
Transcriptomic Analysis of Medicago truncatula under Long-Day Conditions

Abstract
:1. Introduction
2. Materials and Methods
2.1. Test Material
2.2. Methods
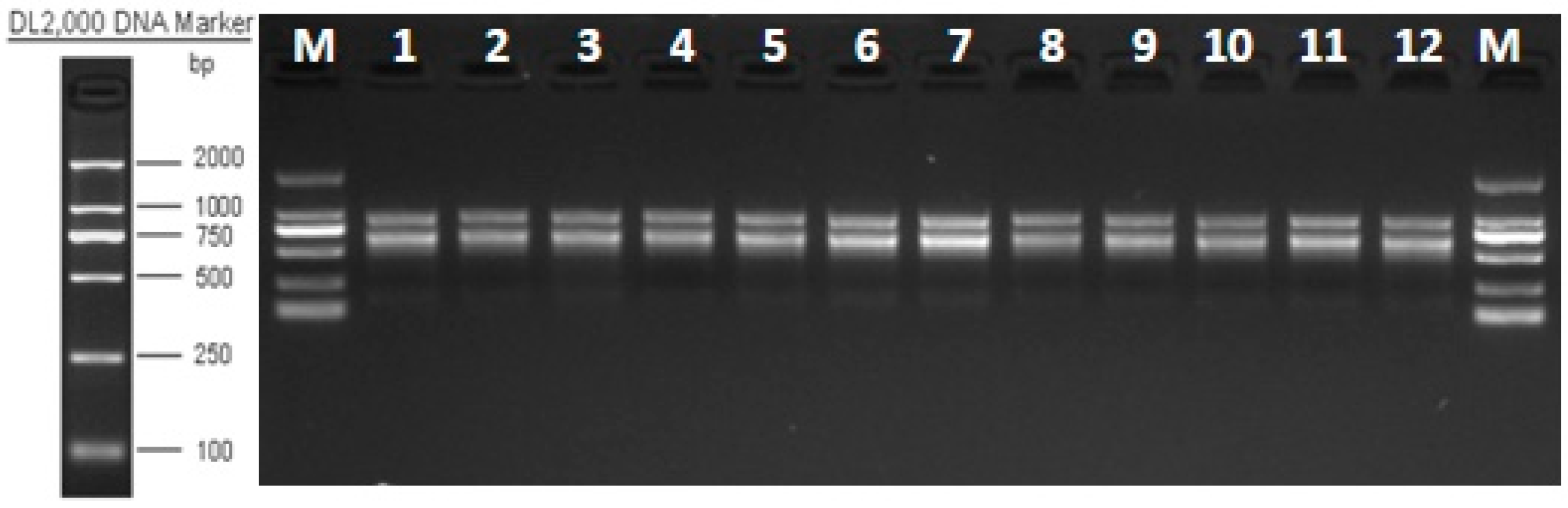
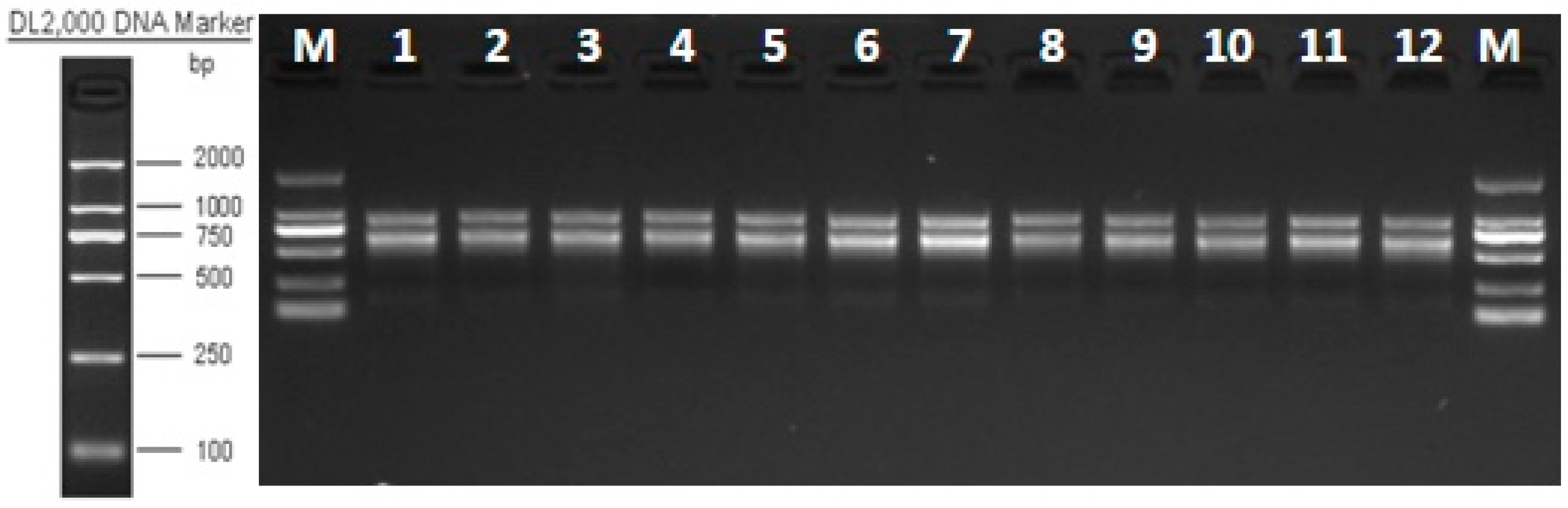
2.2.1. Total RNA Extraction and Detection, cDNA Library Construction, and Transcriptome Sequencing
2.2.2. Raw Data Processing and Comparison with Reference Genome Sequence
2.2.3. Analysis of Differential Gene Expressions (DEGs)
2.2.4. Analysis of GO Functional Enrichment
2.2.5. Analysis of KEGG Pathway Enrichment
2.2.6. Analysis of cSNP Structure
2.2.7. Analysis of eggNOG
3. Results
3.1. Analysis of RNA-Seq Sequence
3.2. Functional Annotation of Genes
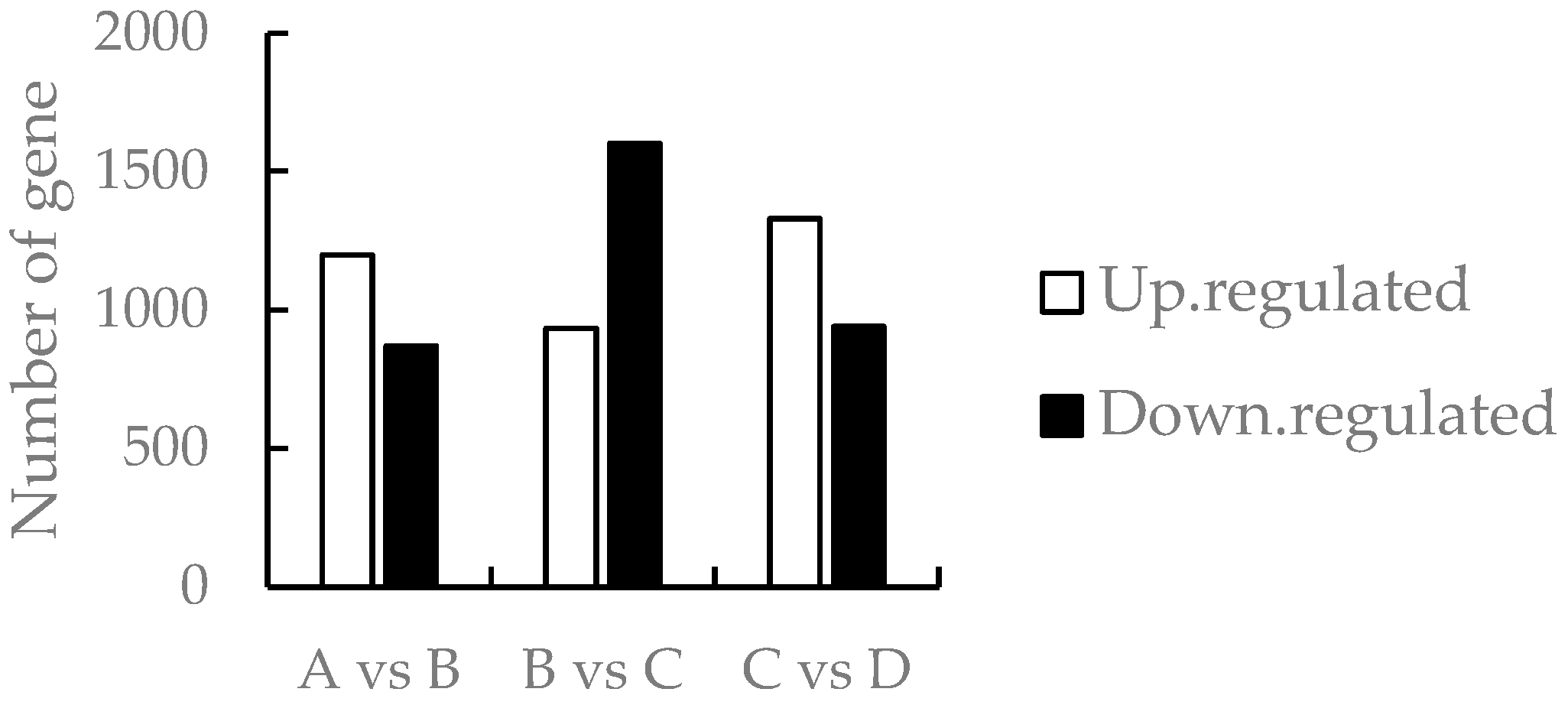
3.3. Comparison of Differentially Expressed Genes
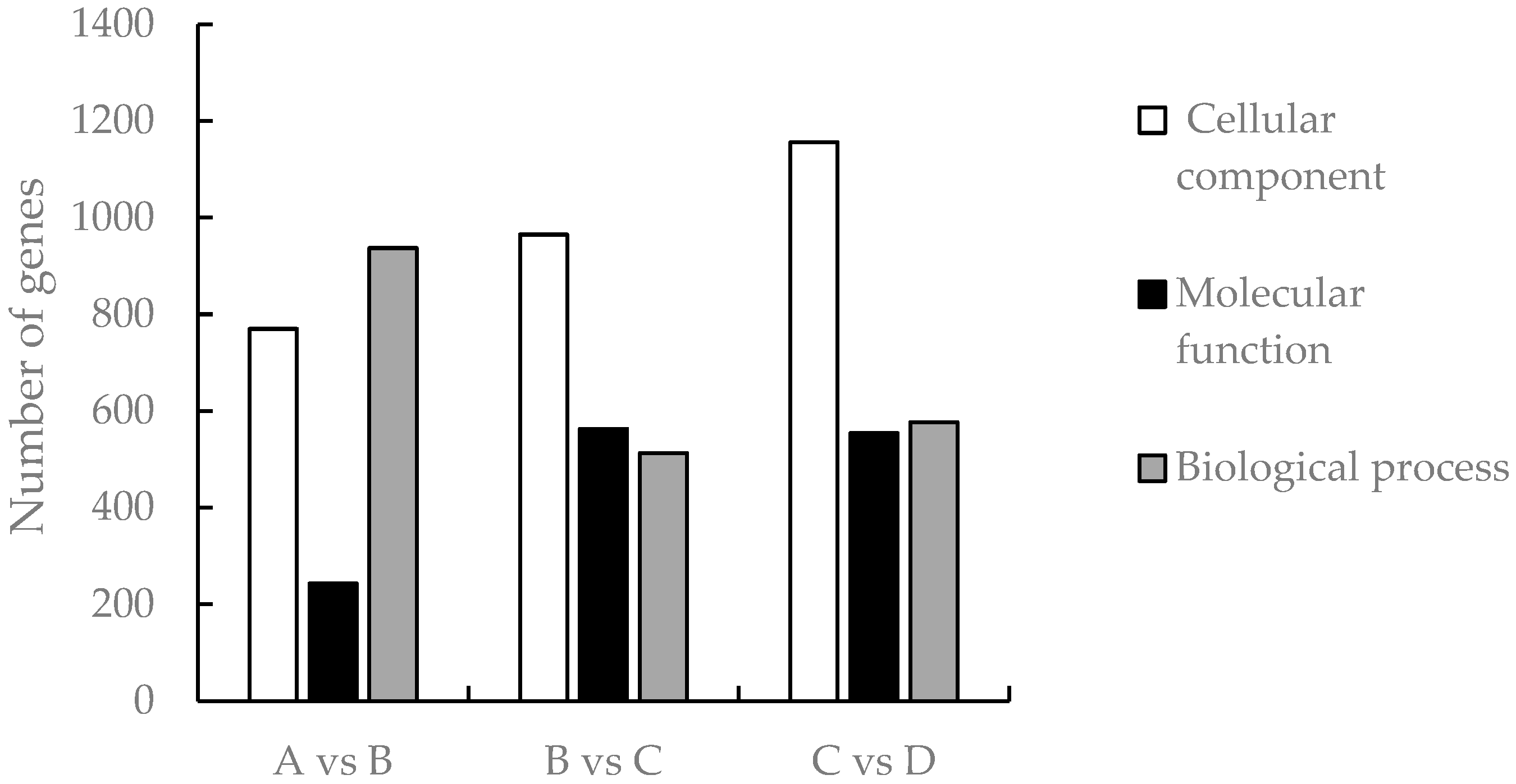
3.4. Analysis of GO Function Enrichment of Differential Genes
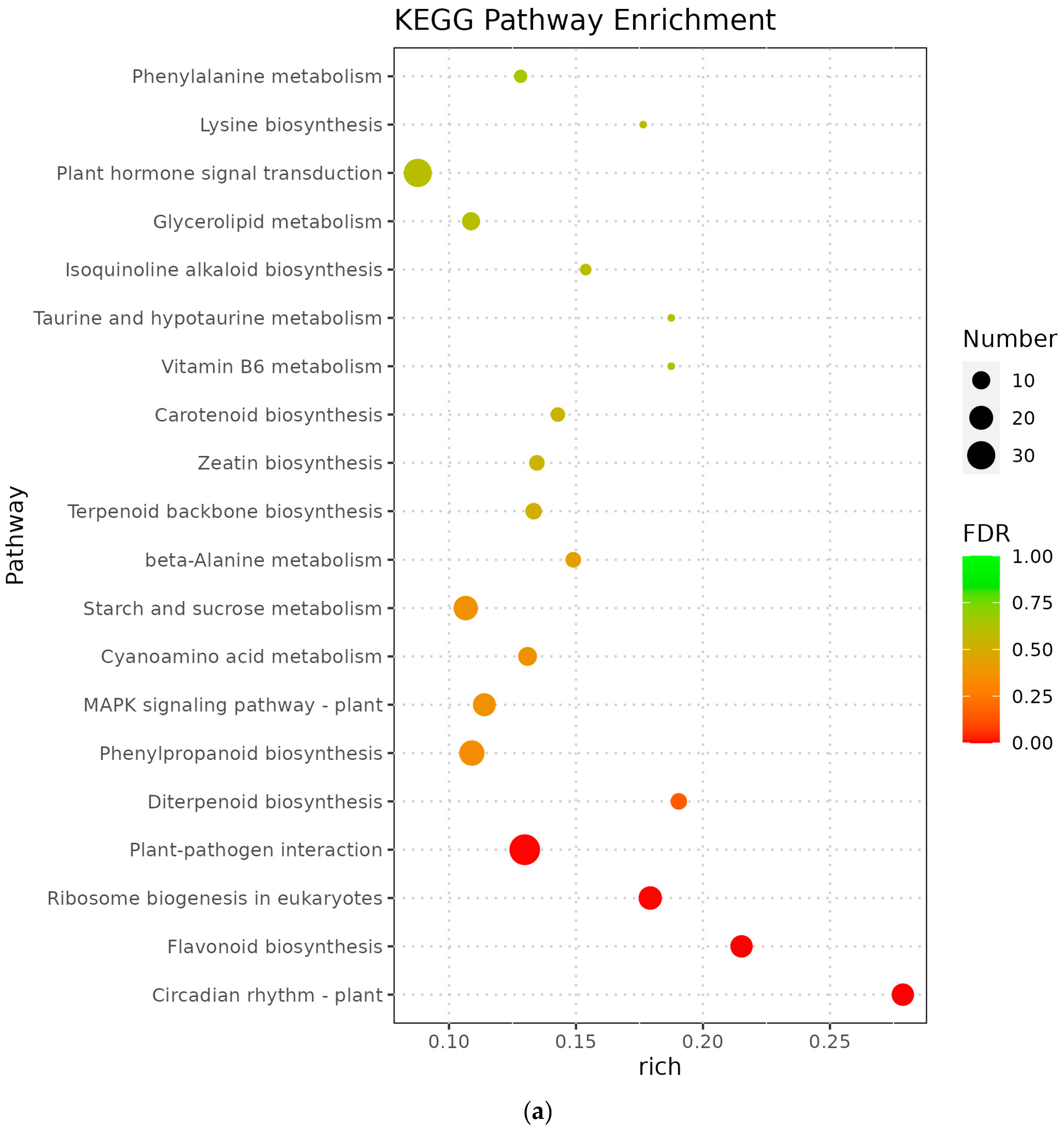
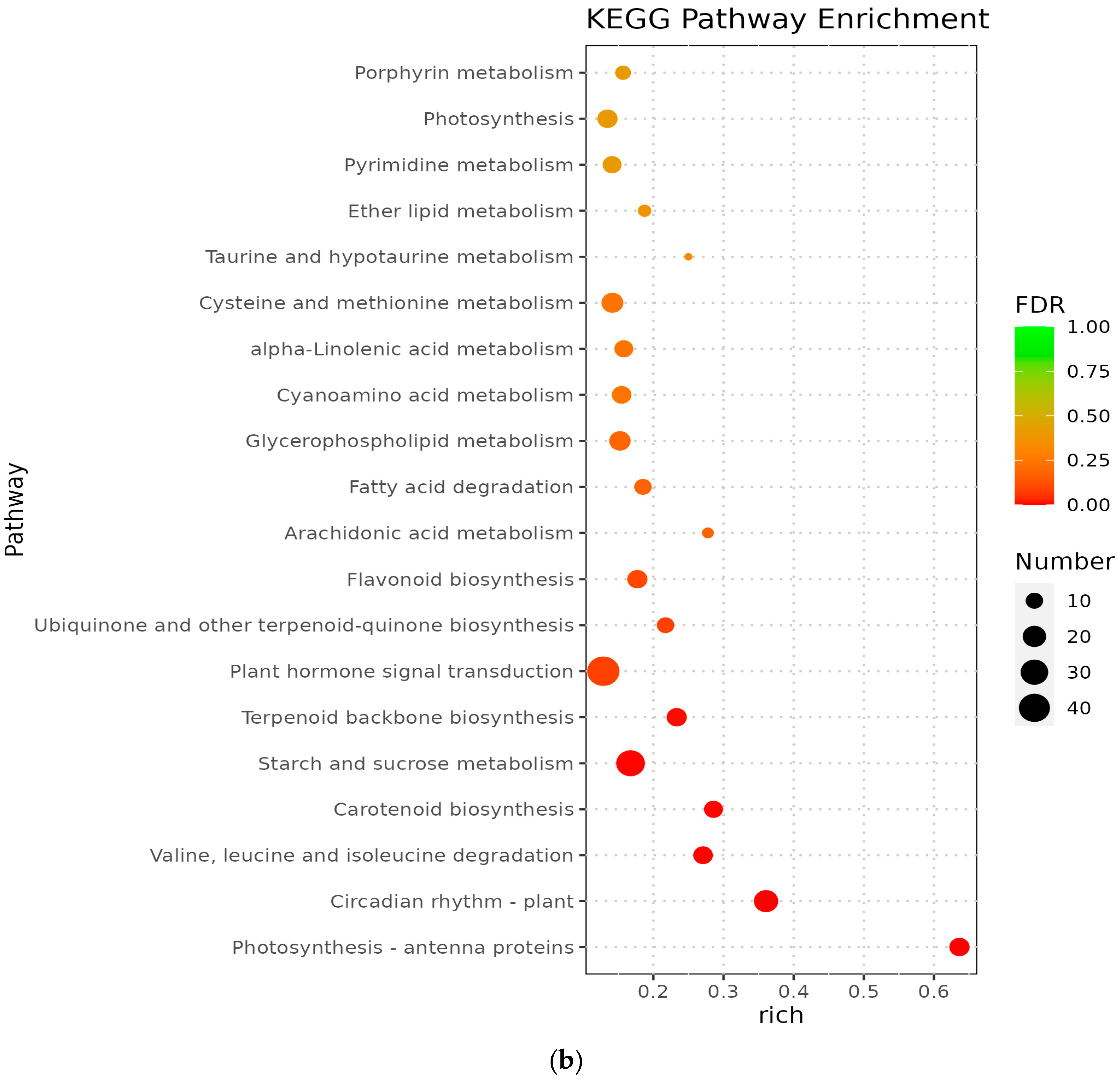
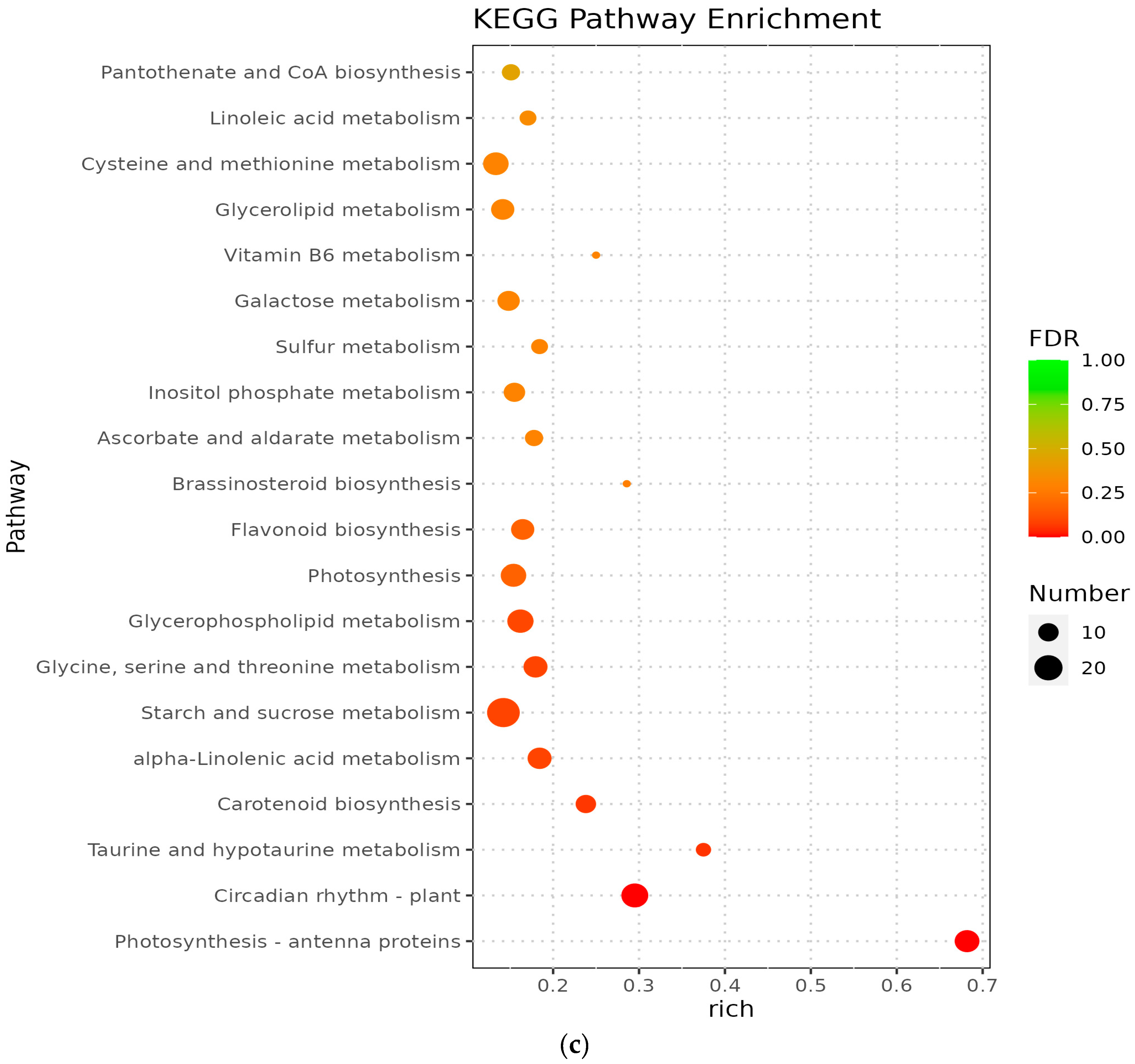
3.5. Analysis of KEGG Pathway of Differential Genes
3.6. Analysis of cSNP
3.7. Functional Classification of eggNOG
3.8. Photoperiod-Related Differentially Expressed Genes
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Xiao, H.S.; Lei, T.; Shun, X.W.; Zhou, J.L.; Zhang, J.; Chen, Z.; Wu, L.J.; Ku, L.X.; Chen, Y.H. Integrating transcriptomic and proteomic analyses of photoperiod-sensitive in near isogenic maize line under long-day conditions. J. Integr. Agric. 2019, 18, 1211–1221. [Google Scholar] [CrossRef]
- Kathleen, G.; Robertson, C.M. Integrating circadian dynamics with physiological processes in plants. Nat. Rev. Genet. 2015, 16, 598–610. [Google Scholar] [CrossRef]
- Fernando, A.; George, C. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef]
- Sung, J.S.; Akane, K.; Takato, I. Circadian Clock and Photoperio5-15dic Flowering in Arabidopsis: CONSTANS Is a Hub for Signal Integration. Plant Physiol. 2017, 173, 5–15. [Google Scholar] [CrossRef]
- Soledad, P.; Nathanael, N.; Fran, R.; Weller, J.L.; Bond, D.M.; Macknight, R.C. A Point Mutation in Phytochromobilin synthase Alters the Circadian Clock and Photoperiodic Flowering of Medicago truncatula. Plants 2022, 11, 239. [Google Scholar] [CrossRef]
- Sanchez, E.S.; Rugnone, L.M.; Kay, A.S. Light Perception: A Matter of Time. Mol. Plant 2020, 13, 363–385. [Google Scholar] [CrossRef] [PubMed]
- Viker, K.B.; Steele, M.B.; Iankov, I.D.; Concilio, S.C.; Ammayappan, A.; Bolon, B.; Jenks, N.J.; Goetz, M.P.; Panagioti, E.; Federspiel, M.J.; et al. Preclinical safety assessment of MV-s-NAP, a novel oncolytic measles virus strain armed with an H. pylori immunostimulatory bacterial transgene. Mol. Ther.-Meth. D 2022, 26, 532–546. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Zheng, J.; Xu, Z.; Zhang, X.H.; Zhang, K.; Wang, G.Y. Functional analysis of ZmDWF1, a maize homolog of the Arabidopsis brassinosteroids biosynthetic DWF1/DIM gene. Plant Sci. 2004, 167, 743–751. [Google Scholar] [CrossRef]
- Laurie, R.E.; Diwadkar, P.; Jaudal, M.; Zhang, L.L.; Hecht, V.; Wen, J.Q.; Tadege, M.; Mysore, K.S.; Putterill, J.; Weller, J.L.; et al. The Medicago FLOWERING LOCUS T homolog, MtFTa1, is a key regulator of flowering time. Plant Physiol. 2011, 156, 2207–2224. [Google Scholar] [CrossRef]
- Putterill, J.; Varkonyi-Gasic, E. FT and florigen long-distance flowering control in plants. Curr. Opin. Plant Biol. 2016, 33, 77–82. [Google Scholar] [CrossRef]
- Zhang, L.L.; Jiang, A.; Thomson, G.; Kerr-Phillips, M.; Phan, C.; Krueger, T.; Jaudal, M.; Wen, J.Q.; Mysore, K.S.; Putterill, J. Overexpression of Medicago MtCDFd1_1 Causes Delayed Flowering in Medicago via Repression of MtFTa1 but Not MtCO-Like Genes. Front. Plant Sci. 2019, 10, 1148. [Google Scholar] [CrossRef] [PubMed]
- Jaudal, M.; Wen, J.Q.; Mysore, K.S.; Putterill, J. Medicago PHYA promotes flowering, primary stem elongation and expression of flowering time genes in long days. BMC Plant Biol. 2020, 20, 329. [Google Scholar] [CrossRef] [PubMed]
- Thomson, G.; Zhang, L.L.; Wen, J.Q.; Mysore, K.S.; Putterill, J. The Candidate Photoperiod Gene MtFE Promotes Growth and Flowering in Medicago truncatula. Front. Plant Sci. 2021, 12, 634091. [Google Scholar] [CrossRef] [PubMed]
- Liew, L.C.; Hecht, V.; Sussmilch, F.C.; Weller, J.L. The Pea Photoperiod Response Gene STERILE NODES Is an Ortholog of LUX ARRHYTHMO. Plant Physiol. 2014, 165, 648–657. [Google Scholar] [CrossRef]
- Rubenach, A.J.S.; Hecht, V.; Vander, S.J.K.; Liew, L.C.; Aubert, G.; Burstin, J.; Weller, J.L. EARLY FLOWERING3 Redundancy Fine-Tunes Photoperiod Sensitivity. Plant Physiol. 2017, 173, 2253–2264. [Google Scholar] [CrossRef]
- Wong, A.; Hecht, V.F.; Picard, K. Isolation and functional analysis of CONSTANS-LIKE genes suggests that a central role for CONSTANS in flowering time control is not evolutionarily conserved in Medicago truncatula. Front. Plant Sci. 2014, 5, 486. [Google Scholar] [CrossRef] [PubMed]
- Xia, Z.J.; Watanabe, S.; Yamada, T.; Tsubokura, Y.; Nakashima, H.; Zhai, H.; Anai, T.; Sato, S.; Yamazaki, T.; Lv, S.X. Positional cloning and characterization reveal the molecular basis for soybean maturity locus E1 that regulates photoperiodic flowering. Proc. Natl. Acad. Sci. USA 2012, 109, E2155–E2164. [Google Scholar] [CrossRef]
- Liu, W.; Jiang, B.J.; Ma, L.M.; Zhang, S.W.; Zhai, H.; Xu, X.; Hou, W.S.; Xia, Z.J.; Wu, C.X.; Sun, S.; et al. Functional diversification of Flowering Locus T homologs in soybean: GmFT1a and GmFT2a/5a have opposite roles in controlling flowering and maturation. New Phytol. 2018, 217, 1335–1345. [Google Scholar] [CrossRef]
- Xu, M.L.; Yamagishi, N.; Zhao, C.; Takeshima, R.; Kasai, M.; Watanabe, S.; Kanazawa, A.; Yoshikawa, N.; Liu, B.H.; Yamada, T.; et al. The Soybean-Specific Maturity Gene E1 Family of Floral Repressors Controls Night-Break Responses through Down-Regulation of FLOWERING LOCUS T Orthologs. Plant Physiol. 2015, 168, 1735–1746. [Google Scholar] [CrossRef]
- Zhang, X.Z.; Zhai, H.; Wang, Y.Y.; Tian, X.J.; Zhang, Y.P.; Wu, H.Y.; Lv, S.X.; Yang, G.; Li, Y.Q.; Wang, L.; et al. Functional conservation and diversification of the soybean maturity gene E1 and its homologs in legumes. Sci. Rep. 2016, 6, 29548. [Google Scholar] [CrossRef]
- Jin, M.L.; Liu, X.G.; Jia, W.; Liu, H.J.; Li, W.Q.; Peng, Y.; Du, Y.F.; Wang, Y.B.; Yin, Y.J.; Zhang, X.H.; et al. ZmCOL3 a CCT gene represses flowering in maize by interfering with the circadian clock and activating expression of ZmCCT. J. Integr. Plant Biol. 2018, 60, 465–480. [Google Scholar] [CrossRef]
- Meng, X.; Muszynski, M.G.; Danilevskaya, O.N. The FT-like ZCN8 Gene Functions as a Floral Activator and Is Involved in Photoperiod Sensitivity in Maize. Plant Cell 2011, 23, 942–960. [Google Scholar] [CrossRef] [PubMed]
- Chen, A.; Li, C.X.; Hu, W.; Lau, M.Y.; Lin, H.Q.; Rockwell, N.C.; Martin, S.S.; Jernstedt, J.A.; Lagarias, J.C.; Dubcovsky, J. Phytochrome C plays a major role in the acceleration of wheat flowering under long-day photoperiod. Proc. Natl. Acad. Sci. USA 2014, 111, 10037–10044. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef] [PubMed]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Michael, A.; Catherine, A.B.; Judith, A.B.; David, B.; Heather, B.; Cherry, J.M.; Allan, P.D.; Kara, D.; Selina, S.D.; Janan, T.E.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef]
- Minoru, K.; Susumu, G.; Shuichi, K.; Yasushi, O.; Masahiro, H. The KEGG resource for deciphering the genome. Nucleic Acids Res. 2004, 32, D277–D280. [Google Scholar]
- Yu, H.H.; Xie, W.B.; Li, J.; Zhou, F.S.; Zhang, Q.F. A whole-genome SNP array (RICE6K) for genomic breeding in rice. Plant Biotechnol. J. 2014, 12, 28–37. [Google Scholar] [CrossRef]
- Grabherr, M.G.; Haas, B.J.; Yassour, M.; Levin, J.Z.; Thompson, D.A.; Amit, I.; Adiconis, X.; Fan, L.; Raychowdhury, R.; Zeng, Q.D.; et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 2011, 29, 644–652. [Google Scholar] [CrossRef]
- Panigrahi, S.C.K.; EMishra, P. GIGANTEA—An Emerging Story. Front. Plant Sci. 2015, 6, 8. [Google Scholar] [CrossRef]
- Simpson, G.G.; Dean, C. Arabidopsis, the Rosetta Stone of Flowering Time? Science 2002, 296, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Chentao, L. Blue light receptors and signal transduction. Plant Cell 2002, 14, S207–S225. [Google Scholar]
- Li, R.N.; Li, T.; Wu, X.; Yao, X.Y.; Ai, H.; Zhang, Y.J.; Gan, Z.C.; Huang, X.Z. Genome-Wide Identification, Characterization and Expression Profiling of the CONSTANS-like Genes in Potato (Solanum tuberosum L.). Genes 2023, 14, 1174. [Google Scholar] [CrossRef]
- Endre, G.; Kereszt, A.; Kevei, Z.; Mihacea, S.; Kaló, P.; Kiss, G.B. A receptor kinase gene regulating symbiotic nodule development. Nature 2002, 417, 962–966. [Google Scholar] [CrossRef] [PubMed]
- Stacey, G.; Libault, M.; Brechenmacher, L.; Wan, J.; May, G.D. Genetics and functional genomics of legume nodulation. Curr. Opin. Plant Biol. 2006, 9, 110–121. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Reads No. | Clean Reads No. | Q30 (%) | Total Mapped (%) | Multiple Mapped (%) | Uniquely Mapped (%) |
|---|---|---|---|---|---|---|
| A_1 | 45425876 | 45101984 | 94.84 | 92.87% | 4.00% | 96.00% |
| A_2 | 44676462 | 44323034 | 94.58 | 93.74% | 4.46% | 95.54% |
| A_3 | 46069654 | 45525622 | 93.61 | 91.77% | 4.31% | 95.69% |
| B_1 | 42730730 | 42144712 | 93.01 | 91.69% | 15.13% | 94.87% |
| B_2 | 46843152 | 46205678 | 93.06 | 91.11% | 3.31% | 96.69% |
| B_3 | 42296358 | 41995038 | 94.49 | 92.05% | 3.81% | 96.19% |
| C_1 | 43099064 | 42532114 | 93.16 | 90.75% | 4.20% | 95.80% |
| C_2 | 44530066 | 44066860 | 94.06 | 90.39% | 3.74% | 96.26% |
| C_3 | 44945554 | 44437312 | 93.76 | 90.85% | 3.99% | 96.01% |
| D_1 | 46751856 | 46319452 | 94.13 | 92.00% | 4.03% | 95.97% |
| D_2 | 43762322 | 43326008 | 94.04 | 92.00% | 5.02% | 94.98% |
| D_3 | 52817612 | 52325294 | 93.96 | 91.76% | 3.33% | 96.67% |
| Database | Number | Percentage (%) |
|---|---|---|
| NR | 11,151 | 92.46 |
| GO | 6490 | 53.81 |
| KEGG | 4351 | 36.07 |
| eggNOG | 9780 | 81.09 |
| Swissprot | 8269 | 68.56 |
| In all database | 3436 | 28.49 |
| Functional Classification | Number of Genes/Each | Percentage/% |
|---|---|---|
| RNA processing and modification | 753 | 2.82 |
| Chromatin structure and dynamics | 270 | 1.01 |
| Energy production and conversion | 674 | 2.52 |
| Cell cycle control, cell division, and chromosome partitioning | 296 | 1.11 |
| Amino acid transport and metabolism | 851 | 3.18 |
| Nucleotide transport and metabolism | 207 | 0.77 |
| Carbohydrate transport and metabolism | 1312 | 4.91 |
| Coenzyme transport and metabolism | 275 | 1.03 |
| Lipid transport and metabolism | 618 | 2.31 |
| Translation, ribosomal structure, and biogenesis | 998 | 3.73 |
| Transcription | 2194 | 8.20 |
| Replication, recombination, and repair | 1236 | 4.62 |
| Cell wall/membrane/envelope biogenesis | 183 | 0.68 |
| Post-translational modification, protein turnover, and chaperones | 2123 | 7.94 |
| Inorganic ion transport and metabolism | 608 | 2.27 |
| Secondary metabolites biosynthesis, transport, and catabolism | 1126 | 4.21 |
| Function unknown | 9008 | 33.68 |
| Signal transduction mechanisms | 2669 | 9.98 |
| Intracellular trafficking, secretion, and vesicular transport | 794 | 2.97 |
| Defense mechanisms | 302 | 1.13 |
| Extracellular structures | 8 | 0.03 |
| Nuclear structure | 7 | 0.03 |
| Cytoskeleton | 233 | 0.87 |
| Gene | Unigene Code | FPKM | Gene Expression Model | |||||
|---|---|---|---|---|---|---|---|---|
| A Branching Stage | B Bud Stage | C First Flowering Stage | D Blooming Stage | A vs. B | B vs. C | C vs. D | ||
| GI | gene-LOC11410562 | 22.45 | 52.22 | 1.45 | 27.22 | Up | Down | Up |
| CO | gene-LOC11435974 | 0.19 | 1.17 | 4.61 | 0.43 | Up | - | Down |
| PHYA | gene-LOC25500742 | 12.89 | 26.25 | 6.01 | 12.42 | - | Down | Up |
| PHYB | gene-LOC11420025 | 12.71 | 11.89 | 16.79 | 13.27 | - | - | - |
| CRY1 | gene-LOC11428875 | 138.89 | 162.06 | 165.35 | 134.21 | - | - | - |
| CRY2 | gene-LOC25484452 | 51.70 | 69.89 | 80.66 | 56.78 | - | - | - |
| TOC1 | gene-LOC11422615 | 32.28 | 56.06 | 66.98 | 33.86 | Up | - | - |
| ELF3 | gene-LOC11431402 | 1.47 | 2.93 | 16.78 | 1.20 | - | Up | Down |
| LHY | gene-LOC11432385 | 469.08 | 192.19 | 806.17 | 415.10 | Down | Up | - |
| Col2 | gene-LOC25497637 | 129.53 | 122.78 | 174.20 | 134.80 | - | - | - |
| Col13 | gene-LOC11434778 | 22.92 | 31.47 | 56.71 | 17.66 | - | - | Down |
| Col4 | gene-LOC11431452 | 169.85 | 104.34 | 163.58 | 146.27 | - | - | - |
| Col5 | gene-LOC11425462 | 131.48 | 191.78 | 251.04 | 195.10 | - | - | - |
| Col6 | gene-LOC25498015 | 20.00 | 17.68 | 5.79 | 16.2 | - | Down | Up |
| Col9 | gene-LOC11415514 | 3.25 | 2.32 | 57.72 | 2.85 | - | Up | Down |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Luo, W.; Li, Y.; Ma, P.; Chen, C.; Yang, L.; He, D.; Wang, X. Transcriptomic Analysis of Medicago truncatula under Long-Day Conditions. Diversity 2023, 15, 1020. https://doi.org/10.3390/d15091020
Luo W, Li Y, Ma P, Chen C, Yang L, He D, Wang X. Transcriptomic Analysis of Medicago truncatula under Long-Day Conditions. Diversity. 2023; 15(9):1020. https://doi.org/10.3390/d15091020
Chicago/Turabian StyleLuo, Wenju, Yajiao Li, Peijie Ma, Caijun Chen, Longjiao Yang, Dengming He, and Xiaoli Wang. 2023. "Transcriptomic Analysis of Medicago truncatula under Long-Day Conditions" Diversity 15, no. 9: 1020. https://doi.org/10.3390/d15091020
APA StyleLuo, W., Li, Y., Ma, P., Chen, C., Yang, L., He, D., & Wang, X. (2023). Transcriptomic Analysis of Medicago truncatula under Long-Day Conditions. Diversity, 15(9), 1020. https://doi.org/10.3390/d15091020