
Population Structure and Phylogeography of Marine Gastropods Monodonta labio and M. confusa (Trochidae) along the Northwestern Pacific Coast


Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and Sequencing
2.2. Sequence Alignment and Data Analysis
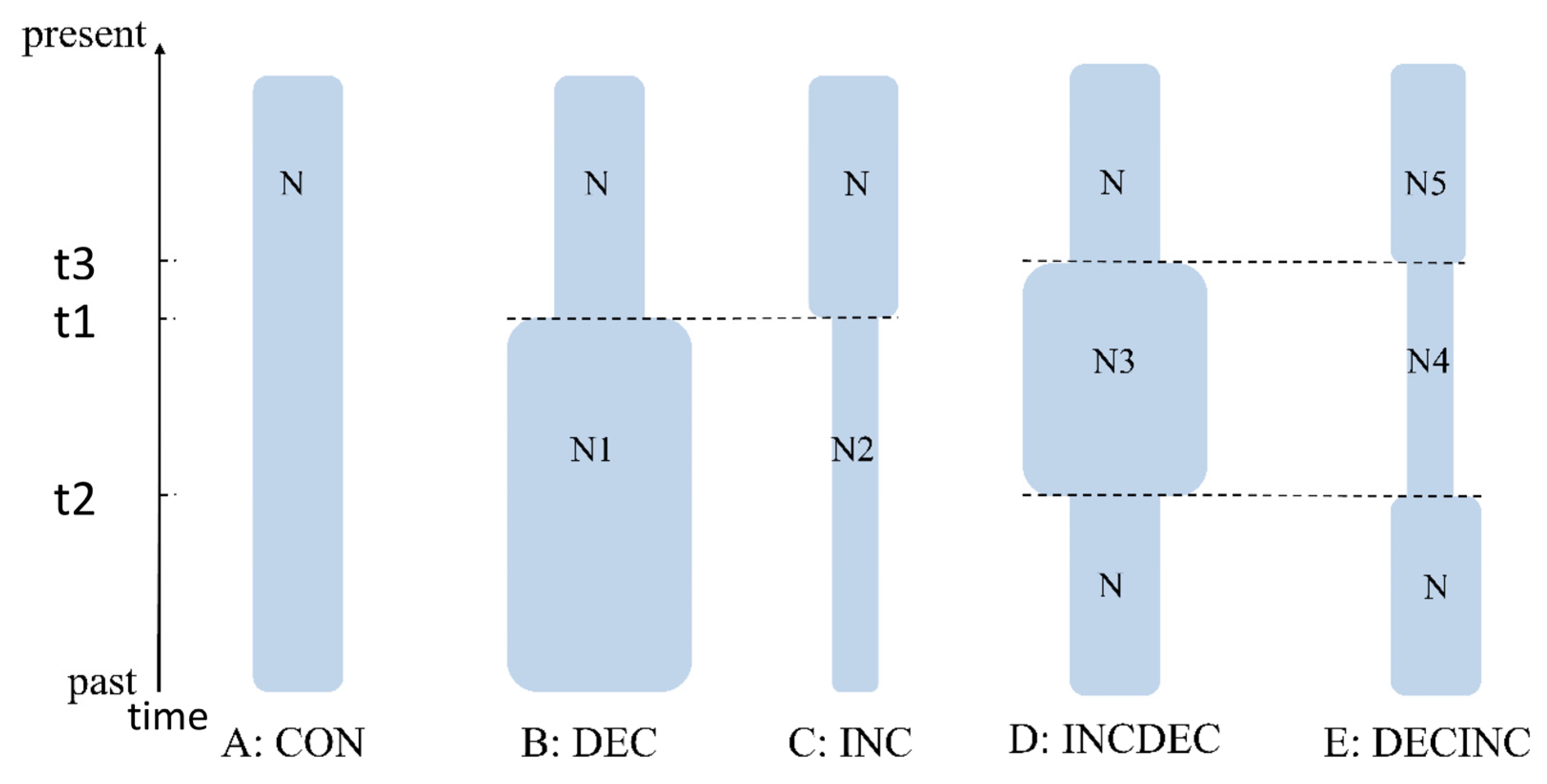
2.3. Historical Demography, Divergence Time Estimation and Biogeographic Analysis
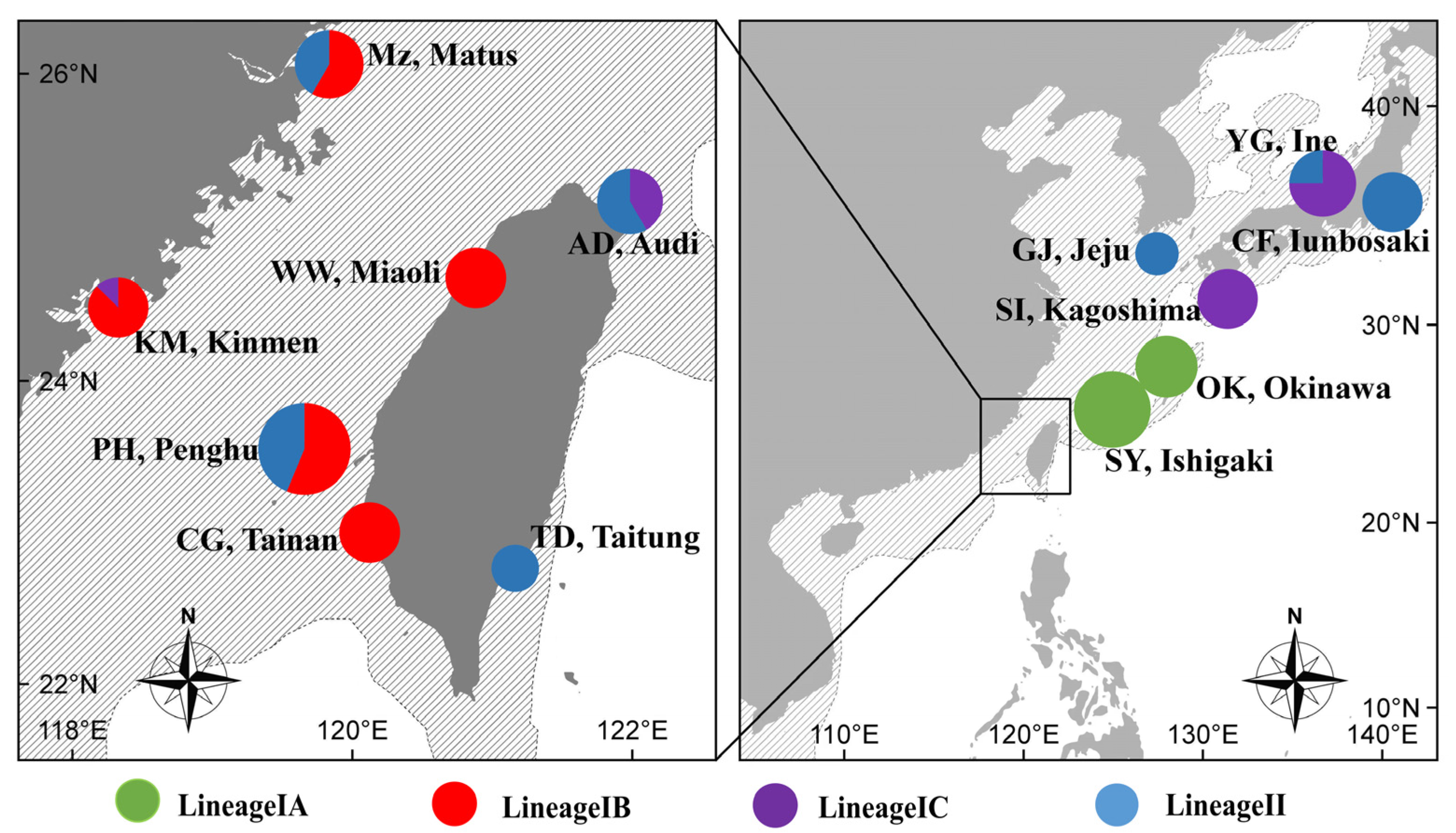
3. Results
3.1. Genetic Diversity
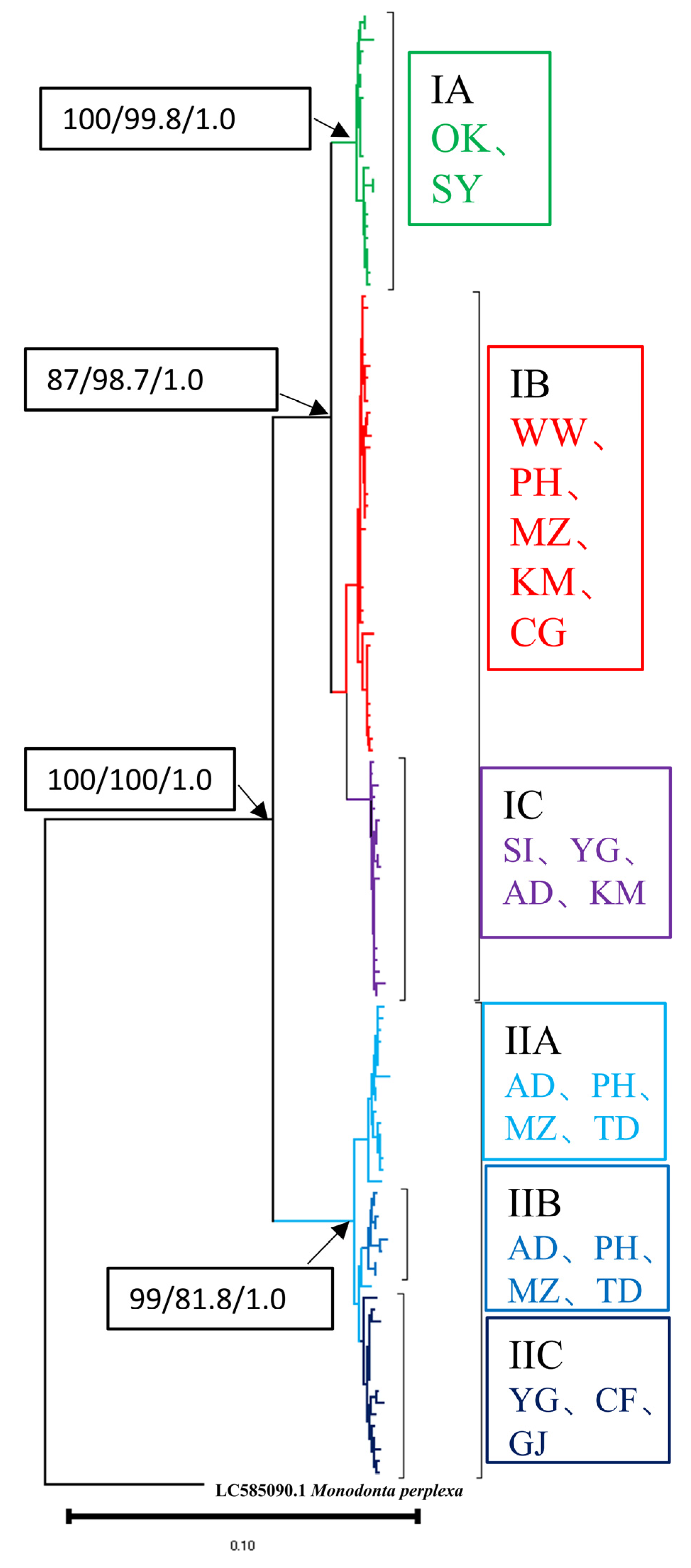
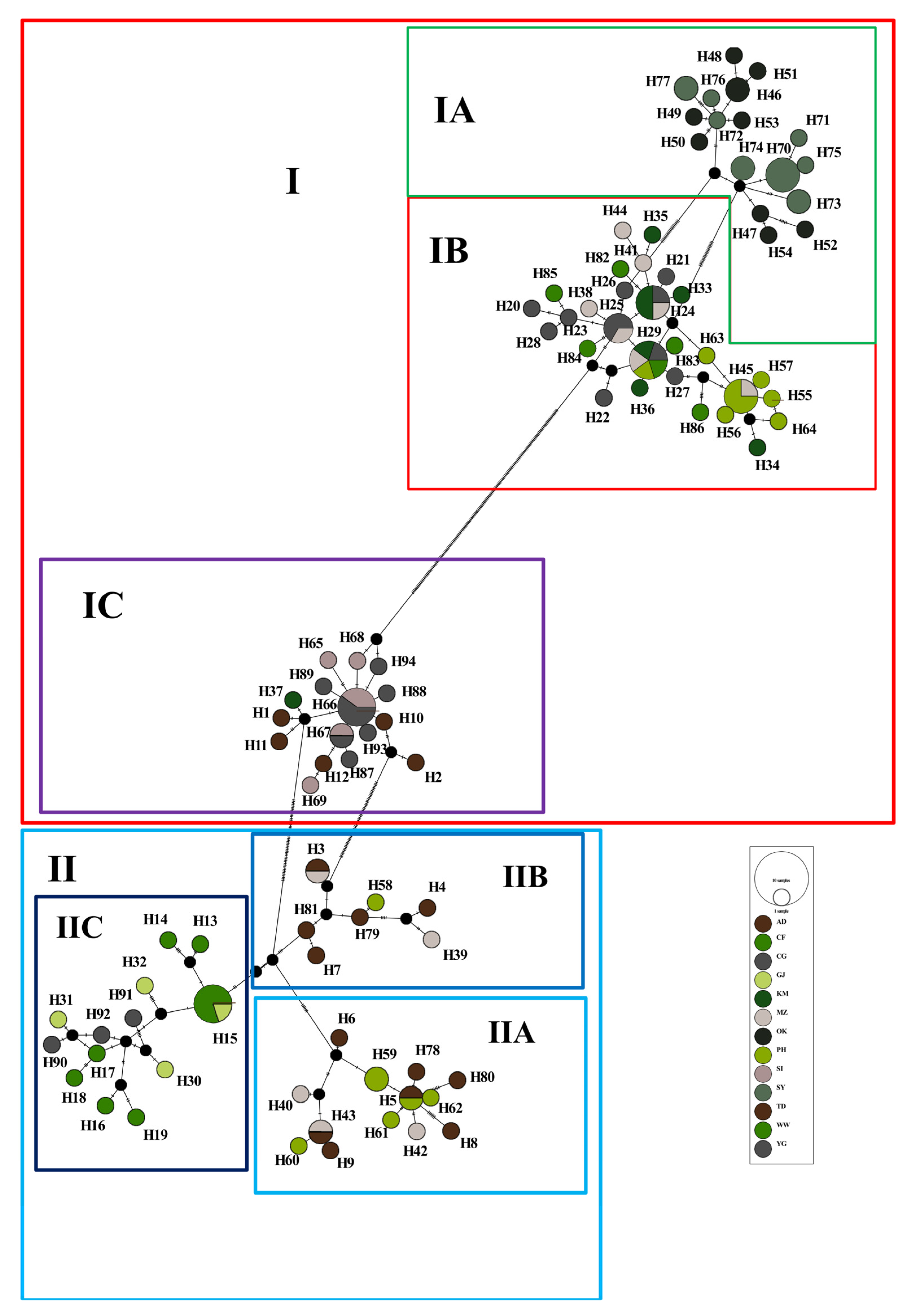
3.2. Phylogenetic Reconstruction and Genetic Structure

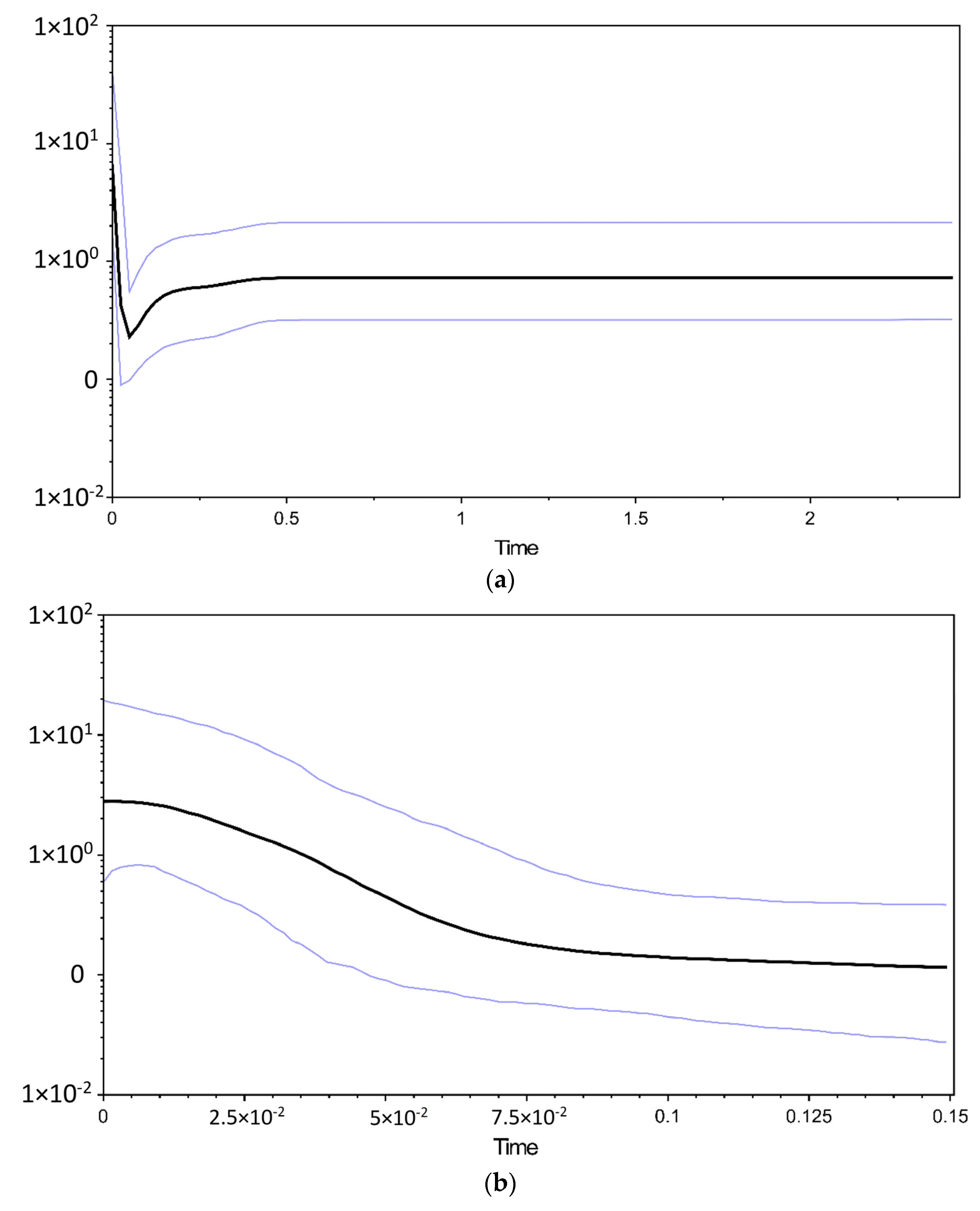
3.3. Molecular Dating and Historical Demographic Expansion
4. Discussion
4.1. Genetic Diversity
4.2. Population Structure and Demographic History
4.3. Phylogeography of Monodonta labio
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hewitt, G. The genetic legacy of the Quaternary ice ages. Nature 2000, 405, 907–913. [Google Scholar] [CrossRef] [PubMed]
- Galindo, H.M.; Olson, D.B.; Palumbi, S.R. Seascape genetics: A coupled oceanographic-genetic model predicts population structure of Caribbean corals. Curr. Biol. 2006, 16, 1622–1626. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.C.; Bowen, B.W. A realignment of marine biogeographic provinces with particular reference to fish distributions. J. Biogeogr. 2012, 39, 12–30. [Google Scholar] [CrossRef]
- Yang, Y.; Qi, L.; Kong, L.; Li, Q. Phylogeography of the marine gastropod Reticunassa festiva complex (Nassariidae) in the coast of China. J. Shellfish Res. 2020, 39, 419–431. [Google Scholar] [CrossRef]
- Shen, K.N.; Jamandre, B.W.; Hsu, C.C.; Tzeng, W.N.; Durand, J.D. Plio-Pleistocene sea level and temperature fluctuations in the northwestern Pacific promoted speciation in the globally-distributed flathead mullet Mugil cephalus. BMC Evol. Biol. 2011, 11, 83. [Google Scholar] [CrossRef]
- Yan, Y.R.; Hsu, K.C.; Yi, M.R.; Li, B.; Wang, W.K.; Kang, B.; Lin, H.D. Cryptic diversity of the spotted scat Scatophagus argus (Perciformes: Scatophagidae) in the South China Sea: Pre-or post-production isolation. Mar. Freshw. Res. 2020, 71, 1640–1650. [Google Scholar] [CrossRef]
- Gu, S.; Yi, M.R.; He, X.B.; Lin, P.S.; Liu, W.H.; Luo, Z.S.; Lin, H.D.; Yan, Y.R. Genetic diversity and population structure of cutlassfish (Lepturacanthus savala) along the coast of mainland China, as inferred by mitochondrial and microsatellite DNA markers. Reg. Stud. Mar. Sci. 2021, 43, 101702. [Google Scholar] [CrossRef]
- Wang, L.; Wan, Z.Y.; Lim, H.S.; Yue, G.H. Genetic variability, local selection and demographic history: Genomic evidence of evolving towards allopatric speciation in Asian seabass. Mol. Ecol. 2016, 25, 3605–3621. [Google Scholar] [CrossRef]
- Chiang, H.C.; Hsu, C.C.; Lin, H.D.; Ma, G.C.; Chiang, T.Y.; Yang, H.Y. Population structure of bigeye tuna (Thunnus obesus) in the South China Sea, Philippine Sea and western Pacific Ocean inferred from mitochondrial DNA. Fish. Res. 2006, 79, 219–225. [Google Scholar] [CrossRef]
- Li, H.; Lin, H.; Li, J.; Ding, S. Phylogeography of the Chinese beard eel, Cirrhimuraena chinensis Kaup, inferred from mitochondrial DNA: A range expansion after the last glacial maximum. Int. J. Mol. Sci. 2014, 15, 13564–13577. [Google Scholar] [CrossRef]
- Liu, Q.; Lin, H.; Chen, J.; Ma, J.; Liu, R.; Ding, S. Genetic variation and population genetic structure of the large yellow croaker (Larimichthys crocea) based on genome-wide single nucleotide polymorphisms in farmed and wild populations. Fish. Res. 2020, 232, 105718. [Google Scholar] [CrossRef]
- Donald, K.M.; Kennedy, M.; Spencer, H.G. The phylogeny and taxonomy of austral monodontine topshells (Mollusca: Gastropoda: Trochidae), inferred from DNA sequences. Mol. Phylogenet. Evol. 2005, 37, 474–483. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, R. Assessment and appearance of planktonic larvae of Haliotis (Nordotis) discus hannai around Kesennuma Bay. Aquaculture 1985, 32, 199. [Google Scholar]
- Palumbi, S.R. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Evol. Syst. 1994, 25, 547–572. [Google Scholar] [CrossRef]
- Ju, Y.M.; Hsu, C.H.; Fang, L.S.; Lin, H.D.; Wu, J.H.; Han, C.C.; Chen, I.S.; Chiang, T.Y. Population structure and demographic history of Sicyopterus japonicus (Perciformes; Gobiidae) in Taiwan inferred from mitochondrial control region sequences. Genet. Mol. Res. 2013, 12, 4046–4059. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, D.; Jung, D.; Li, Q.; Kong, L.F.; Ni, G.; Nakano, T.; Matsukuma, A.; Kim, S.; Park, C.; et al. Phylogeography of the rock shell Thais clavigera (Mollusca): Evidence for long-distance dispersal in the northwestern Pacific. PLoS ONE 2015, 10, e0129715. [Google Scholar] [CrossRef]
- Kojima, S.; Segawa, R.; Hayashi, I. Genetic differentiation among populations of the Japanese turban shell Turbo (Batillus) cornutus corresponding to warm currents. Mar. Ecol. Prog. Ser. 1997, 150, 149–155. [Google Scholar] [CrossRef]
- Chiu, Y.W.; Bor, H.; Tan, M.S.; Lin, H.D.; Jean, C.T. Phylogeography and genetic differentiation among populations of the moon turban snail Lunella granulata Gmelin, 1791 (Gastropoda: Turbinidae). Int. J. Mol. Sci. 2013, 14, 9062–9079. [Google Scholar] [CrossRef]
- Zhao, D.; Kong, L.F.; Sasaki, T.; Li, Q. Shell variations in the gastropod, Monodonta labio, in the North-western Pacific: The important role of temperature in the evolution process. J. Mar. Biolog. Assoc. UK 2019, 99, 1591–1599. [Google Scholar] [CrossRef]
- Zhao, D.; Li, Q.; Kong, L.; Yu, H. Cryptic diversity of marine gastropod Monodonta labio (Trochidae): Did the early Pleistocene glacial isolation and sea surface temperature gradient jointly drive diversification of sister species and/or subspecies in the Northwestern Pacific? Mar. Ecol. 2017, 38, e12443. [Google Scholar] [CrossRef]
- Yamazaki, D.; Miura, O.; Ikeda, M.; Kijima, A.; Van Tu, D.; Sasaki, T.; Chiba, S. Genetic diversification of intertidal gastropoda in an archipelago: The effects of islands, oceanic currents, and ecology. Mar. Biol. 2017, 164, 184. [Google Scholar] [CrossRef]
- Yamazaki, D.; Miura, O.; Uchida, S.; Ikeda, M.; Chiba, S. Comparative seascape genetics of co-distributed intertidal snails Monodonta spp. in the Japanese and Ryukyu archipelagoes. Mar. Ecol. Prog. Ser. 2021, 657, 135–146. [Google Scholar] [CrossRef]
- Folmer, O.; Black, M.; Hoeh, W.; Lutz, R.; Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–297. [Google Scholar] [PubMed]
- Simon, C.; Franke, A.; Martin, A. The polymerase chain reaction: DNA extraction and amplification. Mol. Tech. Taxon. 1991, 57, 329–355. [Google Scholar] [CrossRef]
- Thompson, J.D.; Gibson, T.J.; Plewniak, F.; Jeanmougin, F.; Higgins, D.G. The CLUSTAL X windows interface: Flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 1997, 25, 4876–4882. [Google Scholar] [CrossRef]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef]
- Jukes, T.H.; Cantor, C.R. Evolution of protein molecules. Mamm. Protein Metab. 1969, 3, 21–132. [Google Scholar]
- Excoffier, L.; Lischer, H.E.L. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Posada, D. jModelTest: Phylogenetic model averaging. Mol. Biol. Evol. 2008, 25, 1253–1256. [Google Scholar] [CrossRef]
- Lefort, V.; Longueville, J.E.; Gascuel, O. SMS: Smart model selection in PhyML. Mol. Biol. Evol. 2017, 34, 2422–2424. [Google Scholar] [CrossRef]
- Leigh, J.W.; Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 2015, 6, 1110–1116. [Google Scholar] [CrossRef]
- Tajima, F. The effect of change in population size on DNA polymorphism. Genetics 1989, 123, 597–601. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.X. Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics 1997, 147, 915–925. [Google Scholar] [CrossRef]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Rambaut, A.; Suchard, M.A.; Xie, D.; Drummond, A.J. Tracer v1.6. Available online: http://beast.bio.ed.ac.uk/Tracer (accessed on 26 March 2021).
- Hellberg, M.E.; Vacquier, V.D. Rapid evolution of fertilization selectivity and lysin cDNA sequences in teguline gastropods. Mol. Biol. Evol. 1999, 16, 839–848. [Google Scholar] [CrossRef]
- Beerli, P.; Felsenstein, J. Maximum likelihood estimation of a migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 2001, 98, 4563–4568. [Google Scholar] [CrossRef]
- Cornuet, J.M.; Pudlo, P.; Veyssier, J.; Dehne-Garcia, A.; Gautier, M.; Leblois, R.; Marin, J.M.; Estoup, A. DIYABC v2.0: A software to make approximate Bayesian computation inferences about population history using single nucleotide polymorphism, DNA sequence and microsatellite data. Bioinformatics 2014, 30, 1187–1189. [Google Scholar] [CrossRef]
- Cabrera, A.A.; Palsbøll, P.J. Inferring past demographic changes from contemporary genetic data: A simulation-based evaluation of the ABC methods implemented in diyabc. Mol. Ecol. Resour. 2017, 17, e94–e110. [Google Scholar] [CrossRef] [PubMed]
- Pons, O.; Petit, R.J. Measuring and testing genetic differentiation with ordered versus unordered alleles. Genetics 1996, 144, 1237–1245. [Google Scholar] [CrossRef] [PubMed]
- Benzie, J.A.H.; Ballment, E.; Forbes, A.T.; Demetriades, N.T.; Sugama, K.; Moria, S. Mitochondrial DNA variation in Indo-Pacific populations of the giant tiger prawn, Penaeus monodon. Mol. Ecol. 2002, 11, 2553–2569. [Google Scholar] [CrossRef] [PubMed]
- Kojima, Y. On the spawning of a top-shell, Monodonta labio (Linne). Venus (Jpn. J. Malacol.) 1962, 22, 200–203. [Google Scholar]
- Hu, Z.M.; Zhang, J.; Lopez-Bautista, J.; Duan, D.L. Asymmetric genetic exchange in the brown seaweed Sargassum fusiforme (Phaeophyceae) driven by oceanic currents. Mar. Biol. 2013, 160, 1407–1414. [Google Scholar] [CrossRef]
- Grant, W.A.S.; Bowen, B.W. Shallow population histories in deep evolutionary lineages of marine fishes: Insights from sardines and anchovies and lessons for conservation. J. Hered. 1998, 89, 415–426. [Google Scholar] [CrossRef]
- Chen, X.; Wang, J.J.; Ai, W.M.; Chen, H.; Lin, H.D. Phylogeography and genetic population structure of the spadenose shark (Scoliodon macrorhynchos) from the Chinese coast. Mitochondrial DNA Part A 2018, 29, 1100–1107. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.R.; Hsu, K.C.; Wang, J.X.; Feng, B.; Lin, H.D.; Yan, Y.R. Genetic structure and diversity of the yellowbelly threadfin bream Nemipterus bathybius in the Northern South China Sea. Diversity 2021, 13, 324. [Google Scholar] [CrossRef]
- Gu, S.; Yan, Y.R.; Yi, M.R.; Luo, Z.S.; Wen, H.; Jiang, C.P.; Lin, H.D.; He, X.B. Genetic pattern and demographic history of cutlassfish (Trichiurus nanhaiensis) in South China Sea by the influence of Pleistocene climatic oscillations. Sci. Rep. 2022, 12, 14716. [Google Scholar] [CrossRef] [PubMed]
- Chen, P.C.; Shih, C.H.; Chu, T.J.; Wang, D.; Lee, Y.C.; Tzeng, T.D. Population structure and historical demography of the oriental river prawn (Macrobrachium nipponense) in Taiwan. PLoS ONE 2015, 10, e0145927. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.Y.V.; Huang, I.H.; Liu, M.Y.; Lin, H.D.; Wang, F.Y.; Liao, T.Y. Genetic stock structure of Terapon jarbua in Taiwanese waters. Mar. Coast. Fish. 2015, 7, 464–473. [Google Scholar] [CrossRef]
- Ding, S.; Mishra, M.; Wu, H.; Liang, S.; Miyamoto, M.M. Characterization of hybridization within a secondary contact region of the inshore fish, Bostrychus sinensis, in the East China Sea. Heredity 2018, 120, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Hirase, S.; Kanno, M.; Ikeda, M.; Kijima, A. Evidence of the restricted gene flow within a small spatial scale in the Japanese common intertidal goby Chaenogobius annularis. Mar. Ecol. 2012, 33, 481–489. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locations | Sampling Location | Code | Coordinates | N | Nh | h | θπ | θω |
|---|---|---|---|---|---|---|---|---|
| Monodonta labio | 85 | 61 | 0.987 | 0.0131 | 0.0151 | |||
| Taiwan Island | 22 | 20 | 0.991 | 0.0.0448 | 0.0376 | |||
| Audi | AD | 121.92 25.08 | 5 | 5 | 1.000 | 0.0032 | 0.0035 | |
| Miaoli | WW | 120.73 24.61 | 6 | 6 | 1.000 | 0.0041 | 0.0052 | |
| Tainan | CG | 120.10 23.12 | 11 | 10 | 0.982 | 0.0019 | 0.0028 | |
| Penghu | 9 | 7 | 0.917 | 0.0017 | 0.0027 | |||
| Penghu | PH | 119.58 23.58 | 9 | 7 | 0.917 | 0.0017 | 0.0027 | |
| Coast of mainland China | 15 | 12 | 0.962 | 0.0179 | 0.0387 | |||
| Kinmen | KM | 118.35 24.43 | 8 | 7 | 0.964 | 0.0315 | 0.0480 | |
| Matsu | MZ | 119.98 26.22 | 7 | 7 | 1.000 | 0.0027 | 0.0030 | |
| Japan | 15 | 10 | 0.895 | 0.0015 | 0.0029 | |||
| Ine | YG | 135.29 35.70 | 9 | 7 | 0.917 | 0.0012 | 0.0019 | |
| Kagoshima | SI | 130.46 32.51 | 6 | 5 | 0.933 | 0.0018 | 0.0022 | |
| Ryukyu | 24 | 17 | 0.964 | 0.0037 | 0.0056 | |||
| Okinawa | OK | 127.79 24.25 | 10 | 9 | 0.978 | 0.0035 | 0.0044 | |
| Ishigaki | SY | 124.11 24.36 | 14 | 8 | 0.901 | 0.0032 | 0.0035 | |
| Monodonta confusa | 41 | 33 | 0.983 | 0.0082 | 0.0115 | |||
| Taiwan Island | 12 | 12 | 1.000 | 0.0.0086 | 0.0096 | |||
| Audi | AD | 121.92 25.08 | 7 | 7 | 1.000 | 0.0093 | 0.0097 | |
| Taitung | TD | 121.19 22.82 | 5 | 5 | 1.000 | 0.0089 | 0.0086 | |
| Penghu | 5 | 5 | 1.000 | 0.0093 | 0.0093 | |||
| Penghu | PH | 119.58 23.58 | 5 | 5 | 1.000 | 0.0093 | 0.0093 | |
| Coast of mainland China | 7 | 6 | 0.952 | 0.0050 | 0.0067 | |||
| Matsu | MZ | 119.98 26.22 | 7 | 6 | 0.952 | 0.0050 | 0.0067 | |
| Japan | 13 | 10 | 0.923 | 0.0037 | 0.0050 | |||
| Inubosaki | CF | 140.87 25.70 | 10 | 7 | 0.867 | 0.0031 | 0.0034 | |
| Ine | YG | 135.29 35.70 | 3 | 3 | 1.000 | 0.0049 | 0.0049 | |
| Korea | GY | 4 | 4 | 1.000 | 0.0042 | 0.0044 | ||
| Total | 126 | 94 | 0.992 | 0.0825 | 0.0441 | |||
| (A) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| AD | WW | CG | PH | KM | MZ | YG | SI | OK | SY | |
| AD | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.270 | 0.000 | 0.000 | |
| WW | 0.968 | 0.414 | 0.000 | 0.891 | 0.774 | 0.000 | 0.000 | 0.000 | 0.000 | |
| CG | 0.980 | 0.000 | 0.000 | 0.252 | 0.513 | 0.000 | 0.000 | 0.000 | 0.000 | |
| PH | 0.981 | 0.455 | 0.594 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | 0.000 | |
| KM | 0.799 | −0.041 | 0.034 | 0.145 | 0.981 | 0.000 | 0.000 | 0.000 | 0.000 | |
| MZ | 0.975 | −0.047 | −0.008 | 0.519 | −0.030 | 0.000 | 0.000 | 0.000 | 0.000 | |
| YG | 0.152 | 0.979 | 0.986 | 0.987 | 0.849 | 0.984 | 0.558 | 0.000 | 0.000 | |
| SI | 0.029 | 0.974 | 0.983 | 0.985 | 0.816 | 0.980 | −0.015 | 0.000 | 0.000 | |
| OK | 0.973 | 0.795 | 0.849 | 0.850 | 0.483 | 0.819 | 0.981 | 0.977 | 0.000 | |
| SY | 0.975 | 0.797 | 0.843 | 0.844 | 0.522 | 0.817 | 0.981 | 0.978 | 0.154 | |
| (B) | ||||||||||
| AD | TD | PH | MZ | CF | YG | GJ | ||||
| AD | 0.972 | 0.396 | 0.936 | 0.000 | 0.036 | 0.027 | ||||
| TD | −0.133 | 0.594 | 0.990 | 0.000 | 0.009 | 0.072 | ||||
| PH | 0.013 | −0.042 | 0.342 | 0.000 | 0.000 | 0.009 | ||||
| MZ | −0.137 | −0.138 | −0.002 | 0.000 | 0.036 | 0.036 | ||||
| CF | 0.395 | 0.442 | 0.605 | 0.459 | 0.036 | 0.279 | ||||
| YG | 0.332 | 0.351 | 0.053 | 0.356 | 0.193 | 0.576 | ||||
| GJ | 0.260 | 0.284 | 0.528 | 0.306 | 0.018 | −0.049 | ||||
| Source of Variation | Percentage Variation | p | |
|---|---|---|---|
| Monodonta labio | |||
| 1. Scenario I: two groups (WW, CG, PH, KM, MZ, YG) (AD, OK, SY, SI) | |||
| Among groups | −5.50 | FCT = −0.055 | 0.461 |
| Among populations within groups | 72.41 | FSC = 0.686 | 0.000 |
| Among individuals within populations | 33.10 | FST = 0.669 | 0.000 |
| 2. Scenario II: three groups (AD, WW, CG, PH, KM, MZ) (OK, SY) (YG, SI) | |||
| Among groups | 68.40 | FCT = 0.683 | 0.016 |
| Among populations within groups | 24.36 | FSC = 0.770 | 0.000 |
| Among individuals within populations | 7.24 | FST = 0.927 | 0.000 |
| 3. Scenario III: five groups (AD, WW, CG, PH) (KM, MZ) (OK, SY) (YG, SI) | |||
| Among groups | 59.93 | FCT = 0.599 | 0.000 |
| Among populations within groups | 31.74 | FSC = 0.792 | 0.000 |
| Among individuals within populations | 8.31 | FST = 0.916 | 0.000 |
| Monodonta confusa | |||
| 1. Scenario I: two groups (PH, MZ, YG, GJ) (AD, TD, CF) | |||
| Among groups | −10.46 | FCT = −0.104 | 1.000 |
| Among populations within groups | 35.55 | FSC = 0.321 | 0.000 |
| Among individuals within populations | 74.90 | FST = 0.250 | 0.000 |
| 2. Scenario II: three groups (AD, TD, PH, MZ) (CF, YG, GJ) | |||
| Among groups | 43.25 | FCT = 0.432 | 0.032 |
| Among populations within groups | −2.04 | FSC = −0.035 | 0.686 |
| Among individuals within populations | 58.78 | FST =0.412 | 0.000 |
| 3. Scenario III: five groups (AD, TD, PH) (MZ) (CF, YG) (GJ) | |||
| Among groups | 33.51 | FCT = 0.335 | 0.000 |
| Among populations within groups | −0.27 | FSC = −0.004 | 0.546 |
| Among individuals within populations | 66.75 | FST = 0.332 | 0.000 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiu, Y.-W.; Bor, H.; Wu, J.-X.; Shieh, B.-S.; Lin, H.-D. Population Structure and Phylogeography of Marine Gastropods Monodonta labio and M. confusa (Trochidae) along the Northwestern Pacific Coast. Diversity 2023, 15, 1021. https://doi.org/10.3390/d15091021
Chiu Y-W, Bor H, Wu J-X, Shieh B-S, Lin H-D. Population Structure and Phylogeography of Marine Gastropods Monodonta labio and M. confusa (Trochidae) along the Northwestern Pacific Coast. Diversity. 2023; 15(9):1021. https://doi.org/10.3390/d15091021
Chicago/Turabian StyleChiu, Yuh-Wen, Hor Bor, Jin-Xian Wu, Bao-Sen Shieh, and Hung-Du Lin. 2023. "Population Structure and Phylogeography of Marine Gastropods Monodonta labio and M. confusa (Trochidae) along the Northwestern Pacific Coast" Diversity 15, no. 9: 1021. https://doi.org/10.3390/d15091021
APA StyleChiu, Y.-W., Bor, H., Wu, J.-X., Shieh, B.-S., & Lin, H.-D. (2023). Population Structure and Phylogeography of Marine Gastropods Monodonta labio and M. confusa (Trochidae) along the Northwestern Pacific Coast. Diversity, 15(9), 1021. https://doi.org/10.3390/d15091021