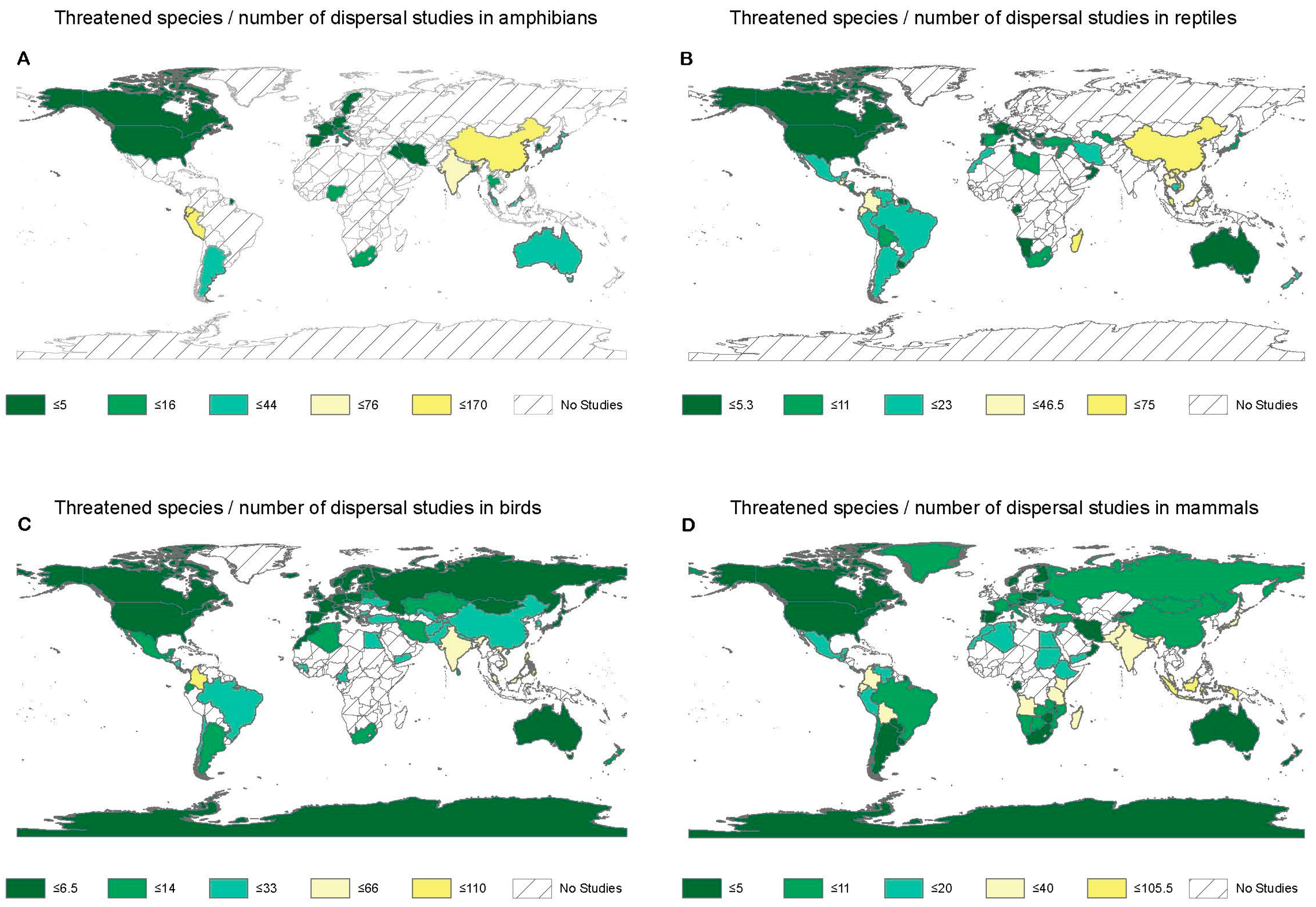
3.2. Geographic Coverage of Dispersal Research in Amphibians
3.3. Geographic Coverage of Dispersal Research in Reptiles
3.4. Geographic Coverage of Dispersal Research in Birds
3.5. Geographic Coverage of Dispersal Research in Mammals
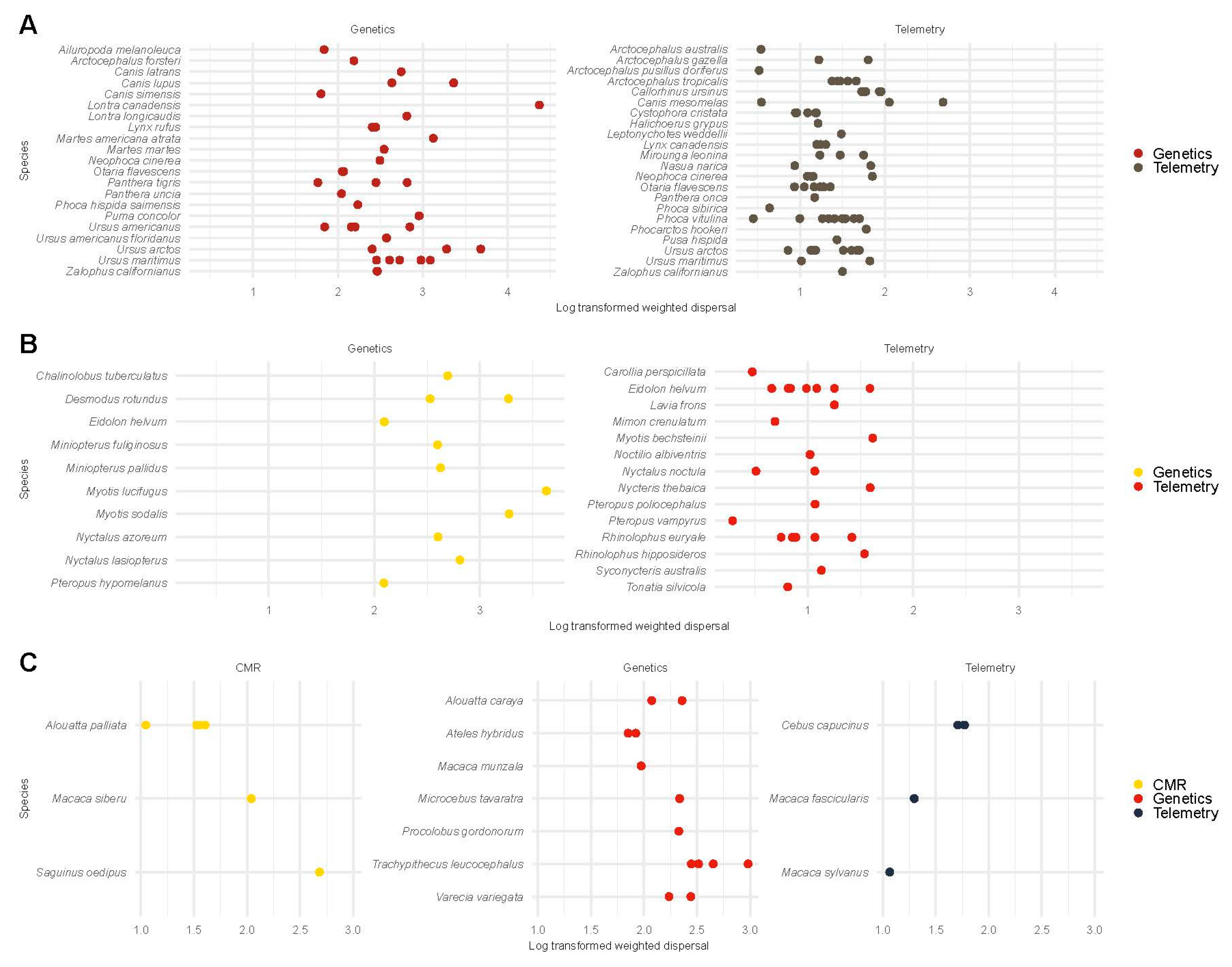
3.6. Weighted Dispersal by Study Type
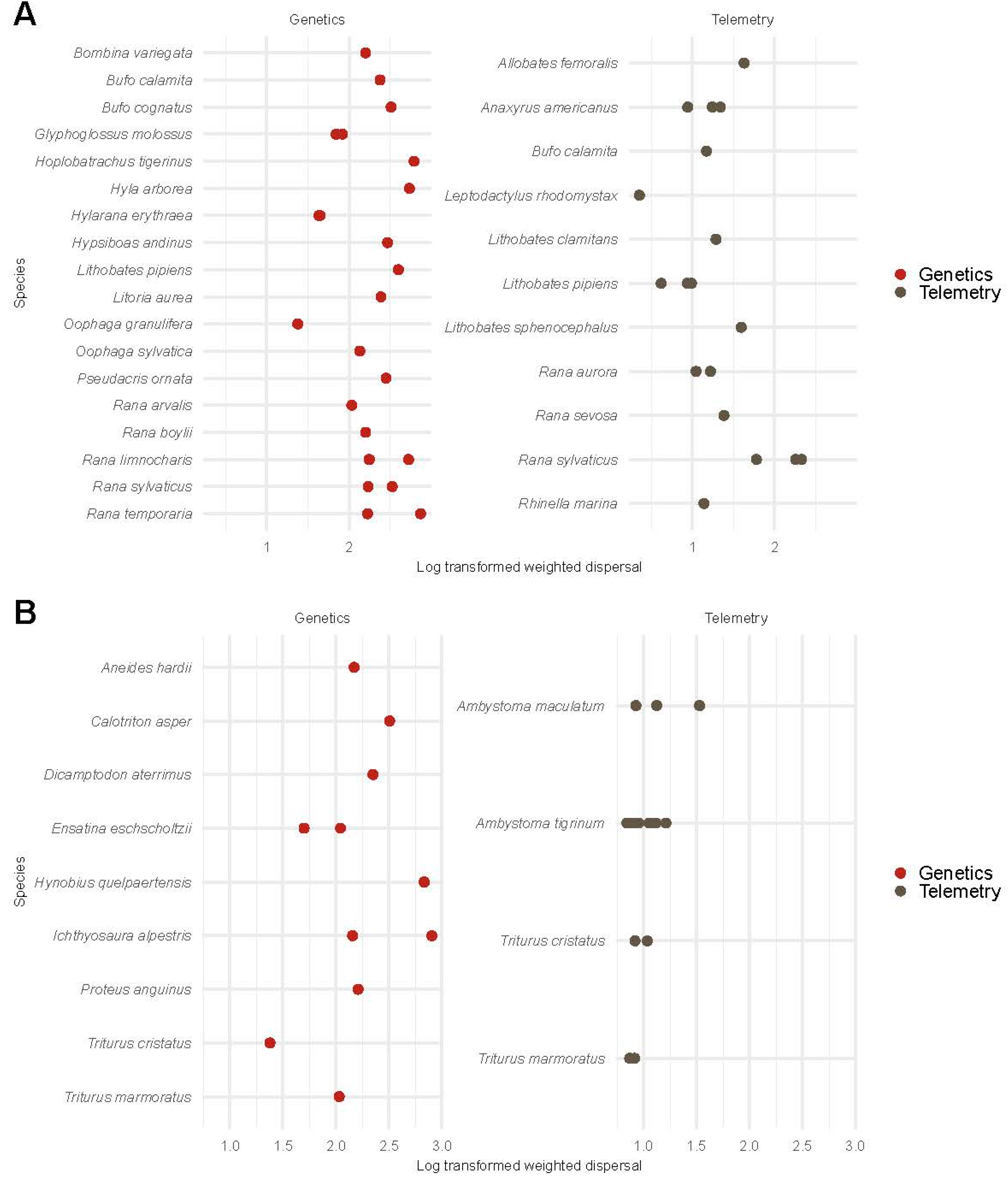
3.6.1. Weighted Dispersal in Amphibians
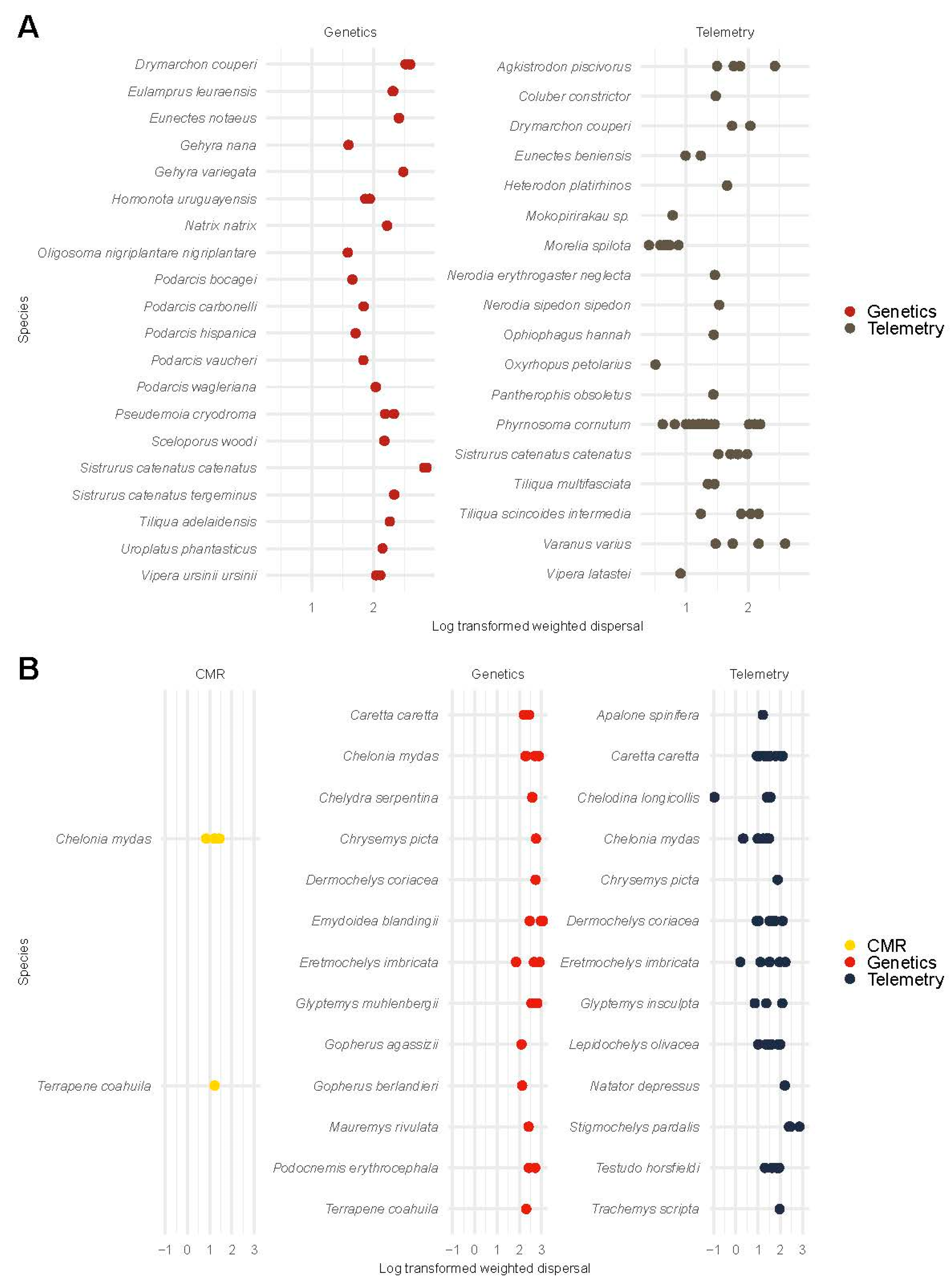
3.6.2. Weighted Dispersal in Reptiles
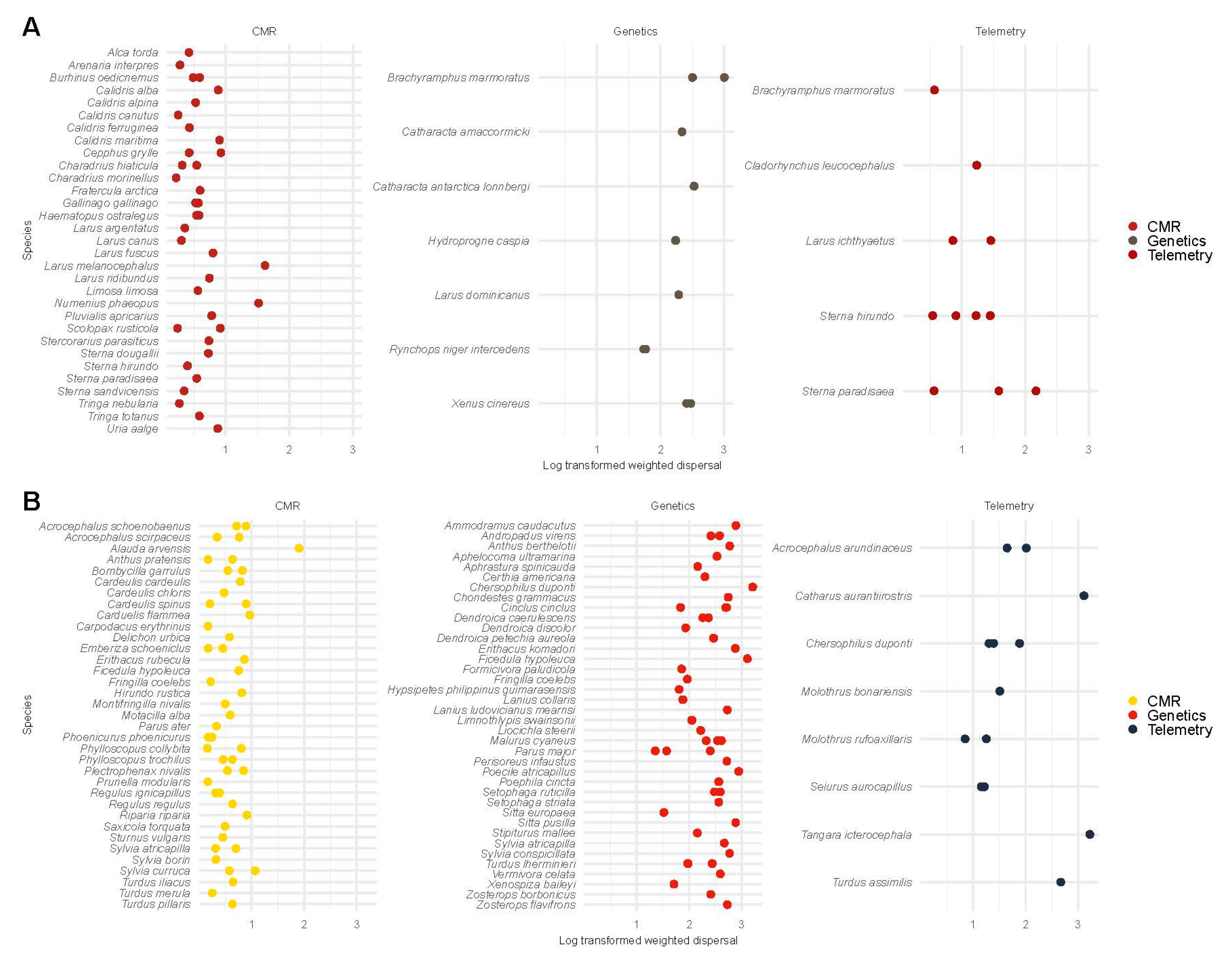
3.6.3. Weighted Dispersal in Birds
3.6.4. Weighted Dispersal in Mammals
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Díaz, S.; Settele, J.; Brondízio, E.S.; Ngo, H.T.; Agard, J.; Arneth, A.; Balvanera, P.; Brauman, K.A.; Butchart, S.H.M.; Chan, K.M.A.; et al. Pervasive human-driven decline of life on Earth points to the need for transformative change. Science 2019, 366, eaax3100. [Google Scholar] [CrossRef] [PubMed]
- Milner-Gulland, E.; Addison, P.; Arlidge, W.N.; Baker, J.; Booth, H.; Brooks, T.; Bull, J.W.; Burgass, M.J.; Ekstrom, J.; zu Ermgassen, S.O. Four steps for the Earth: Mainstreaming the post-2020 global biodiversity framework. One Earth 2021, 4, 75–87. [Google Scholar] [CrossRef]
- Jetz, W.; McGowan, J.; Rinnan, D.S.; Possingham, H.P.; Visconti, P.; O’Donnell, B.; Londoño-Murcia, M.C. Include biodiversity representation indicators in area-based conservation targets. Nat. Ecol. Evol. 2022, 6, 123–126. [Google Scholar] [CrossRef] [PubMed]
- Saura, S.; Bastin, L.; Battistella, L.; Mandrici, A.; Dubois, G. Protected areas in the world’s ecoregions: How well connected are they? Ecol. Indic. 2017, 76, 144–158. [Google Scholar] [CrossRef] [PubMed]
- Santini, L.; Saura, S.; Rondinini, C. Connectivity of the global network of protected areas. Divers. Distrib. 2016, 22, 199–211. [Google Scholar] [CrossRef]
- Minor, E.S.; Lookingbill, T.R. A multiscale network analysis of protected-area connectivity for mammals in the United States. Conserv. Biol. 2010, 24, 1549–1558. [Google Scholar] [CrossRef]
- Cayuela, H.; Rougemont, Q.; Prunier, J.G.; Moore, J.S.; Clobert, J.; Besnard, A.; Bernatchez, L. Demographic and genetic approaches to study dispersal in wild animal populations: A methodological review. Mol. Ecol. 2018, 27, 3976–4010. [Google Scholar] [CrossRef]
- Driscoll, D.A.; Banks, S.C.; Barton, P.S.; Ikin, K.; Lentini, P.; Lindenmayer, D.B.; Smith, A.L.; Berry, L.E.; Burns, E.L.; Edworthy, A. The trajectory of dispersal research in conservation biology. Systematic review. PLoS ONE 2014, 9, e95053. [Google Scholar] [CrossRef]
- Beese, W.J.; Deal, J.; Dunsworth, B.G.; Mitchell, S.J.; Philpott, T.J. Two decades of variable retention in British Columbia: A review of its implementation and effectiveness for biodiversity conservation. Ecol. Process. 2019, 8, 1–22. [Google Scholar] [CrossRef]
- Koenig, W.D.; Van Vuren, D.; Hooge, P.N. Detectability, philopatry, and the distribution of dispersal distances in vertebrates. Trends Ecol. Evol. 1996, 11, 514–517. [Google Scholar] [CrossRef]
- Caci, G.; Biscaccianti, A.B.; Cistrone, L.; Bosso, L.; Garonna, A.P.; Russo, D. Spotting the right spot: Computer-aided individual identification of the threatened cerambycid beetle Rosalia alpina. J. Insect Conserv. 2013, 17, 787–795. [Google Scholar] [CrossRef]
- Muñoz, D.J.; Miller, D.A.; Sutherland, C.; Grant, E.H.C. Using spatial capture–recapture to elucidate population processes and space-use in herpetological studies. J. Herpetol. 2016, 50, 570–581. [Google Scholar] [CrossRef]
- Broquet, T.; Petit, E.J. Molecular estimation of dispersal for ecology and population genetics. Annu. Rev.Ecol.Evol. Syst. 2009, 40, 193–216. [Google Scholar] [CrossRef]
- Slatkin, M. Gene flow and the geographic structure of natural populations. Science 1987, 236, 787–792. [Google Scholar] [CrossRef]
- Griesser, M.; Halvarsson, P.; Sahlman, T.; Ekman, J. What are the strengths and limitations of direct and indirect assessment of dispersal? Insights from a long-term field study in a group-living bird species. Behav. Ecol. Sociobiol. 2014, 68, 485–497. [Google Scholar] [CrossRef]
- Alex Smith, M.; Green, D.M. Dispersal and the metapopulation paradigm in amphibian ecology and conservation: Are all amphibian populations metapopulations? Ecography 2005, 28, 110–128. [Google Scholar] [CrossRef]
- Brannelly, L.A.; Chatfield, M.W.; Richards-Zawacki, C. Visual implant elastomer (VIE) tags are an unreliable method of identification in adult anurans. Herpetol. J. 2013, 23, 125–129. [Google Scholar]
- Alford, R.; Rowley, J. Techniques for tracking amphibians: The effects of tag attachment, and harmonic direction finding versus radio telemetry. Amphib.-Reptil. 2007, 28, 367–376. [Google Scholar]
- Moore, J.S.; Harris, L.N.; Le Luyer, J.; Sutherland, B.J.; Rougemont, Q.; Tallman, R.F.; Fisk, A.T.; Bernatchez, L. Genomics and telemetry suggest a role for migration harshness in determining overwintering habitat choice, but not gene flow, in anadromous Arctic Char. Mol. Ecol. 2017, 26, 6784–6800. [Google Scholar] [CrossRef]
- Hebblewhite, M.; Haydon, D.T. Distinguishing technology from biology: A critical review of the use of GPS telemetry data in ecology. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 2303–2312. [Google Scholar] [CrossRef] [PubMed]
- Andrews, K.R.; Good, J.M.; Miller, M.R.; Luikart, G.; Hohenlohe, P.A. Harnessing the power of RADseq for ecological and evolutionary genomics. Nat. Rev. Genet. 2016, 17, 81–92. [Google Scholar] [CrossRef]
- Berry, O.; Tocher, M.D.; Sarre, S.D. Can assignment tests measure dispersal? Mol. Ecol. 2004, 13, 551–561. [Google Scholar] [CrossRef]
- Taberlet, P.; Waits, L.P.; Luikart, G. Noninvasive genetic sampling: Look before you leap. Trends Ecol. Evol. 1999, 14, 323–327. [Google Scholar] [CrossRef]
- Titley, M.A.; Snaddon, J.L.; Turner, E.C. Scientific research on animal biodiversity is systematically biased towards vertebrates and temperate regions. PLoS ONE 2017, 12, e0189577. [Google Scholar] [CrossRef]
- Leidner, A.K.; Haddad, N.M. Combining measures of dispersal to identify conservation strategies in fragmented landscapes. Conserv. Biol. 2011, 25, 1022–1031. [Google Scholar] [CrossRef] [PubMed]
- IUCN. The IUCN Red List of Threatened Species. Version 2023-1. Available online: https://www.iucnredlist.org (accessed on 22 May 2024).
- Gusenbauer, M.; Haddaway, N.R. Which academic search systems are suitable for systematic reviews or meta-analyses? Evaluating retrieval qualities of Google Scholar, PubMed, and 26 other resources. Res. Synth. Methods 2020, 11, 181–217. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2023. [Google Scholar]
- Bohonak, A.J. IBD (isolation by distance): A program for analyses of isolation by distance. J. Hered. 2002, 93, 153–154. [Google Scholar] [CrossRef]
- Jensen, J.L.; Bohonak, A.J.; Kelley, S.T. Isolation by distance, web service. BMC Genet. 2005, 6, 13. [Google Scholar] [CrossRef] [PubMed]
- Rousset, F. Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 1997, 145, 1219–1228. [Google Scholar] [CrossRef]
- Beebee, T.J. Conservation genetics of amphibians. Heredity 2005, 95, 423–427. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, D.G.; Carey, M.; Czerniewska, J.; Fletcher, J.; Hether, T.; Jones, A.; Knight, S.; Knox, J.; Long, T.; Mannino, M. A meta-analysis of isolation by distance: Relic or reference standard for landscape genetics? Ecography 2010, 33, 315–320. [Google Scholar] [CrossRef]
- Puebla, O.; Bermingham, E.; Guichard, F. Estimating dispersal from genetic isolation by distance in a coral reef fish (Hypoplectrus puella). Ecology 2009, 90, 3087–3098. [Google Scholar] [CrossRef]
- Jones, K.E.; Bielby, J.; Cardillo, M.; Fritz, S.A.; O’Dell, J.; Orme, C.D.L.; Safi, K.; Sechrest, W.; Boakes, E.H.; Carbone, C. PanTHERIA: A species-level database of life history, ecology, and geography of extant and recently extinct mammals: Ecological Archives E090-184. Ecology 2009, 90, 2648. [Google Scholar] [CrossRef]
- Santini, L.; Isaac, N.J.; Ficetola, G.F. TetraDENSITY: A database of population density estimates in terrestrial vertebrates. Glob. Ecol. Biogeogr. 2018, 27, 787–791. [Google Scholar] [CrossRef]
- Bürkner, P.C. brms: An R package for Bayesian multilevel models using Stan. J. Stat. Softw. 2017, 80, 1–28. [Google Scholar] [CrossRef]
- Doherty, T.S.; Balouch, S.; Bell, K.; Burns, T.J.; Feldman, A.; Fist, C.; Garvey, T.F.; Jessop, T.S.; Meiri, S.; Driscoll, D.A. Reptile responses to anthropogenic habitat modification: A global meta-analysis. Glob. Ecol. Biogeogr. 2020, 29, 1265–1279. [Google Scholar] [CrossRef]
- Gelman, A.; Rubin, D.B. Inference from iterative simulation using multiple sequences. Stat. Sci. 1992, 7, 457–472. [Google Scholar] [CrossRef]
- Antonelli, A.; Ariza, M.; Albert, J.; Andermann, T.; Azevedo, J.; Bacon, C.; Faurby, S.; Guedes, T.; Hoorn, C.; Lohmann, L.G.; et al. Conceptual and empirical advances in Neotropical biodiversity research. PeerJ 2018, 6, e5644. [Google Scholar] [CrossRef]
- Marcus Rowcliffe, J.; Carbone, C.; Kays, R.; Kranstauber, B.; Jansen, P.A. Bias in estimating animal travel distance: The effect of sampling frequency. Methods Ecol. Evol. 2012, 3, 653–662. [Google Scholar] [CrossRef]
- Dawson, G.A. The use of time and space by the Panamanian tamarin, Saguinus oedipus. Folia Primatol. 1979, 31, 253–284. [Google Scholar] [CrossRef]
- McClintock, B.T.; White, G.C. A less field-intensive robust design for estimating demographic parameters with mark–resight data. Ecology 2009, 90, 313–320. [Google Scholar] [CrossRef] [PubMed]
- Palumbi, S.R. Population genetics, demographic connectivity, and the design of marine reserves. Ecol. Appl. 2003, 13, 146–158. [Google Scholar] [CrossRef]
- Manel, S.; Holderegger, R. Ten years of landscape genetics. Trends Ecol. Evol. 2013, 28, 614–621. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.J.; Bradburd, G.S. Isolation by environment. Mol. Ecol. 2014, 23, 5649–5662. [Google Scholar] [CrossRef] [PubMed]
- Comte, L.; Olden, J.D. Fish dispersal in flowing waters: A synthesis of movement- and genetic-based studies. Fish Fish. 2018, 19, 1063–1077. [Google Scholar] [CrossRef]
- Epps, C.W.; Wehausen, J.D.; Bleich, V.C.; Torres, S.G.; Brashares, J.S. Optimizing dispersal and corridor models using landscape genetics. J. Appl. Ecol. 2007, 44, 714–724. [Google Scholar] [CrossRef]
- Thatte, P.; Joshi, A.; Vaidyanathan, S.; Landguth, E.; Ramakrishnan, U. Maintaining tiger connectivity and minimizing extinction into the next century: Insights from landscape genetics and spatially-explicit simulations. Biol. Conserv. 2018, 218, 181–191. [Google Scholar] [CrossRef]
- Martín-López, B.; Montes, C.; Ramírez, L.; Benayas, J. What drives policy decision-making related to species conservation? Biol. Conserv. 2009, 142, 1370–1380. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sirsi, S.; Halstead, B.J.; Morrison, M.L.; Green, M.C.; Rodriguez, D.; Forstner, M.R.J. Comparing the Utility of Capture–Mark–Recapture, Telemetry, and Genetic Data in Assessing Population-Level Dispersal. Diversity 2025, 17, 163. https://doi.org/10.3390/d17030163
Sirsi S, Halstead BJ, Morrison ML, Green MC, Rodriguez D, Forstner MRJ. Comparing the Utility of Capture–Mark–Recapture, Telemetry, and Genetic Data in Assessing Population-Level Dispersal. Diversity. 2025; 17(3):163. https://doi.org/10.3390/d17030163
Chicago/Turabian StyleSirsi, Shashwat, Brian J. Halstead, Michael L. Morrison, M. Clay Green, David Rodriguez, and Michael R. J. Forstner. 2025. "Comparing the Utility of Capture–Mark–Recapture, Telemetry, and Genetic Data in Assessing Population-Level Dispersal" Diversity 17, no. 3: 163. https://doi.org/10.3390/d17030163
APA StyleSirsi, S., Halstead, B. J., Morrison, M. L., Green, M. C., Rodriguez, D., & Forstner, M. R. J. (2025). Comparing the Utility of Capture–Mark–Recapture, Telemetry, and Genetic Data in Assessing Population-Level Dispersal. Diversity, 17(3), 163. https://doi.org/10.3390/d17030163