
Comparative Metagenomic Analysis of Marine eDNA Investigating the Production Crisis of Aquacultured Saccharina japonica

 ,
,  , , ,
, , ,  and
and {kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
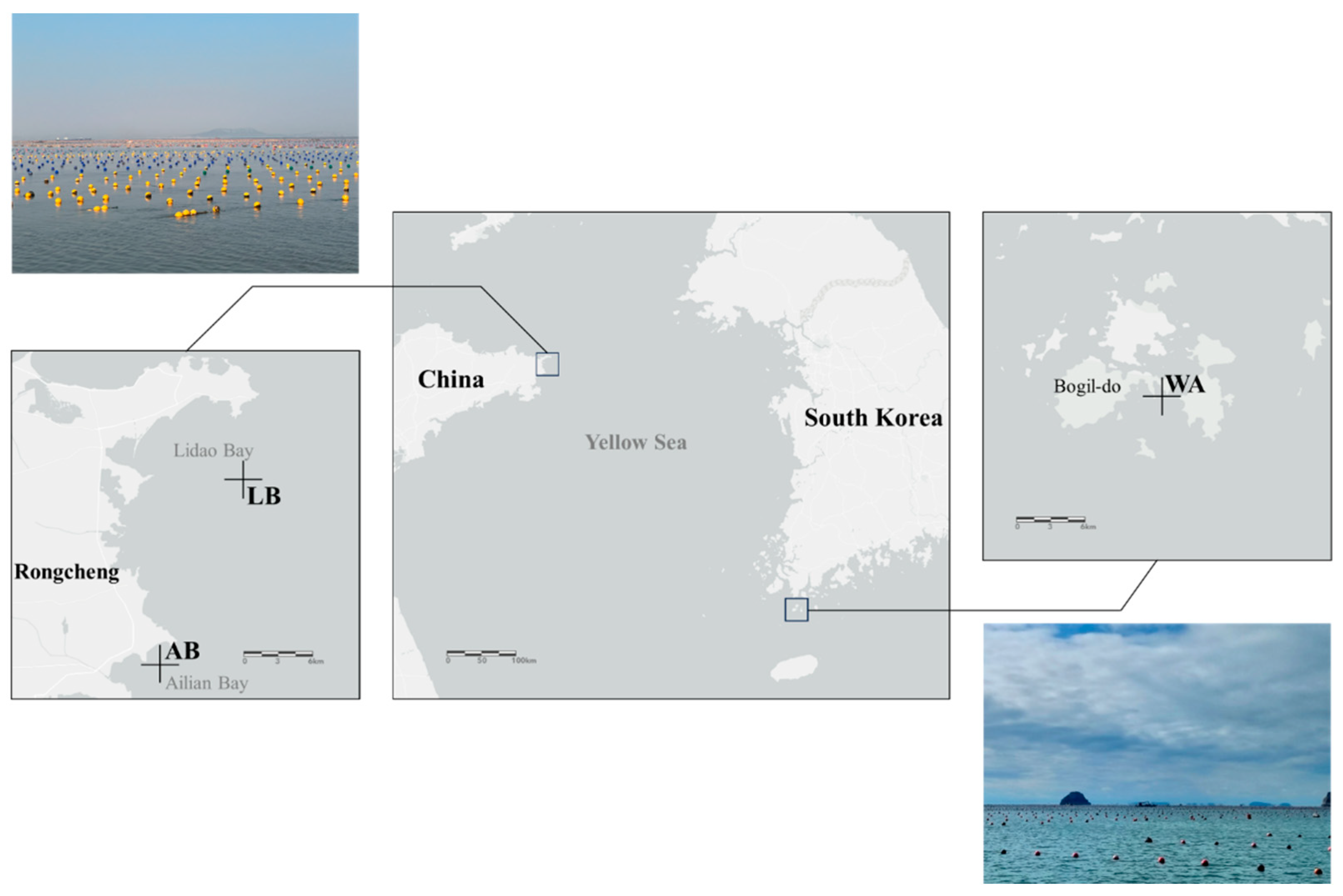
2.1. Sample Collection and Extraction of eDNA
2.2. Amplification of 16S rRNA and Sequencing
2.3. Bioinformatic Data Analysis
3. Results
3.1. Sequencing Results of 16S rRNA Obtained from eDNA
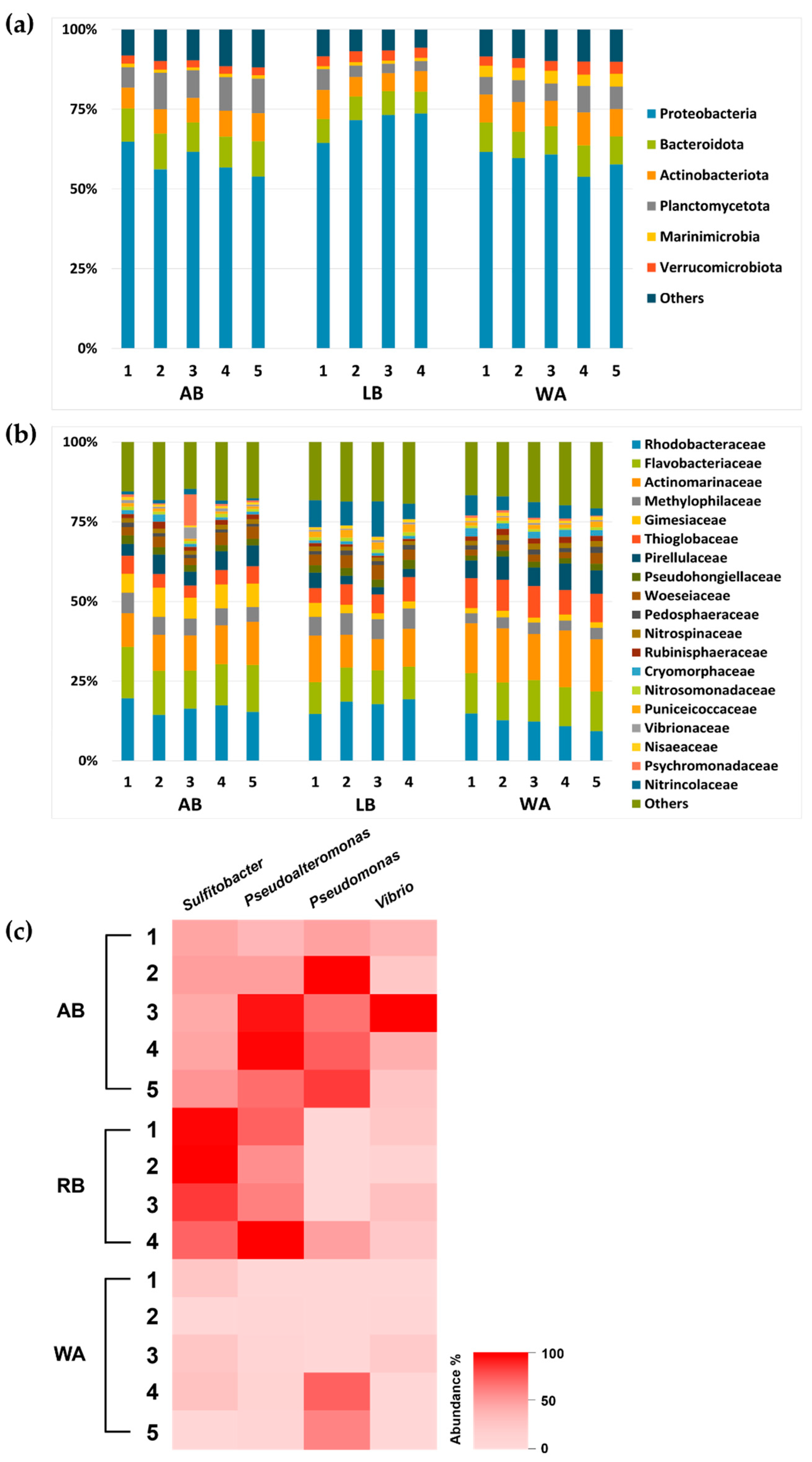
3.2. Taxonomic Microbial Communities of Seawater from Wando and Rongcheng
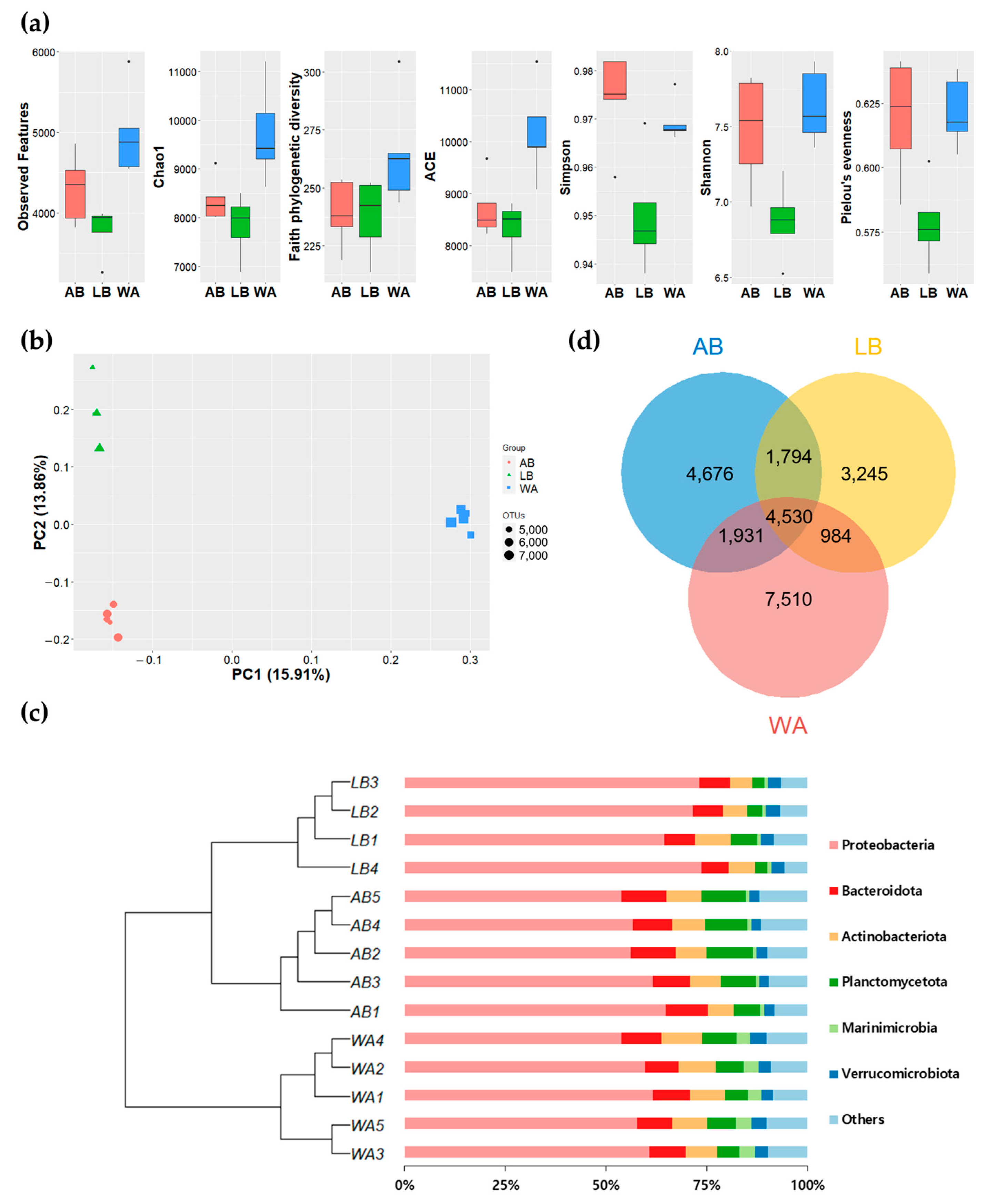
3.3. Comparison of Microbial Diversity between the Seawater Samples
3.4. Molecular Clustering Analysis of Seawater Microorganisms
4. Discussion
4.1. Taxonomical Differences of Seawater Microbial Communities between Wando and Rongcheng
4.2. Microbial Biodiversity and Its Implications
4.3. Pathogenic Bacteria Related to Kelp Diseases
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hwang, E.K.; Liu, F.; Lee, K.H.; Ha, D.S.; Park, C.S.; Hwang, E.K.; Liu, F.; Lee, K.H.; Ha, D.S.; Park, C.S. Comparison of the cultivation performance between Korean (Sugwawon No. 301) and Chinese strains (Huangguan No. 1) of kelp Saccharina japonica in an aquaculture farm in Korea. Algae 2018, 33, 101–108. [Google Scholar] [CrossRef]
- Zhang, J.; Wang, X.; Yao, J.; Li, Q.; Liu, F.; Yotsukura, N.; Krupnova, T.N.; Duan, D. Effect of domestication on the genetic diversity and structure of Saccharina japonica populations in China. Sci. Rep. 2017, 7, 42158. [Google Scholar] [CrossRef] [PubMed]
- Park, C.S.; Hwang, E.K. Seasonality of epiphytic development of the hydroid Obelia geniculata on cultivated Saccharina japonica (Laminariaceae, Phaeophyta) in Korea. J. Appl. Phycol. 2012, 24, 433–439. [Google Scholar] [CrossRef]
- Xu, S.; Yu, Z.; Zhou, Y.; Wang, F.; Yue, S.; Zhang, X. Insights into spatiotemporal distributions of trace elements in kelp (Saccharina japonica) and seawater of the western Yellow Sea, northern China. Sci. Total Environ. 2021, 774, 145544. [Google Scholar] [CrossRef]
- Kim, J.-O.; Kim, W.-S.; Jeong, H.-N.; Choi, S.-J.; Seo, J.-S.; Park, M.-A.; Oh, M.-J. A survey of epiphytic organisms in cultured kelp Saccharina japonica in Korea. Fish. Aquat. Sci. 2017, 20, 1. [Google Scholar] [CrossRef]
- Xu, S.; Yu, Z.; Zhou, Y.; Yue, S.; Liang, J.; Zhang, X. The potential for large-scale kelp aquaculture to counteract marine eutrophication by nutrient removal. Mar. Pollut. Bull. 2023, 187, 114513. [Google Scholar] [CrossRef]
- Wang, G.; Lu, B.; Shuai, L.; Li, D.; Zhang, R. Microbial diseases of nursery and field-cultivated Saccharina japonica (Phaeophyta) in China. Algol. Stud. 2014, 145–146, 39–51. [Google Scholar] [CrossRef]
- Hwang, E.K.; Ha, D.S.; Park, C.S. Strain selection and initiation timing influence the cultivation period of Saccharina japonica and their impact on the abalone feed industry in Korea. J. Appl. Phycol. 2017, 29, 2297–2305. [Google Scholar] [CrossRef]
- Li, X.D.; Zhong, C.H.; Jin, Z.H.; Lin, Q.; Shan, T.F.; Pang, S.J. Genetic structure, major chemical component contents and agronomic characters of the principal cultivars of the kelp Saccharina japonica in the largest kelp production province in China. J. Appl. Phycol. 2023, 35, 763–772. [Google Scholar] [CrossRef]
- Nie, J.; Fu, X.; Wang, L.; Xu, J.; Gao, X. A systematic review of fermented Saccharina japonica: Fermentation conditions, metabolites, potential health benefits and mechanisms. Trends Food Sci. Technol. 2022, 123, 15–27. [Google Scholar] [CrossRef]
- Lee, C.-W.; Kim, H.-A.; Yoon, H.-R.; Jeon, T.-Y. Establishment of seaweed fermentation process for cosmetic material research. J. Korea Acad.-Ind. Coop. Soc. 2019, 20, 14–19. [Google Scholar]
- Suraiya, S.; Park, H.D.; Jang, W.J.; Choi, Y.B.; Rafiquzzaman, S.M.; Sarker, M.K.; Kong, I.-S. Utilization of Saccharina japonica as a solid-fermented substrate for the production of bioactive ethanolic extract. Waste Biomass Valorization 2020, 11, 1917–1928. [Google Scholar] [CrossRef]
- Gachon, C.M.; Sime-Ngando, T.; Strittmatter, M.; Chambouvet, A.; Kim, G.H. Algal diseases: Spotlight on a black box. Trends Plant Sci. 2010, 15, 633–640. [Google Scholar] [CrossRef] [PubMed]
- Kim, G.H.; Moon, K.-H.; Kim, J.-Y.; Shim, J.; Klochkova, T.A. A revaluation of algal diseases in Korean Pyropia (Porphyra) sea farms and their economic impact. Algae 2014, 29, 249–265. [Google Scholar] [CrossRef]
- Tsiresy, G.; Preux, J.; Lavitra, T.; Dubois, P.; Lepoint, G.; Eeckhaut, I. Phenology of farmed seaweed Kappaphycus alvarezii infestation by the parasitic epiphyte Polysiphonia sp. in Madagascar. J. Appl. Phycol. 2016, 28, 2903–2914. [Google Scholar] [CrossRef]
- Zhang, R.; Chang, L.; Xiao, L.; Zhang, X.; Han, Q.; Li, N.; Egan, S.; Wang, G. Diversity of the epiphytic bacterial communities associated with commercially cultivated healthy and diseased Saccharina japonica during the harvest season. J. Appl. Phycol. 2020, 32, 2071–2080. [Google Scholar] [CrossRef]
- Yan, Y.W.; Yang, H.C.; Tang, L.; Li, J.; Mao, Y.X.; Mo, Z.L. Compositional shifts of bacterial communities associated with Pyropia yezoensis and surrounding seawater co-occurring with red rot disease. Front. Microbiol. 2019, 10, 1666. [Google Scholar] [CrossRef] [PubMed]
- Ward, G.M.; Faisan, J.P., Jr.; Cottier-Cook, E.J.; Gachon, C.; Hurtado, A.Q.; Lim, P.E.; Matoju, I.; Msuya, F.E.; Bass, D.; Brodie, J. A review of reported seaweed diseases and pests in aquaculture in Asia. J. World Aquac. Soc. 2020, 51, 815–828. [Google Scholar] [CrossRef]
- Ruppert, K.M.; Kline, R.J.; Rahman, M.S. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Glob. Ecol. Conserv. 2019, 17, e00547. [Google Scholar] [CrossRef]
- Thomsen, P.F.; Willerslev, E. Environmental DNA–An emerging tool in conservation for monitoring past and present biodiversity. Biol. Conserv. 2015, 183, 4–18. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, Y.; Wu, H.; Liu, F.; Peng, W.; Zhang, X.; Chang, F.; Xie, P.; Zhang, H. A review and perspective of eDNA application to eutrophication and HAB control in freshwater and marine ecosystems. Microorganisms 2020, 8, 417. [Google Scholar] [CrossRef] [PubMed]
- Won, N.I.; Kim, K.H.; Kang, J.H.; Park, S.R.; Lee, H.J. Exploring the impacts of anthropogenic disturbance on seawater and sediment microbial communities in Korean coastal waters using metagenomics analysis. Int. J. Environ. Res. Public Health 2017, 14, 130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Wikfors, G.H.; Clark, P.; Pitchford, S.; Krisak, M.; Dixon, M.S.; Li, Y. A deep dive into the epibiotic communities on aquacultured sugar kelp Saccharina latissima in Southern New England. Algal Res. 2022, 63, 102654. [Google Scholar] [CrossRef]
- Peng, Y.; Li, W. A bacterial pathogen infecting gametophytes of Saccharina japonica (Laminariales, Phaeophyceae). Chin. J. Oceanol. Limnol. 2013, 31, 366–373. [Google Scholar] [CrossRef]
- Bolyen, E.; Rideout, J.R.; Dillon, M.R.; Bokulich, N.A.; Abnet, C.C.; Al-Ghalith, G.A.; Alexander, H.; Alm, E.J.; Arumugam, M.; Asnicar, F.; et al. Reproducible, interactive, scalable and extensible microbiome data science using QIIME 2. Nat. Biotechnol. 2019, 37, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- Rognes, T.; Flouri, T.; Nichols, B.; Quince, C.; Mahé, F. VSEARCH: A versatile open source tool for metagenomics. PeerJ 2016, 4, e2584. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef]
- Michelou, V.K.; Caporaso, J.G.; Knight, R.; Palumbi, S.R. The Ecology of Microbial Communities Associated with Macrocystis pyrifera. PLoS ONE 2013, 8, e67480. [Google Scholar] [CrossRef]
- Li, J.; Li, Q.; Su, L.; Pang, S. Effects of light intensity on the epiphytic bacterial community of sporelings of Saccharina japonica. J. Appl. Phycol. 2021, 33, 1759–1764. [Google Scholar] [CrossRef]
- Singh, R.P.; Reddy, C.R. Seaweed-microbial interactions: Key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 2014, 88, 213–230. [Google Scholar] [CrossRef] [PubMed]
- Rhein-Knudsen, N.; Reyes-Weiss, D.; Horn, S.J. Extraction of high purity fucoidans from brown seaweeds using cellulases and alginate lyases. Int. J. Biol. Macromol. 2023, 229, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Wegner, C.E.; Richter-Heitmann, T.; Klindworth, A.; Klockow, C.; Richter, M.; Achstetter, T.; Glöckner, F.O.; Harder, J. Expression of sulfatases in Rhodopirellula baltica and the diversity of sulfatases in the genus Rhodopirellula. Mar. Genom. 2013, 9, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Zhao, D.; Xu, J.; Xu, X. Bioactivity of fucoidan extracted from Laminaria japonica using a novel procedure with high yield. Food Chem. 2018, 245, 911–918. [Google Scholar] [CrossRef]
- Burgunter-Delamare, B.; Rousvoal, S.; Legeay, E.; Tanguy, G.; Fredriksen, S.; Boyen, C.; Dittami, S.M. The Saccharina latissima microbiome: Effects of region, season, and physiology. Front. Microbiol. 2022, 13, 1050939. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.M.; Wang, Y. Genomic differences within the phylum Marinimicrobia: From waters to sediments in the Mariana Trench. Mar. Genom. 2020, 50, 100699. [Google Scholar] [CrossRef] [PubMed]
- Gross, P.; Smith, R.P. Biologic activity of hydroxylamine: A review. Crit. Rev. Toxicol. 1985, 14, 87–99. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Jimenez, K.; Araya-Lobo, A.; Quesada-Perez, F.; Akerman-Sanchez, J.; Delgado-Duran, B.; Ganzert, L.; Zavialov, P.O.; Alymkulov, S.; Kirillin, G.; Grossart, H.P. Variation of bacterial communities along the vertical gradient in Lake Issyk Kul, Kyrgyzstan. Environ. Microbiol. Rep. 2021, 13, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Tamayo-Leiva, J.; Cifuentes-Anticevic, J.; Aparicio-Rizzo, P.; Arroyo, J.I.; Masotti, I.; Díez, B. Influence of estuarine water on the microbial community structure of Patagonian fjords. Front. Mar. Sci. 2021, 8, 611981. [Google Scholar] [CrossRef]
- Francis, B.; Urich, T.; Mikolasch, A.; Teeling, H.; Amann, R. North Sea spring bloom-associated Gammaproteobacteria fill diverse heterotrophic niches. Environ. Microbiome 2021, 16, 15. [Google Scholar] [CrossRef] [PubMed]
- Koskella, B.; Vos, M. Adaptation in natural microbial populations. Annu. Rev. Ecol. Evol. Syst. 2015, 46, 503–522. [Google Scholar] [CrossRef]
- Steiner, C.F.; Long, Z.T.; Krumins, J.A.; Morin, P.J. Population and community resilience in multitrophic communities. Ecology 2006, 87, 996–1007. [Google Scholar] [CrossRef]
- Kaur, M.; Saini, K.C.; Mallick, A.; Bast, F. Seaweed-associated epiphytic bacteria: Diversity, ecological and economic implications. Aquat. Bot. 2023, 189, 103698. [Google Scholar] [CrossRef]
- Dromgoole, F.I.; Silvester, W.B.; Hicks, B.J. Nitrogenase activity associated with Codium species from New Zealand marine habitats. N. Z. J. Mar. Freshw. Res. 1978, 12, 17–22. [Google Scholar] [CrossRef]
- Ariosa, Y.; Quesada, A.; Aburto, J.; Carrasco, D.; Carreres, R.; Leganés, F.; Fernández Valiente, E. Epiphytic cyanobacteria on Chara vulgaris are the main contributors to N2 fixation in rice fields. Appl. Environ. Microbiol. 2004, 70, 5391–5397. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.T.; Lawrence, A.D.; Raux-Deery, E.; Warren, M.J.; Smith, A.G. Algae acquire vitamin B12 through a symbiotic relationship with bacteria. Nature 2005, 438, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Croft, M.T.; Warren, M.J.; Smith, A.G. Algae need their vitamins. Eukaryot. Cell 2006, 5, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Vieira, R.P.; Gonzalez, A.M.; Cardoso, A.M.; Oliveira, D.N.; Albano, R.M.; Clementino, M.M.; Martins, O.B.; Paranhos, R. Relationships between bacterial diversity and environmental variables in a tropical marine environment, Rio de Janeiro. Environ. Microbiol. 2008, 10, 189–199. [Google Scholar] [CrossRef] [PubMed]
- Hunter-Cevera, J.; Karl, D.; Buckley, M. Marine Microbial Diversity: The Key to Earth’s Habitability: This Report Is Based on a Colloquium, Sponsored by the American Academy of Microbiology, Held April 8–10, 2005, in San Francisco, California; American Society for Microbiology: Washington, DC, USA, 2020. [Google Scholar]
- Wang, G.; Shuai, L.; Li, Y.; Lin, W.; Zhao, X.; Duan, D. Phylogenetic analysis of epiphytic marine bacteria on Hole-Rotten diseased sporophytes of Laminaria japonica. J. Appl. Phycol. 2008, 20, 403–409. [Google Scholar] [CrossRef]
- Sawabe, T.; Makino, H.; Tatsumi, M.; Nakano, K.; Tajima, K.; Iqbal, M.M.; Yumoto, I.; Ezura, Y.; Christen, R. Pseudoalteromonas bacteriolytica sp. nov., a marine bacterium that is the causative agent of red spot disease of Laminaria japonica. Int. J. Syst. Bacteriol. 1998, 48, 769–774. [Google Scholar] [CrossRef]
- Sawabe, T.; Narita, M.; Tanaka, R.; Onji, M.; Tajima, K.; Ezura, Y. Isolation of Pseudoalteromonas elyakovii strains from spot-wounded fronds of Laminaria Japonica. Nippon Suisan Gakkaishi 2000, 66, 249–254. [Google Scholar] [CrossRef]
- Yan, Y.; Wang, S.; Li, J.; Liu, F.; Mo, Z. Alterations in epiphytic bacterial communities during the occurrence of green rot disease in Saccharina japonica seedlings. J. Mar. Sci. Eng. 2022, 10, 730. [Google Scholar] [CrossRef]
- Morris, M.M.; Haggerty, J.M.; Papudeshi, B.N.; Vega, A.A.; Edwards, M.S.; Dinsdale, E.A. Nearshore pelagic microbial community abundance affects recruitment success of giant kelp, Macrocystis pyrifera. Front. Microbiol. 2016, 7, 1800. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Choi, S.; Yang, K.M.; Choi, D.M.; Choi, Y.H.; Wang, X.; Wang, L.; Liu, X.; Duan, D.; Park, H.; Kim, J.H. Comparative Metagenomic Analysis of Marine eDNA Investigating the Production Crisis of Aquacultured Saccharina japonica. Diversity 2024, 16, 245. https://doi.org/10.3390/d16040245
Choi S, Yang KM, Choi DM, Choi YH, Wang X, Wang L, Liu X, Duan D, Park H, Kim JH. Comparative Metagenomic Analysis of Marine eDNA Investigating the Production Crisis of Aquacultured Saccharina japonica. Diversity. 2024; 16(4):245. https://doi.org/10.3390/d16040245
Chicago/Turabian StyleChoi, Soyun, Kwon Mo Yang, Dong Mun Choi, Yang Ho Choi, Xiuliang Wang, Lingxiu Wang, Xiaoyong Liu, Delin Duan, Hyun Park, and Jeong Ha Kim. 2024. "Comparative Metagenomic Analysis of Marine eDNA Investigating the Production Crisis of Aquacultured Saccharina japonica" Diversity 16, no. 4: 245. https://doi.org/10.3390/d16040245
APA StyleChoi, S., Yang, K. M., Choi, D. M., Choi, Y. H., Wang, X., Wang, L., Liu, X., Duan, D., Park, H., & Kim, J. H. (2024). Comparative Metagenomic Analysis of Marine eDNA Investigating the Production Crisis of Aquacultured Saccharina japonica. Diversity, 16(4), 245. https://doi.org/10.3390/d16040245