
Gamete Recognition Gene Divergence Yields a Robust Eutherian Phylogeny across Taxonomic Levels

 , , and
, , and
Abstract
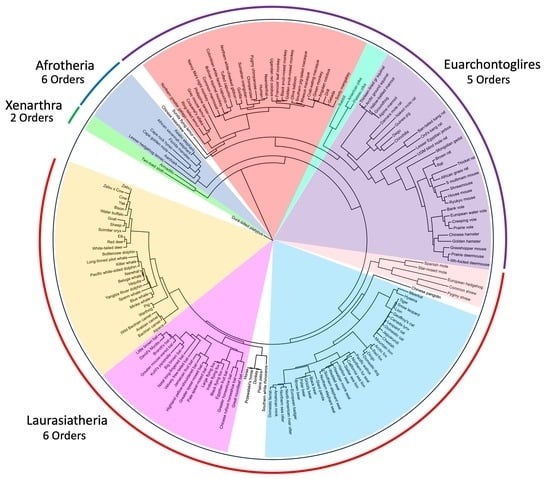
1. Introduction
2. Materials and Methods
2.1. Phylogenetic Analysis of Zan
2.2. Testing the Phylogenetic Utility of Zan
2.3. Tanglegram Comparisons
2.4. Topological Statistical Comparisons
2.5. Divergence/Selection Comparisons
3. Results


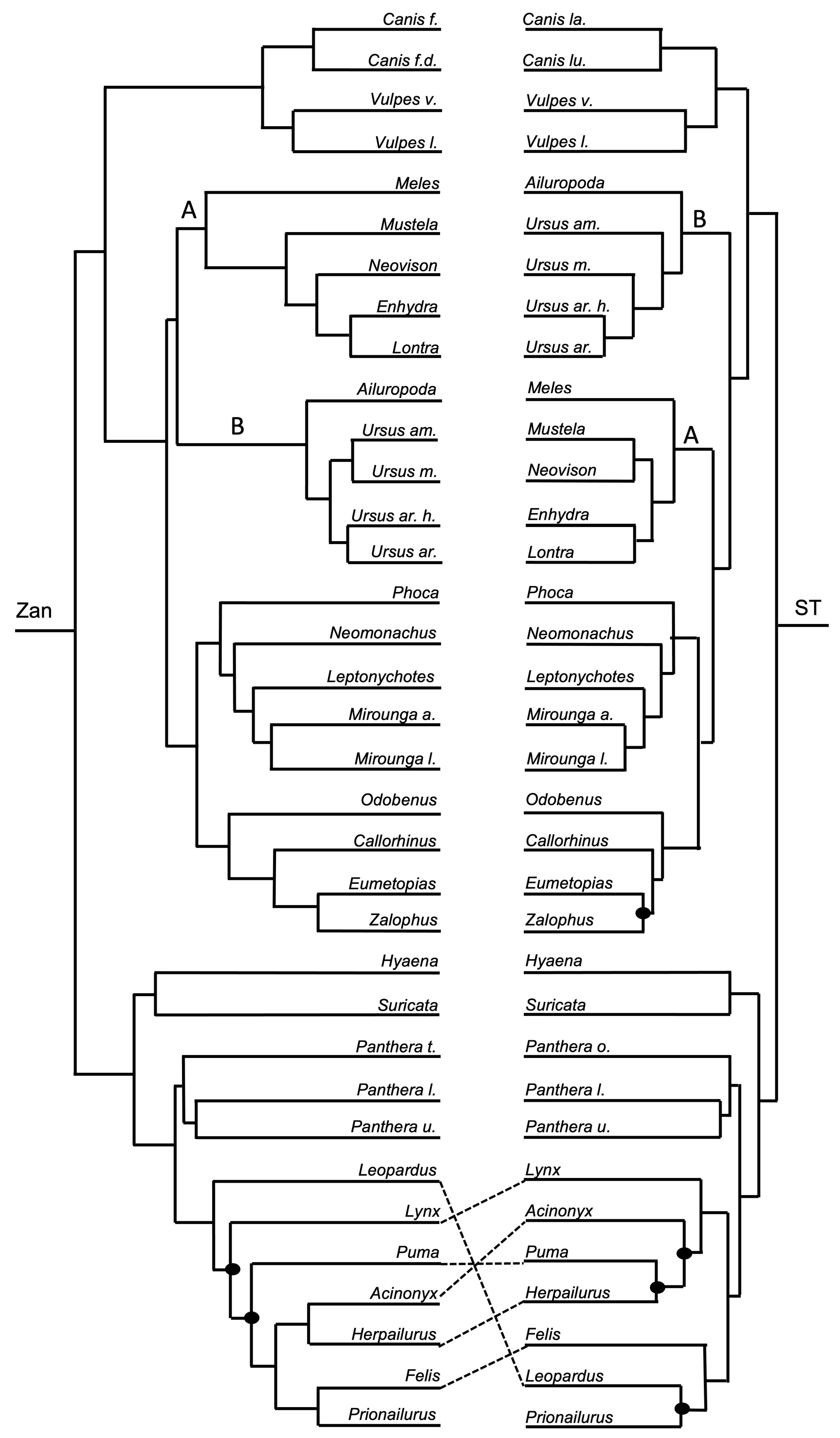
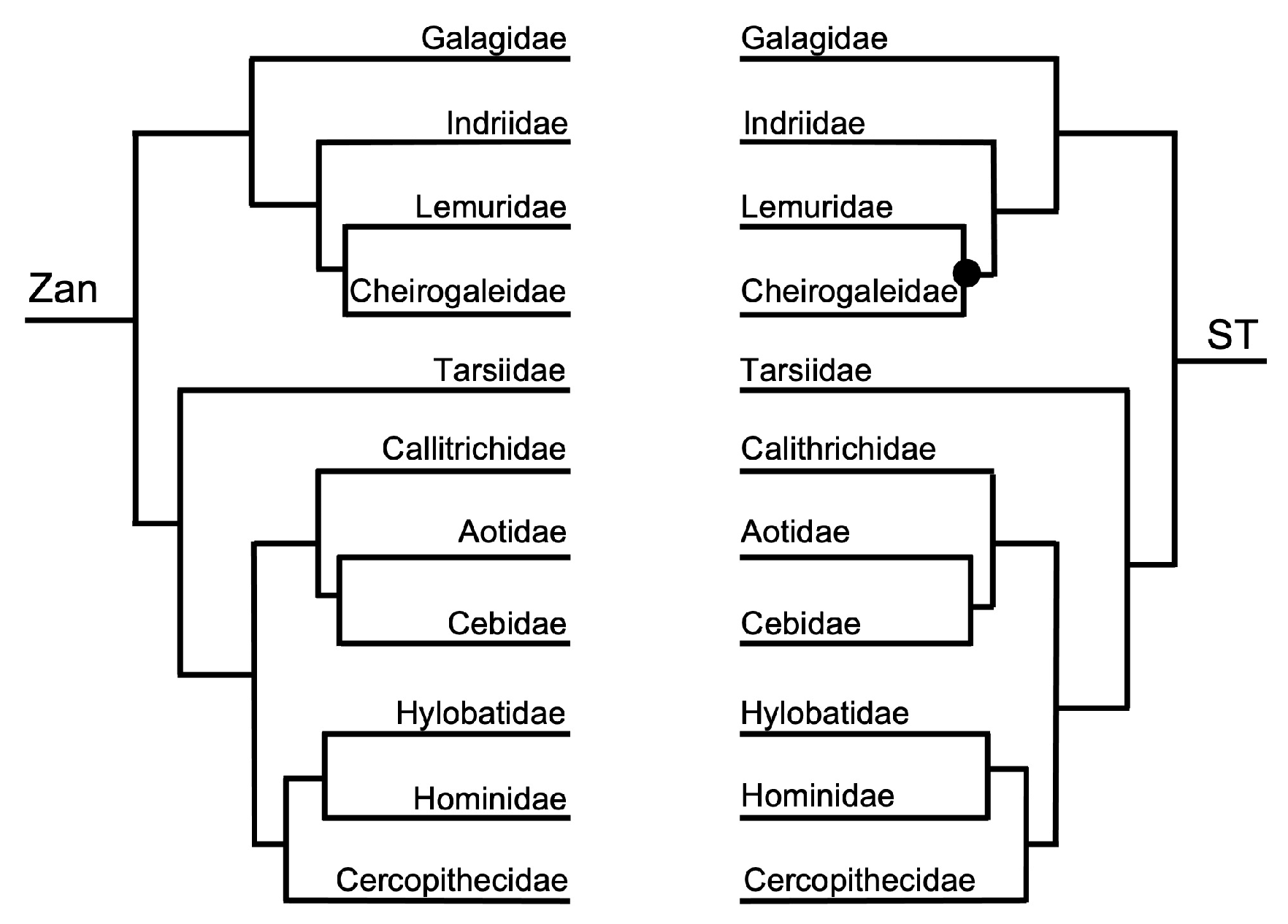
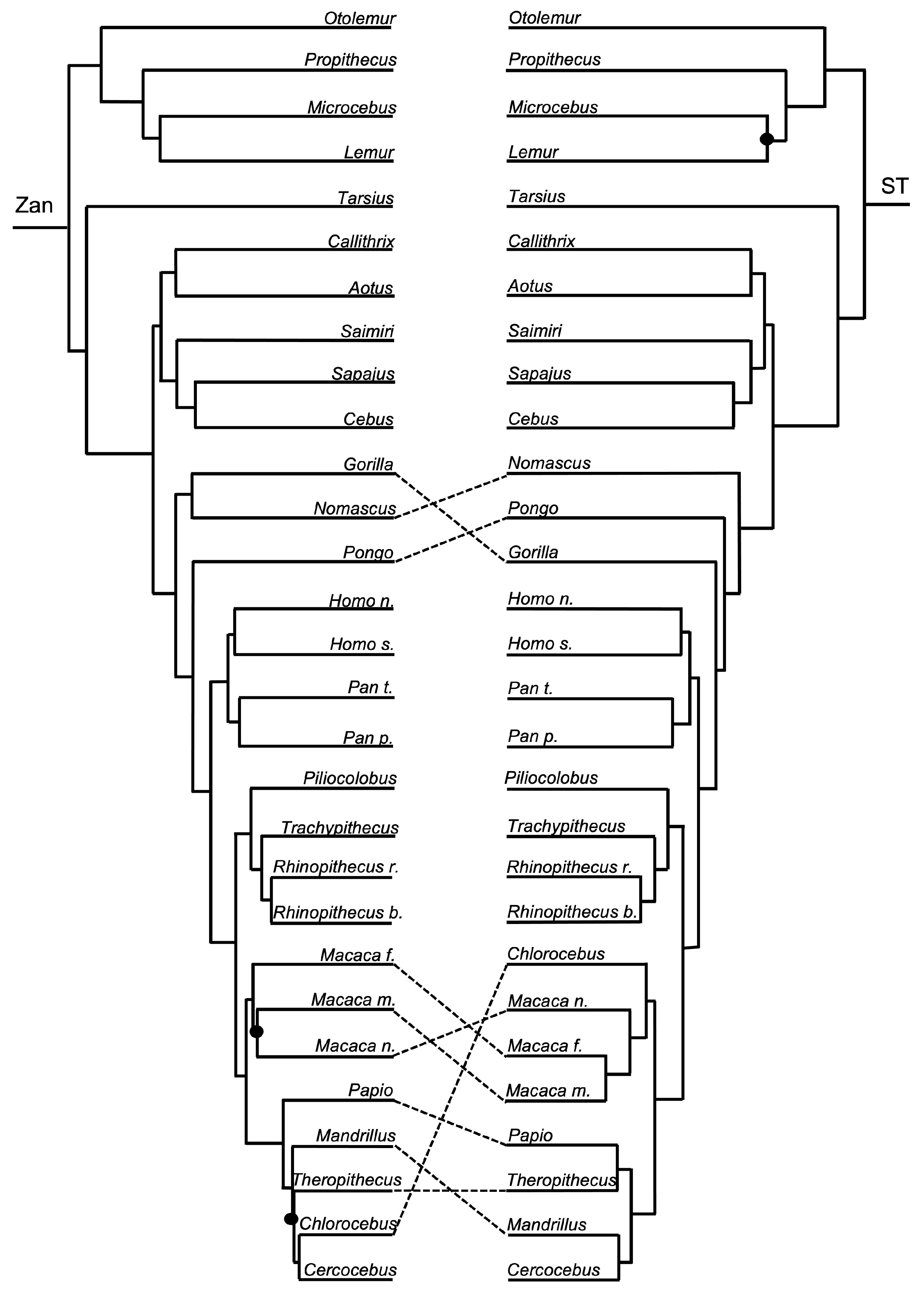
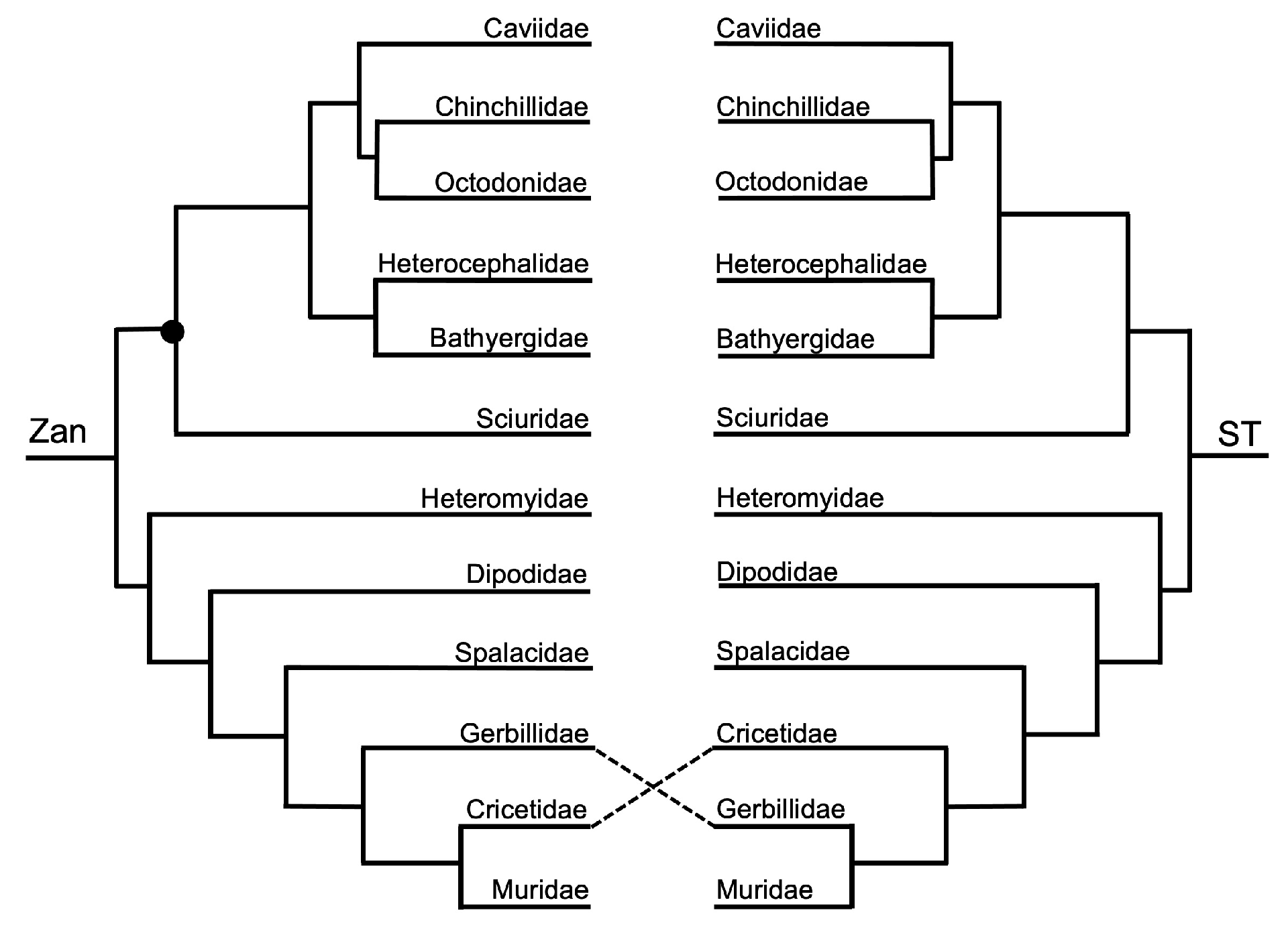
3.1. Tanglegram Comparisons
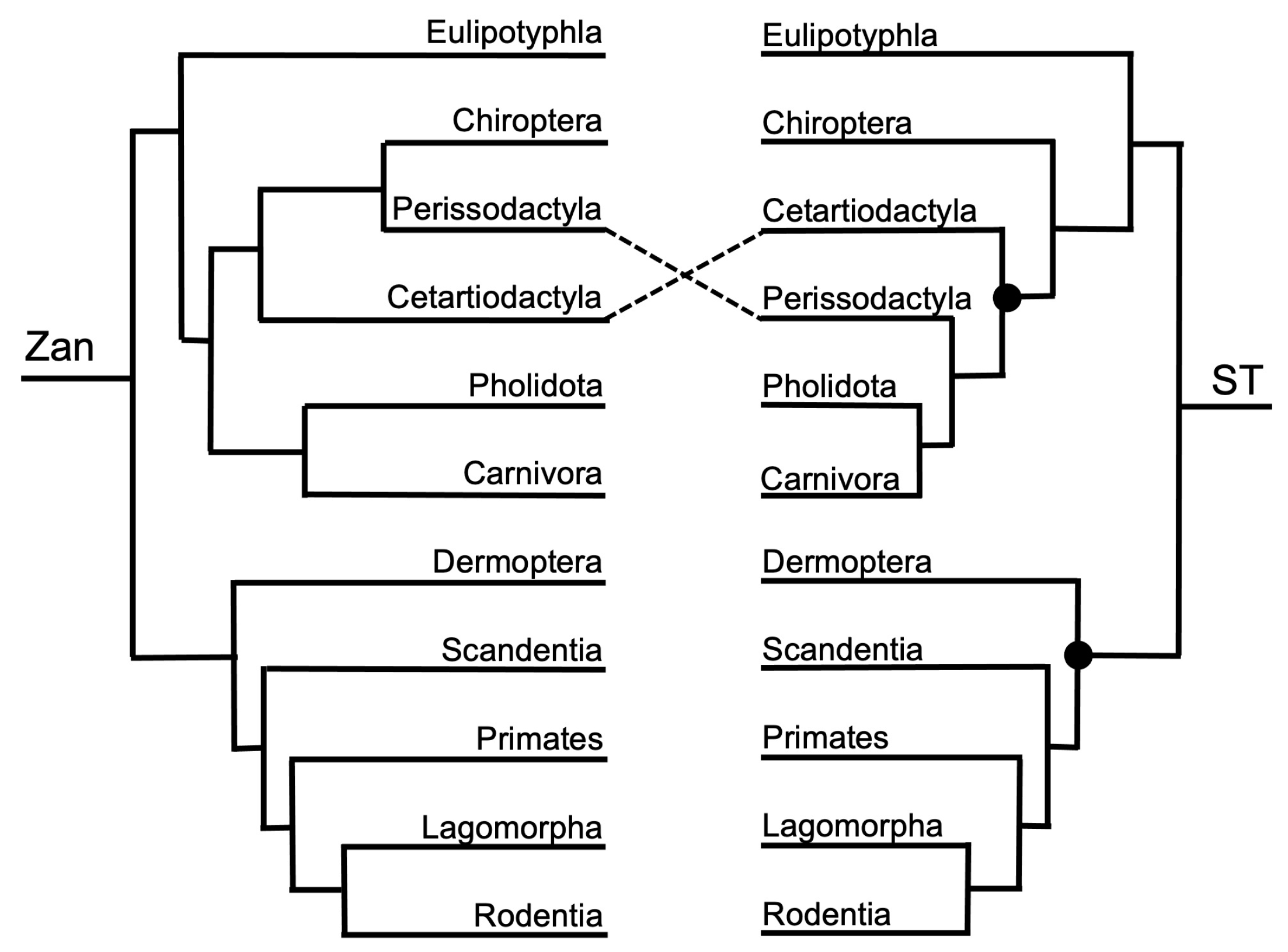
3.1.1. Magnordinal- and Superordinal-Levels
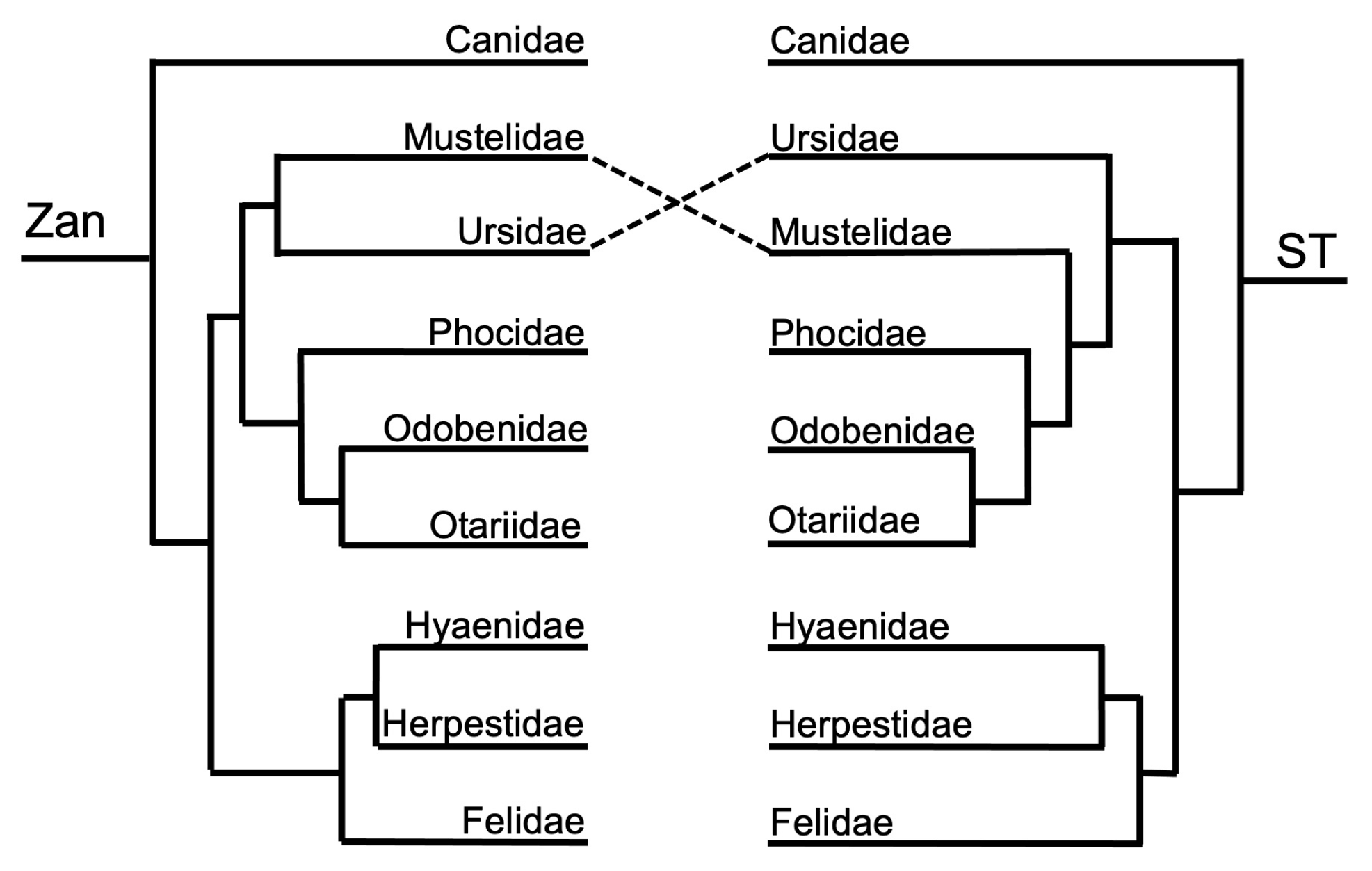
3.1.2. Ordinal-Level
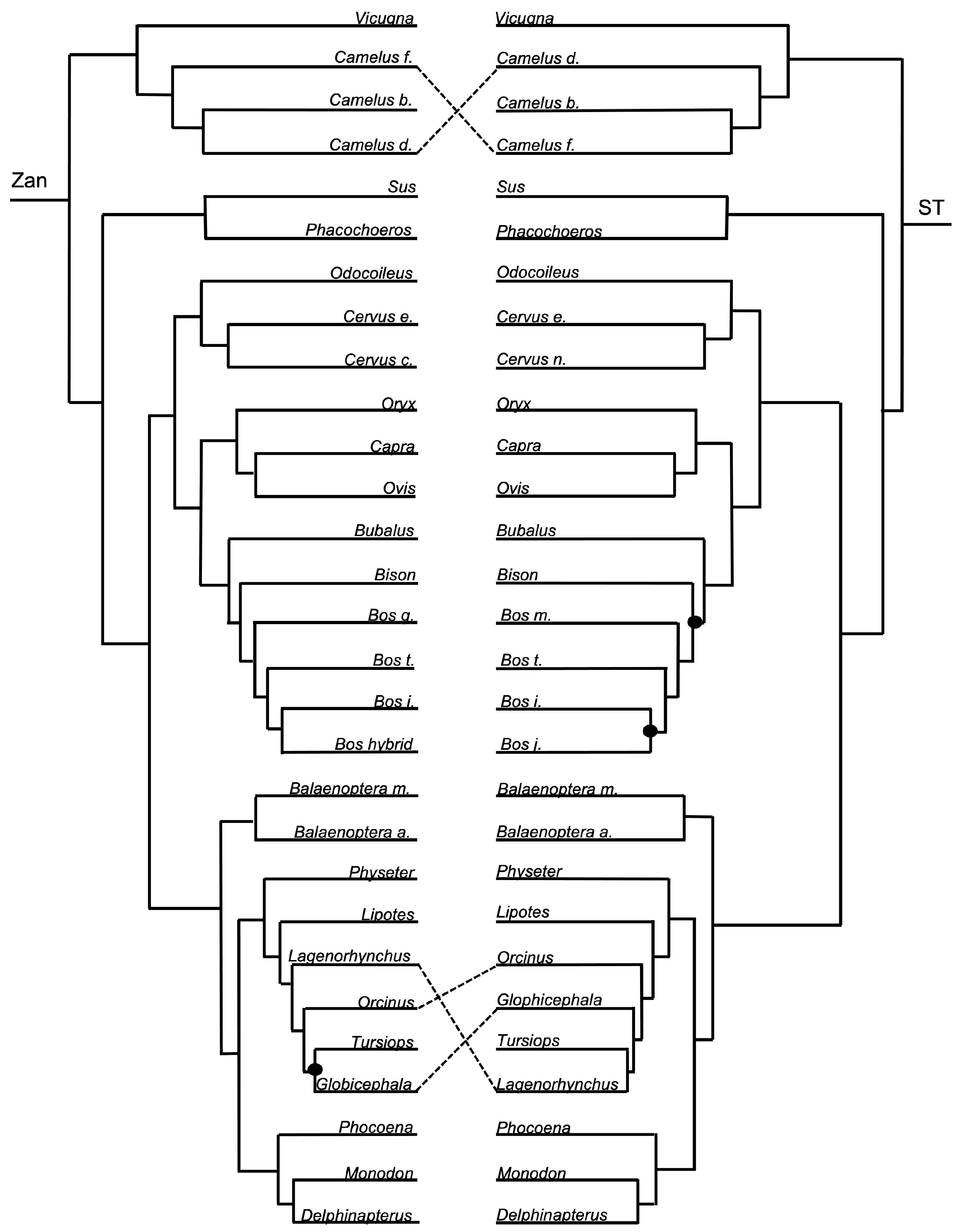
3.1.3. Intra-Ordinal Level
3.2. Topology Statistical Comparisons
3.3. Divergence/Selection Comparisons
4. Discussion
4.1. Tanglegram Comparisons
4.1.1. Inter-Ordinal
4.1.2. Intra-Ordinal
4.1.3. Resolving Controversial Relationships
4.1.4. Zan as a Speciation Gene Phylogenetic Marker
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Meredith, R.W.; Janecka, J.E.; Gatesy, J.; Ryder, O.A.; Fisher, C.A.; Teeling, E.C.; Goodbla, A.; Eizirik, E.; Simão, T.L.L.; Stadler, T.; et al. Impacts of the Cretaceous Terrestrial Revolution and KPg extinction on mammal diversification. Science 2011, 334, 521–524. [Google Scholar] [CrossRef]
- dos Reis, M.; Inoue, J.; Hasegawa, M.; Asher, R.J.; Donoghue, P.C.; Yang, Z. Phylogenomic datasets provide both precision and accuracy in estimating the timescale of placental mammal phylogeny. Proc. R. Soc. B 2012, 279, 3491–3500. [Google Scholar] [CrossRef]
- Song, S.; Liu, L.; Edwards, S.V.; Wu, S. Resolving conflict in eutherian mammal phylogeny using phylogenomics and the multispecies coalescent model. Proc. Natl. Acad. Sci. USA 2012, 109, 14942–14947. [Google Scholar] [CrossRef]
- O’Leary, M.A.; Bloch, J.I.; Flynn, J.J.; Gaudin, T.J.; Giallombardo, A.; Giannini, N.P.; Goldberg, S.L.; Kraatz, B.P.; Luo, Z.; Meng, J.; et al. The placental mammal ancestor and the post-KPg radiation of placentals. Science 2013, 339, 662–667. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Meredith, R.W.; Teeling, E.C.; Murphy, W.J. Technical comment on ‘The placental mammal ancestor and the post-KPg radiation of placentals’. Science 2013, 341, 613. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Gatesy, J. The gene tree delusion. Mol. Phy. Evol. 2016, 94, 1–33. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G. The Principles of Classification and a Classification of Mammals; American Museum of Natural History: New York, NY, USA, 1945; Volume 85. [Google Scholar]
- Shoshani, J.; McKenna, M.C. Higher Taxonomic Relationships among Extant Mammals Based on Morphology, with Selected Comparisons of Results from Molecular Data. Mol. Phy. Evol. 1998, 9, 572–584. [Google Scholar] [CrossRef]
- Novacek, M.J.; Wyss, A.R.; McKenna, M.C. The major groups of Eutherian mammals. In The Phylogeny and Classification of the Tetrapods; Benton, M.J., Ed.; Clarendon Press: Oxford, UK, 1988; Volume 2, pp. 31–71. [Google Scholar]
- Novacek, M.J. Higher mammal phylogeny: The morphological-molecular synthesis. In The Hierarchy of Life; Fernholm, B., Bremer, K., Jornvall, H., Eds.; Elsevier: Amsterdam, The Netherlands, 1989; pp. 421–435. [Google Scholar]
- Novacek, M.J. Mammalian phylogeny: Shaking the tree. Nature 1992, 356, 121–125. [Google Scholar] [CrossRef]
- MacPhee, R.D.E.; Novacek, M.J. Definition and relationships of Lipotyphla. In Mammal Phylogeny: Placentals; Szalay, F.S., Novacek, M.J., McKenna, M.C., Eds.; Springer: New York, NY, USA, 1993; pp. 13–31. [Google Scholar]
- Gaudin, T.J.; Wible, J.R.; Hopson, J.A.; Turnbull, W.D. Reexamination of the morphological evidence for the cohort Epitheria (Mammalia, Eutheria). J. Mamm. Evol. 1996, 3, 31–79. [Google Scholar] [CrossRef]
- Madsen, O.; Scally, M.; Douady, C.J.; Kao, D.J.; DeBry, R.W.; Adkins, R.; Amrine, H.M.; Stanhope, M.J.; de Jong, W.W.; Springer, M.S. Parallel adaptive radiations in two major clades of placental mammals. Nature 2001, 409, 610–614. [Google Scholar] [CrossRef]
- Murphy, W.J.; Eizirik, E.; Johnson, W.E.; Zhang, Y.P.; Ryder, O.A.; O’Brien, S.J. Molecular phylogenetics and the origins of placental mammals. Nature 2001, 409, 614–618. [Google Scholar] [CrossRef]
- Springer, M.S.; Cleven, G.C.; Madsen, O.; de Jong, W.W.; Waddell, V.G.; Amrine, H.M.; Stanhope, M.J. Endemic African mammals shake the phylogenetic tree. Nature 1997, 388, 61–64. [Google Scholar] [CrossRef] [PubMed]
- De Jong, W.W.; van Dijk, M.A.M.; Poux, C.; Kappé, G.; van Rheede, T.; Madsen, O. Indels in protein-coding sequences of Euarchontoglires constrain the rooting of the eutherian tree. Mol. Phy. Evol. 2003, 28, 328–340. [Google Scholar] [CrossRef] [PubMed]
- Stanhope, M.J.; Waddell, V.G.; Madsen, O.; de Jong, W.W.; Hedges, S.B.; Cleven, G.C.; Kao, D.; Springer, M.S. Molecular evidence for multiple origins of Insectivora and for a new order of endemic African insectivore mammals. Proc. Natl. Acad. Sci. USA 1998, 95, 9967–9972. [Google Scholar] [CrossRef] [PubMed]
- Waddell, P.J.; Okada, N.; Hasegawa, M. Towards resolving the interordinal relationships of placental mammals. Syst. Biol. 1999, 48, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Amrine-Madsen, H.; Koepfli, K.P.; Wayne, R.K.; Springer, M.S. A new phylogenetic marker, apoliprotein B, provides compelling evidence for eutherian relationships. Mol. Phylogenet. Evol. 2003, 28, 225–240. [Google Scholar] [CrossRef]
- Nishihara, H.; Hasegawa, M.; Okada, N. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc. Natl. Acad. Sci. USA 2006, 103, 9929–9934. [Google Scholar] [CrossRef]
- Hallstrom, B.; Kullberg, M.; Nilsson, M.A.; Janke, A. Phylogenomic data analyses provide evidence that Xenarthra and Afrotheria are sister groups. Mol. Biol. Evol. 2007, 24, 2059–2068. [Google Scholar] [CrossRef]
- Wildman, D.E.; Uddin, M.; Opazo, J.C.; Liu, G.; Lefort, V.; Guindon, S.; Gascuel, O.; Grossman, L.I.; Romero, R.; Goodman, M. Genomics, biogeography, and the diversification of placental mammals. Proc. Natl. Acad. Sci. USA 2007, 104, 14395–14400. [Google Scholar] [CrossRef]
- Asher, R.J.; Bennett, N.; Lehmann, T. The new framework for understanding placental mammal evolution. Bioessays 2009, 31, 853–864. [Google Scholar] [CrossRef]
- Liu, F.G.R.; Miyamoto, M.M.; Freire, N.P.; Ong, P.Q.; Tennant, M.R.; Young, T.S.; Gugel, K.F. Molecular and morphological supertrees for eutherian (placental) mammals. Science 2001, 291, 1786–1789. [Google Scholar] [CrossRef]
- Beck, R.M.D.; Bininda-Emonds, O.R.P.; Cardillo, M.; Liu, F.; Purvis, A. A higher-level MRP supertree of placental mammals. BMC Evol. Biol. 2006, 6, 93. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.P.; Cardillo, M.; Jones, K.E.; MacPhee, R.D.E.; Beck, R.M.D.; Grenyer, R.; Price, S.A.; Vos, R.A.; Gittleman, J.L.; Purvis, A. The delayed rise of present-day mammals. Nature 2007, 446, 507–512. [Google Scholar] [CrossRef]
- Upham, N.S.; Esselstyn, J.A.; Jetz, W. Inferring the mammal tree: Species-level sets of phylogenies for questions in ecology, evolution, and conservation. PLoS Biol. 2019, 17, e3000494. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Pevzner, P.A.; O’Brien, S.J. Mammalian phylogenomics comes of age. Tren. Genet. 2004, 20, 631–639. [Google Scholar] [CrossRef] [PubMed]
- Fabre, P.; Hautier, L.; Dimitrov, D.; Douzery, E.J.P. A glimpse on the pattern of rodent diversification a phylogenetic approach. BMC Evol. Biol. 2012, 12, 88. [Google Scholar] [CrossRef] [PubMed]
- Halliday, T.J.D.; Upchurch, P.; Goswami, A. Resolving the relationships of Paleocene placental mammals. Biol. Rev. 2015, 92, 521–550. [Google Scholar] [CrossRef] [PubMed]
- Springer, M.S.; Stanhope, M.J.; Madsen, O.; de Jong, W.W. Molecules consolidate the placental mammal tree. Tren. Ecol. Evol. 2004, 19, 430–438. [Google Scholar] [CrossRef]
- Springer, M.S.; Murphy, W.J.; Eizirik, E.; Madsen, O.; Scally, M.; Douady, C.J.; Teeling, E.C.; Stanhope, M.J.; de Jong, W.W.; O’Brien, S.J. A molecular classification for the living orders of placental mammals and the phylogenetic placement of primates. In Primate Origins: Adaptations and Evolution; Springer: Boston, MA, USA, 2007; pp. 1–28. [Google Scholar]
- Huelsenbeck, J.P.; Bull, J.J.; Cunningham, C.W. Combining data in phylogenetic analysis. Tren. Ecol. Evol. 1996, 11, 152–158. [Google Scholar] [CrossRef]
- Paradis, E. Analysis of diversification: Combining phylogenetic and taxonomic data. Proc. R. Soc. Lond. B 2003, 270, 2499–2505. [Google Scholar] [CrossRef]
- Hahn, M.W.; Nakhleh, L. Irrational exuberance for resolved species trees. Evolution 2015, 70, 7–17. [Google Scholar] [CrossRef]
- Stadler, T.; Bokma, F. Estimating speciation and extinction rates for phylogenies of higher taxa. Syst. Biol. 2013, 62, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Murphy, W.J.; Eizirik, E.; O’Brian, S.J.; Madsen, O.; Scally, M.; Douady, C.J.; Teeling, E.; Ryder, O.A.; Stanhope, M.J.; de Jong, W.E.; et al. Resolution of the Early Placental Mammalian Radiation Using Bayesian Phylogenetics. Science 2001, 294, 2348–2351. [Google Scholar] [CrossRef] [PubMed]
- Scally, M.; Madsen, O.; Douady, C.J.; de Jong, W.W.; Stanhope, M.J.; Springer, M.S. Molecular evidence for the major clades of placental mammals. J. Mamm. Evol. 2001, 8, 239–277. [Google Scholar] [CrossRef]
- Delsuc, F.; Scally, M.; Madsen, O.; Stanhope, M.J.; de Jong, W.W.; Catzeflis, F.M.; Springer, M.S.; Douzery, E.J.P. Molecular phylogeny of living Xenarthrans and the impact of character and taxon sampling on the placental tree rooting. Mol. Biol. Evol. 2002, 19, 1656–1671. [Google Scholar] [CrossRef][Green Version]
- Springer, M.S.; Murphy, W.J.; Eizirik, E.; O’Brien, S.J. Placental mammal diversification and the Cretaceous-Tertiary boundary. Proc. Natl. Acad. Sci. USA 2003, 100, 1056–1061. [Google Scholar] [CrossRef]
- Springer, M.S.; DeBry, R.W.; Amrine, H.M.; Madsen, O.; de Jong, W.W.; Stanhope, M.J. Mitochondrial versus nuclear gene sequences in deep-level mammalian phylogeny reconstruction. Mol. Biol. Evol. 2001, 18, 132–143. [Google Scholar] [CrossRef]
- Tarver, J.E.; dos Reis, M.; Mirarab, S.; Moran, R.J.; Parker, S.; O’Reilly, J.E.; King, B.L.; O’Connell, M.J.; Asher, R.J.; Warnow, T.; et al. The interrelationships of placental mammals and the limits of phylogenetic inference. Gen. Biol. Evol. 2016, 8, 330–344. [Google Scholar] [CrossRef]
- Foley, N.M.; Springer, M.S.; Teeling, E.C. Mammal madness: Is the mammal tree of life not yet resolved? Phil. Trans. R. Soc. B 2016, 371, 20150140. [Google Scholar] [CrossRef]
- Novacek, M.J. Fossils, topologies, missing data, and the higher level phylogeny of Eutherian mammals. Syst. Biol. 1992, 41, 58–73. [Google Scholar] [CrossRef]
- Swanson, W.J.; Yang, Z.; Wolfner, M.F.; Aquadro, C.F. Positive Darwinian selection drives the evolution of several female reproductive proteins in mammals. Proc. Natl. Acad. Sci. USA 2001, 98, 2509–2514. [Google Scholar] [CrossRef]
- Torgerson, D.G.; Kulathinal, R.J.; Singh, R.S. Mammalian Sperm Proteins Are Rapidly Evolving: Evidence of Positive Selection in Functionally Diverse Genes. Mol. Biol. Evol. 2002, 19, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef]
- Resch, A.M.; Carmel, L.; Mariño-Ramírez, L.; Ogurtsoc, A.Y.; Shabalina, S.A.; Rogozin, I.B. Widespread Positive Selection in Synonymous Sites of Mammalian Genes. Mol. Biol. Evol. 2007, 24, 1821–1831. [Google Scholar] [CrossRef] [PubMed]
- Massey, S.E.; Churbanov, A.; Tastogi, S.; Liberles, D.A. Characterizing positive and negative selection and their phylogenetic effects. Gene 2008, 418, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Koisol, C.; Vinař, T.; da Fonseca, R.R.; Hubisz, M.J.; Bustamante, C.D.; Nielsen, R.; Siepel, A. Patterns of Positive Selection in Six Mammalian Genomes. PLoS Genet. 2008, 4, e1000144. [Google Scholar] [CrossRef]
- Pollard, K.S.; Hubisz, M.J.; Rosembloom, K.R.; Siepel, A. Detection of nonneutral substitution rates on mammalian phylogenies. Gen. Res. 2010, 20, 110–121. [Google Scholar] [CrossRef]
- McCandlish, D.M.; Stoltzfus, A.; Dykhuizen, D.E. Modeling Evolution Using the Probability of Fixation: History and Implications. Quart. Rev. Biol. 2014, 89, 225–252. [Google Scholar] [CrossRef]
- Young, A.D.; Gillung, J.P. Phylogenomics—Principles, opportunities, and pitfalls of big-data phylogenetics. Syst. Entom. 2019, 45, 225–247. [Google Scholar] [CrossRef]
- Molloy, E.K.; Warnow, T. To Include or Not to Include: The Impact of Gene Filtering on Species Tree Estimation Methods. Syst. Biol. 2017, 67, 285–303. [Google Scholar] [CrossRef]
- Latrille, T.; Rodrigue, N.; Lartillot, N. Genes and sites under adaptation at the phylogenetic scale also exhibit adaptation at the population-genetic scale. Proc. Natl. Acad. Sci. USA 2023, 120, e2214977120. [Google Scholar] [CrossRef] [PubMed]
- Yi, S. Neutrality and Molecular Clocks. Nat. Educ. Knowl. 2013, 4, 3. [Google Scholar]
- Ting, C.T.; Takahashi, A.; Wu, C.I. Incipient speciation by sexual isolation in Drosophila: Concurrent evolution at multiple loci. Proc. Natl. Acad. Sci. USA 2001, 98, 6709–6713. [Google Scholar] [CrossRef]
- Wolf, J.B.W.; Lindell, J.; Backstrom, N. Speciation genetics: Current status and evolving approaches. Phil. Trans. R. Soc. Lond. B Biol. Sci. 2010, 365, 1717–1733. [Google Scholar] [CrossRef] [PubMed]
- Phifer-Rixley, M. Searching for the genes that separate species. eLife 2014, 3, e05377. [Google Scholar] [CrossRef] [PubMed]
- Roberts, E.K.; Tardif, S.; Wright, E.A.; Platt II, R.N.; Bradley, R.D.; Hardy, D.M. Rapid divergence of a gamete recognition gene promoted macroevolution of Eutheria. Gen. Biol. 2022, 23, 155. [Google Scholar] [CrossRef]
- Hardy, D.M.; Garbers, D.L. Species-specific binding of sperm proteins to the extracellular matrix (zona pellucida) of the egg. J. Biol. Chem. 1994, 269, 19000–19004. [Google Scholar] [CrossRef]
- Hardy, D.M.; Garbers, D.L. A sperm membrane protein that binds in a species-specific manner to the egg extracellular matrix is homologous to von Willebrand factor. J. Biol. Chem. 1995, 270, 26025–26028. [Google Scholar] [CrossRef]
- Gao, Z.; Garbers, D.L. Species diversity in the structure of zonadhesin, a sperm-specific membrane protein containing multiple cell adhesion molecule-like domains. J. Biol. Chem. 1998, 273, 3415–3421. [Google Scholar] [CrossRef]
- Herlyn, H.; Zischler, H. Tandem repetitive D domains of the sperm ligand zonadhesin evolve faster in the paralogue than in the orthologue comparison. J. Mol. Evol. 2006, 63, 602–611. [Google Scholar] [CrossRef][Green Version]
- Tardif, S.; Wilson, M.D.; Wagner, R.; Hunt, P.; Gertsenstein, M.; Nagy, A.; Lobe, C.; Koop, B.F.; Hardy, D.M. Zonadhesin is essential for species specificity of sperm adhesion to the egg zona pellucida. J. Biol. Chem. 2010, 285, 24863–24870. [Google Scholar] [CrossRef]
- Legan, P.K.; Rau, A.; Kee, J.N.; Richardson, G.P. The mouse tectorins modular matrix proteins of the inner ear homologous to components of the sperm-egg adhesion system. J. Biol. Chem. 1997, 272, 8791–8801. [Google Scholar] [CrossRef] [PubMed]
- Wyckoff, G.J.; Wang, W.; Wu, C.-I. Rapid evolution of male reproductive genes in the descent of man. Nature 2000, 403, 304–309. [Google Scholar] [CrossRef]
- Swanson, W.J.; Vacquier, V.D. The rapid evolution of reproductive proteins. Nat. Rev. Genet. 2002, 3, 137–144. [Google Scholar] [CrossRef]
- Swanson, W.J.; Nielsen, R.; Yang, Q. Pervasive adaptive evolution in mammalian fertilization proteins. Mol. Biol. Evol. 2003, 20, 18–20. [Google Scholar] [CrossRef] [PubMed]
- Dufourny, L.; Levasseur, A.; Martine, M.; Callebaut, I.; Pontarotti, P.; Malpaux, B.; Monget, P. GPR50 is the mammalian ortholog of Mel1c: Evidence of rapid evolution in mammals. BMC Evol. Biol. 2008, 8, 105. [Google Scholar] [CrossRef]
- Raterman, D.; Springer, M.S. The molecular evolution of acrosin in placental mammals. Mol. Reprod. Dev. 2008, 75, 1196–1207. [Google Scholar] [CrossRef]
- Turner, L.M.; Chuong, E.B.; Hoekstra, H.E. Comparative analysis of testis protein evolution in rodents. Genetics 2008, 179, 2075–2089. [Google Scholar] [CrossRef]
- Waddell, L.A.; Lefevre, L.; Bush, S.J.; Raper, A.; Young, R.; Lisowski, Z.M.; McCulloch, M.E.B.; Muriuki, C.; Sauter, K.A.; Clark, E.L.; et al. ADGRE1 (EMR1, F4/80) is a rapidly-evolving gene expressed in mammalian monocyte-macrophages. Front. Immunol. 2008, 9, 2246. [Google Scholar] [CrossRef]
- Oliver, P.L.; Goodstadt, L.; Bayes, J.J.; Birtle, Z.; Roach, K.C.; Phadnis, N.; Beatson, S.A.; Lunter, G.; Malik, H.S.; Ponting, C.P. Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet. 2009, 5, e1000753. [Google Scholar] [CrossRef] [PubMed]
- Finn, S.; Civetta, A. Sexual selection and the molecular evolution of ADAM proteins. J. Mol. Evol. 2010, 71, 231–240. [Google Scholar] [CrossRef] [PubMed]
- Grayson, P.; Civetta, A. Positive selection in the adhesion domain of Mus sperm Adam genes through gene duplications and function-driven gene complex formations. BMC Evol. Biol. 2013, 13, 217. [Google Scholar] [CrossRef] [PubMed]
- Grayson, P. Izumo1 and Juno: The evolutionary origins and coevolution of essential sperm-egg binding partners. R. Soc. Open Sci. 2015, 2, 150296. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, H.Q.; Yao, Y.F.; Liu, W.; Ni, Q.Y.; Zhang, M.W.; Xu, H.L. Uneven evolutionary rate of the melatonin-related receptor gene (GPR50) in primates. Genet. Mol. Res. 2015, 14, 680–690. [Google Scholar] [CrossRef]
- Goodwin, Z.A.; de Guzman, S.C. Recent positive selection in genes of the mammalian epidermal differentiation complex locus. Front. Genet. 2017, 7, 227. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Campbell, M.C.; Li, N.; Lee, D.S.W.; Zhang, Z.; Townsend, J.P. Detection of regional variation in selection intensity within protein-coding genes using DNA sequence polymorphism and divergence. Mol. Biol. Evol. 2017, 34, 3006–3022. [Google Scholar] [CrossRef] [PubMed]
- Cantsilieris, S.; Nelson, B.J.; Huddleston, J.; Baker, C.; Harshman, L.; Penewit, K.; Munson, K.M.; Sorenson, M.; Welch, A.E.; Dang, V.; et al. Recurrent structural variation, clustered sites of selection, and disease risk for the complement factor H (CFH) gene family. Proc. Natl. Acad. Sci. USA 2018, 115, E4433–E4442. [Google Scholar] [CrossRef]
- Moros-Nicolás, C.; Fouchécourt, S.; Goudet, G.; Monget, P. Genes encoding mammalian oviductal proteins involved in fertilization are subjected to gene death and positive selection. J. Mol. Evol. 2018, 86, 655–667. [Google Scholar] [CrossRef]
- Campbell, V.; Lapointe, F. An application of supertree methods to Mammalian mitogenomic sequences. Evol. Bioinform. Online 2010, 6, 57–71. [Google Scholar] [CrossRef]
- Scornavacca, C.; Zickmann, F.; Huson, D.H. Tanglegrams for rooted phylogenetic trees and networks. Bioinformatics 2011, 27, i248–i256. [Google Scholar] [CrossRef]
- Venkatachalam, B.; Gusfield, D. Generalizing Tanglegrams. 2018. Available online: https://scholarship.org/uc/item/0bg5p8ch (accessed on 3 October 2023).
- Shimodaira, H. An Approximately Unbiased Test of Phylogenetic Tree Selection. Syst. Biol. 2002, 51, 492–508. [Google Scholar] [CrossRef]
- Webb, C.O.; Donoghue, M.J. Phylomatic: Tree assembly for applied phylogenetics. Mol. Ecol. Notes 2005, 5, 181–183. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*: Phylogenetic Analysis Using Parsimony, Version 4.0 b10. 2003. Available online: http://phylosolutions.com/paup-test/ (accessed on 11 October 2023).
- Xu, B.; Yang, Z. PAMLX: A graphical user interface for PAML. Mol. Biol. Evol. 2013, 30, 2723–2724. [Google Scholar] [CrossRef]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef]
- Rose, K.D.; Archibald, J.D. The Rise of Placental Mammals: Origins and Relationships of the Major Extant Clades; JHU Press: Baltimore, MD, USA, 2005; p. 65. [Google Scholar]
- Seiffert, E.; Guillon, J.M. A new estimate of afrotherian phylogeny based on simultaneous analysis of genomic, morphological, and fossil evidence. BMC Evol. Biol. 2007, 7, 13. [Google Scholar] [CrossRef] [PubMed]
- Rose, K.D. The Beginning of the Age of Mammals; JHU Press: Baltimore, MD, USA, 2006; pp. 242–243. [Google Scholar]
- Cooper, L.N.; Seiffert, E.R.; Clementz, M.; Madar, S.I.; Bajpai, S.; Hussain, S.T.; Thewissen, J.G.M. Anthracobunids from the Middle Eocene of India and Pakistan Are Stem Perissodactyls. PLoS ONE 2014, 9, e109232. [Google Scholar] [CrossRef]
- Zhou, X.; Xu, S.; Xu, J.; Chen, B.; Zhou, K.; Yang, G. Phylogenomic analysis resolves the interordinal relationships and rapid diversification of the Laurasiatherian mammals. Syst. Biol. 2012, 61, 150–164. [Google Scholar] [CrossRef]
- Ursing, B.M.; Arnason, U. Analyses of mitochondrial genomes strongly support a hippopotamus-whale clade. Proc. R. Soc. B 1998, 265, 2251–2255. [Google Scholar] [CrossRef] [PubMed]
- Asher, R.J.; Helgen, K.M. Nomenclature and placental mammal phylogeny. BMC Evol. Biol. 2010, 6, 93. [Google Scholar] [CrossRef] [PubMed]
- Kriegs, J.O.; Churakov, G.; Jurka, J.; Brosius, J.; Schmitz, J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Tren. Genet. 2007, 23, 158–161. [Google Scholar] [CrossRef]
- Meng, J. The osteology of Rhombomylus (Mammalia, Glires): Implications for phylogeny and evolution of Glires. Bull. Am. Mus. Nat. Hist. 2003, 275, 1–247. [Google Scholar] [CrossRef]
- Meng, J.; Wyss, A.R. The morphology of Tribosphenomys (Rodentiaformes, Mammalia): Phylogenetic Implications for Basal Glires. J. Mam. Evol. 2001, 8, 1–71. [Google Scholar] [CrossRef]
- McKenna, M.C.; Bell, S.K. Classification of Mammals: Above the Species Level; Columbia University Press: New York, NY, USA, 1997. [Google Scholar]
- Burgin, C.J.; Colella, J.P.; Kahn, P.L.; Upham, N.S. How many mammal species are there? J. Mamm. 2019, 99, 1–14. [Google Scholar] [CrossRef]
- Teeling, E.C.; Hedges, S.B. Making the impossible possible: Rooting the tree of placental mammals. Mol. Biol. Evol. 2013, 30, 1999–2000. [Google Scholar] [CrossRef]
- Van Dijk, M.A.M.; Paradis, E.; Catzeflis, F.; de Jong, W.W. The Virtues of Gaps: Xenarthran (Edentate) Monophyly Supported by a Unique Deletion in αA-Crystallin. Syst. Biol. 1999, 48, 94–106. [Google Scholar] [CrossRef]
- Waddell, P.J.; Cao, Y.; Hauf, J.; Hasegawa, M. Using novel phylogenetic methods to evaluate mammalian mtDNA, including amino acid-invariant sites-LogDet plus site stripping, to detect internal conflicts in the data, with special reference to the positions of hedgehog, armadillo, and elephant. Syst. Biol. 1999, 48, 31–53. [Google Scholar] [CrossRef]
- Madsen, O.; Deen, P.M.; Pesole, G.; Saccone, C.; de Jong, W.W. Molecular evolution of mammalian aquaporin-2: Further evidence that elephant shrew and aardvark join the paenungulate clade. Mol. Biol. Evol. 1997, 14, 363–371. [Google Scholar] [CrossRef]
- Nikaido, M.; Nishihara, H.; Hukumoto, Y.; Okada, N. Ancient SINEs from African Endemic Mammals. Mol. Biol. Evol. 2003, 20, 522–527. Erratum in Mol. Biol. Evol. 2003, 20, 1181. https://doi.org/10.1093/molbev/msg161. [CrossRef]
- Murphy, W.J.; O’Brien, S. Designing and optimizing comparative anchor primers for comparative gene mapping and phylogenetic inference. Nat. Protoc. 2007, 2, 3022–3030. [Google Scholar] [CrossRef]
- Yang, F.; Alkalaeva, E.Z.; Perelman, P.L.; Pardini, A.T.; Harrison, W.R.; O’Brien, P.M.C.; Fu, B.; Graphodatsky, A.S.; Ferguson-Smith, M.A.; Robinson, T.J. Reciprocal chromosome painting among human, aardvark, and elephant (superorder Afrotheria) reveals the likely eutherian ancestral karyotype. Proc. Natl. Acad. Sci. USA 2003, 100, 1062–1066. [Google Scholar] [CrossRef] [PubMed]
- Fröenicke, L.; Wienberg, J.; Stone, G.; Adams, L.; Stanyon, R. Towards the delineation of the ancestral eutherian genome organization: Comparative genome maps of human and the African elephant (Loxodonta africana) generated by chromosome painting. Proc. R. Soc. Lond. B 2003, 207, 1331–1340. [Google Scholar] [CrossRef] [PubMed]
- Douady, C.J.; Chatelier, P.I.; Madsen, O.; de Jong, W.W.; Catzeflis, F.; Springer, M.S.; Stanhope, M.J. Molecular phylogenetic evidence confirming the Eulipotyphla concept and in support of hedgehogs as the sister group to shrews. Mol. Phylogenet. Evol. 2002, 25, 200–209. [Google Scholar] [CrossRef] [PubMed]
- Puttick, M.N.; Thomas, G.H. Fossils and living taxa agree on patterns of body mass evolution: A case study with Afrotheria. Proc. R. Soc. Lond. B 2015, 282, 20152023. [Google Scholar] [CrossRef]
- Eizirik, E.; Murphy, W.J.; Koepfli, K.; Johnson, W.E.; Dragoo, J.W.; Wayne, R.K.; O’Brien, S.J. Pattern and timing of diversification of the mammalian order Carnivora inferred from multiple nuclear gene sequences. Mol. Phylogenet. Evol. 2010, 56, 49–63. [Google Scholar] [CrossRef] [PubMed]
- Nash, W.G.; Menninger, J.C.; Wienberg, J.; Padilla-Nash, H.M.; O’Brien, S.J. The pattern of phylogenomic evolution of the Canidae. Cytogenet. Cell Genet. 2001, 95, 210–224. [Google Scholar] [CrossRef]
- Van Rheede, T.; Bastiaans, T.; Boone, D.N.; Hedges, S.B.; de Jong, W.W.; Madsen, O. The Platypus Is in Its Place: Nuclear Genes and Indels Confirm the Sister Group Relation of Monotremes and Therians. Mol. Biol. Evol. 2006, 23, 587–597. [Google Scholar] [CrossRef]
- Murphy, W.J.; Pringle, T.H.; Crider, T.A.; Springer, M.S.; Miller, W. Using genomic data to unravel the root of the placental mammal phylogeny. Genom. Res. 2007, 17, 413–421. [Google Scholar] [CrossRef]
- Waters, P.D.; Dobigny, G.; Waddell, P.J.; Robinson, T.J. Evolutionary History of LINE-1 in the Major Clades of Placental Mammals. PLoS ONE. 2007, 2, e158. [Google Scholar] [CrossRef]
- Cao, Y.; Fujiwara, M.; Nikaido, M.; Okada, N.; Hasegawa, M. Interordinal relationships and timescale of Eutherian evolution as inferred from mitochondrial genome data. Gene 2000, 259, 149–158. [Google Scholar] [CrossRef]
- Easteal, S. The pattern of mammalian evolution and the relative rate of molecular evolution. Genetics 1990, 124, 165–173. [Google Scholar] [CrossRef]
- Kullberg, M.; Nilsson, M.A.; Arnason, U.; Harley, E.H.; Janke, A. Housekeeping Genes for Phylogenetic Analysis of Eutherian Relationships. Mol. Biol. Evol. 2006, 23, 1493–1503. [Google Scholar] [CrossRef] [PubMed]
- Huttley, G.A.; Wakefield, M.J.; Easteal, S. Rates of Genome Evolution and Branching Order from Whole Genome Analysis. Mol. Biol. Evol. 2007, 24, 1722–1730. [Google Scholar] [CrossRef]
- Poux, C.; van Rheede, T.; Madsen, O.; de Jong, W.W. Sequence Gaps Join Mice and Men: Phylogenetic Evidence from Deletions in Two Proteins. Mol. Biol. Evol. 2002, 19, 2035–2037. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Thomas, J.W.; Touchman, J.W.; Blakesley, R.W.; Bouffard, G.G.; Beckstrom-Sternberg, S.M.; Margulies, E.H.; Blanchette, M.; Siepel, A.C.; Thomas, P.J.; McDowell, J.C.; et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature 2003, 424, 788–793. [Google Scholar] [CrossRef] [PubMed]
- Kirkness, E.F.; Bafna, V.; Halpern, A.L.; Levy, S.; Remington, K.; Rusch, D.B. The Dog Genome: Survey Sequencing and Comparative Analysis. Science 2003, 301, 1898–1903. [Google Scholar] [CrossRef]
- Esselstyn, J.A.; Oliveros, C.H.; Swanson, M.T.; Faircloth, B.C. Investigating Difficult Nodes in the Placental Mammal Tree with Expanded Taxon Sampling and Thousands of Ultraconserved Elements. Gen. Biol. Evol. 2017, 9, 2308–2321. [Google Scholar] [CrossRef]
- Zhang, L.; Dong, X.; Tian, X.; Lee, M.; Ablaeva, J.; Firsanov, D.; Lee, S.; Maslov, A.Y.; Gladyshev, V.N.; Seluanov, A.; et al. Maintenance of genome sequence integrity in long- and short-lived rodent species. Sci. Adv. 2021, 7, eabj3284. [Google Scholar] [CrossRef]
- Zhou, X.; Sun, F.; Xu, S.; Yang, G.; Li, M. The position of tree shrews in the mammalian tree: Comparing multi-tree analyses with phylogenomic results leaves monophyly of Euarchonta doubtful. Integr. Zool. 2015, 10, 186–198. [Google Scholar] [CrossRef]
- Kumar, V.; Hallstrom, B.M.; Janke, A. Coalescent-based genome analyses resolve the early branches of the Euarchontoglires. PLoS ONE 2013, 8, e60019. [Google Scholar] [CrossRef]
- Misawa, K.; Nei, M. Reanalysis of Murphy et al.’s Data Gives Various Mammalian Phylogenies and Suggests Overcredibility of Bayesian Trees. J. Mol. Evol. 2003, 57, S290–S296. [Google Scholar] [CrossRef]
- Misawa, K.; Nei, A. Revisiting the Glires concept—Phylogenetic analysis of nuclear sequences. Mol. Phy. Evol. 2003, 28, 320–327. [Google Scholar] [CrossRef]
- Roca, A.L.; Bar-Gal, G.K.; Eizirik, E.; Helgen, K.M.; Maria, R.; Springer, M.S.; O’Brien, S.J.; Murphy, W.J. Mesozoic origin for West Indian insectivores. Nature 2004, 429, 649–651. [Google Scholar] [CrossRef]
- Brace, S.; Thomas, J.A.; Dalén, L.; Burger, J.; MacPhee, R.D.E.; Barnes, I.; Turvey, S.T. Evolutionary History of the Nesophontidae, the Last Unplaced Recent Mammal Family. Mol. Biol. Evol. 2016, 33, 3095–3103. [Google Scholar] [CrossRef]
- Springer, M.S.; Teeling, E.C.; Madsen, O.; de Jong, W.W. Integrated fossil and molecular data reconstruct bat echolocation. Proc. Natl. Acad. Sci. USA 2001, 98, 6241–6246. [Google Scholar] [CrossRef] [PubMed]
- Jones, K.E.; Purvis, A.; MacLarnon, A.; Bininda-Emonds, O.R.P.; Simmons, N.B. A phylogenetic supertree of the bats (Mammalia: Chiroptera). Biol. Rev. 2002, 77, 223–259. [Google Scholar] [CrossRef] [PubMed]
- Teeling, E.C.; Springer, M.S.; Madsen, O.; Bates, P.; O’Brien, S.J.; Murphy, W.J. A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 2005, 307, 580–584. [Google Scholar] [CrossRef] [PubMed]
- Tsagkogeorga, G.; Parker, J.; Stupka, E.; Cotton, J.A.; Rossiter, S.J. Phylogenomic analyses elucidate the evolutionary relationships of bats. Curr. Biol. 2013, 23, 2262–2267. [Google Scholar] [CrossRef]
- Simmons, N.B.; Seymour, K.L.; Habersetzer, J.; Gunnell, G.F. Primitive Early Eocene bat from Wyoming and the evolution of flight and echolocation. Nature 2008, 451, 818–821. [Google Scholar] [CrossRef]
- Pumo, D.E.; Finamore, P.S.; Franek, W.R.; Phillips, C.J.; Tarzami, S.; Balzarano, D. Complete mitochondrial genome of a neotropical fruit bat, Artibeus jamaicensis, and a new hypothesis of the relationships of bats to other eutherian mammals. J. Mol. Evol. 1998, 47, 709–717. [Google Scholar] [CrossRef] [PubMed]
- Mouchaty, S.K.; Gullberg, A.; Janke, A.; Arnason, U. The phylogenetic position of the Talpidae within Eutheria based on analysis of complete mitochondrial sequences. Mol. Biol. Evol. 2000, 17, 60–67. [Google Scholar] [CrossRef]
- Nikaido, M.; Kawai, K.; Cao, Y.; Harado, M.; Tomita, S.; Tomita, S.; Okada, N.; Hasegawa, M. Maximum Likelihood Analysis of the Complete Genomes of Eutherians and a Reevaluation of the Phylogeny of Bats and Insectivores. J. Mol. Evol. 2001, 53, 508–516. [Google Scholar] [CrossRef]
- Lapointe, F.J.; Kirsch, J.A.W.; Hutcheon, J.M. Total evidence, consensus, and bat phylogeny: A distance-based approach. Mol. Phylogenet. Evol. 1999, 11, 55–66. [Google Scholar] [CrossRef] [PubMed]
- Van den Bussche, R.A.; Hoofer, S.R. Phylogenetic relationships among recent chiropteran families and the importance of choosing appropriate out-group taxa. J. Mamm. 2004, 85, 321–330. [Google Scholar] [CrossRef]
- Agnarsson, I.; Zanrana-Torrelio, C.M.; Flores-Saldana, N.P.; May-Collado, L.J. A time-calibrated species-level phylogeny of bats (Chiroptera, Mammalia). PLoS Curr. 2011, 3, RRN1212. [Google Scholar] [CrossRef] [PubMed]
- Price, S.A.; Bininda-Emonds, O.R.P.; Gittleman, J.L. A complete phylogeny of the whales, dolphins and even-toed hoofed mammals (Cetartiodactyla). Biol. Rev. 2005, 80, 445–473. [Google Scholar] [CrossRef]
- Bininda-Emonds, O.R.P.; Gittleman, J.L.; Purvis, A. Building large trees by combining phylogenetic information: A complete phylogeny of the extant Carnivora (Mammalia). Biol. Rev. Camb. Philos. Soc. 1999, 74, 143–175. [Google Scholar] [CrossRef]
- Finarelli, J.A.; Goswami, A. The evolution of orbit orientation and encephalization in the Carnivora (Mammalia). J. Anat. 2009, 214, 671–678. [Google Scholar] [CrossRef]
- Flynn, J.J.; Finarelli, J.A.; Zehr, S.; Hsu, J.; Nedbal, M.A. Molecular phylogeny of the Carnivora (Mammalia): Assessing the impact of increased sampling on resolving enigmatic relationships. Syst. Biol. 2005, 54, 317–337. [Google Scholar] [CrossRef]
- Arnason, U.; Gullberg, A.; Janke, A.; Kullberg, M. Mitogenomic analyses of caniform relationships. Mol. Phylogenet. Evol. 2007, 45, 863–874. [Google Scholar] [CrossRef]
- Barycka, E. Evolution and systematics of the feliform Carnivora. Mamm. Biol. 2007, 72, 257–282. [Google Scholar] [CrossRef]
- Werdelin, L.; Yamaguchi, N.; Johnson, W.E.; O’Brien, S.J. Phylogeny and evolution of cats (Felidae). In Biology and Conservation of Wild Felids; MacDonald, D.W., Loveridge, A.J., Eds.; Oxford University Press: Oxford, UK, 2010; pp. 59–82. [Google Scholar]
- Wozencraft, W.C. Order Carnivora. In Mammal Species of the World: A Taxonomic and Geographic Reference, 3rd ed.; Wilson, D.E., Reeder, D.M., Eds.; Johns Hopkins University Press: Baltimore, MD, USA, 2005; pp. 548–559. [Google Scholar]
- Mattern, M.Y.; McLennan, D.A. Phylogeny and Speciation of Felids. Cladistics 2000, 16, 232–253. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Davis, B.W.; Eizirik, E.; Murphy, W.J. Phylogenomic evidence for ancient hybridization in the genomics of living cats (Felidae). Genome Res. 2016, 26, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Nyakatura, K.; Bininda-Emonds, O.R.P. Updating the evolutionary history of Carnivora (Mammalia): A new species-level supertree complete with divergence time estimates. BMC Biol. 2012, 10, 12. [Google Scholar] [CrossRef]
- Zinner, D.; Arnold, M.L.; Roos, C. The strange blood: Natural hybridization in primates. Evol. Anthropol. 2011, 20, 96–103. [Google Scholar] [CrossRef] [PubMed]
- Finstermeier, K.; Zinner, D.; Brameier, M.; Meyer, M.; Kreuz, E.; Hofreiter, M. A Mitogenomic Phylogeny of Living Primates. PLoS ONE 2013, 8, e69504. [Google Scholar] [CrossRef]
- Pozzi, L.; Hodgson, J.A.; Burrell, A.S.; Sterner, K.N.; Raaum, R.L.; Disotell, T.R. Primate phylogenetic relationships and divergence dates inferred from complete mitochondrial genomes. Mol. Phylogenet. Evol. 2014, 75, 165–183. [Google Scholar] [CrossRef]
- Cao, Y.; Adachi, J.; Yano, T.; Hasegawa, M. Phylogenetic place of guinea pigs: No support of the rodent-polyphyly hypothesis from maximum-likelihood analyses of multiple protein sequences. Mol. Biol. Evol. 1994, 11, 593–604. [Google Scholar] [CrossRef]
- D’Erchia, A.; Gissi, C.; Pesole, G.; Saccone, C.; Arnason, U. The guinea-pig is not a rodent. Nature 1996, 381, 597–600. [Google Scholar] [CrossRef]
- Graur, D.; Hide, W.; Li, W. Is the guinea-pig a rodent? Nature 1991, 351, 649–652. [Google Scholar] [CrossRef]
- Huchon, D.; Catzeflis, F.M.; Douzery, E.J.P. Variance of molecular datings, evolution of rodents and the phylogenetic affinities between Ctenodactylidae and Hystricognathi. Proc. R. Soc. Lond. B 2000, 267, 393–402. [Google Scholar] [CrossRef]
- Montgelard, C.; Forty, E.; Arnal, V.; Matthee, C.A. Suprafamilial relationships among Rodentia and the phylogenetic effect of removing fast-evolving nucleotides in mitochondrial, exon and intron fragments. BMC Evol. Biol. 2008, 8, 321. [Google Scholar] [CrossRef] [PubMed]
- Honeycutt, R.L.; Frabotta, L.J.; Rowe, D.L. Rodent evolution, phylogenetics, and biogeography. In Rodent Societies: An Ecological and Evolutionary Perspective; University of Chicago Press: Chicago, IL, USA, 2007; pp. 8–23. [Google Scholar]
- Honeycutt, R.L. Rodents (Rodentia). In The Timetree of Life; OUP: Oxford, UK, 2009; pp. 490–494. [Google Scholar]
- Alhajeri, B.H.; Hunt, P.J.; Steppan, S.J. Molecular systematics of gerbils and deomyines (Rodentia: Gerbillinae, Deomyinae) and a test of desert adaptation in the tympanic bulla. J. Zoolog. Syst. Evol. Res. 2015, 53, 312–330. [Google Scholar] [CrossRef]
- Li, C.L.; Du, X.Y.; Gao, J.; Wang, C.; Guo, H.G.; Dai, F.W.; Sa, X.Y.; An, W.; Chen, Z.W. Phylogenetic analysis of the Mongolian gerbil (Meriones unguiculatus) from China based on mitochondrial genome. Genet. Mol. Res. 2016, 15, gmr.15037703. [Google Scholar] [CrossRef] [PubMed]
- Ruedi, M.; Mayer, F. Molecular Systematics of Bats of the Genus Myotis (Vespertilionidae) Suggests Deterministic Ecomorphological Convergences. Mol. Phylogenet. Evol. 2001, 21, 436–448. [Google Scholar] [CrossRef]
- Arnold, M.L.; Meyer, A. Natural hybridization in primates: One evolutionary mechanism. Zoology 2006, 109, 261–276. [Google Scholar] [CrossRef]
- Ji, R.; Cui, P.; Ding, F.; Geng, J.; Gao, H.; Zhang, H.; Yu, J.; Hu, S.; Meng, H. Monophyletic origin of domestic Bactrian camel (Camelus bactrianus) and its evolutionary relationship with the extant wild camel (Camelus bactrianus ferus). Anim. Genet. 2009, 40, 377–382. [Google Scholar] [CrossRef]
- Mohandesan, E.; Fitak, R.R.; Corander, J.; Yadamsuren, A.; Chuluunbat, B.; Abdelhadi, O.; Raziq, A.; Nagy, P.; Stalder, G.; Walzer, C.; et al. Mitogenome Sequencing in the Genus Camelus Reveals Evidence for Purifying Selection and Long-term Divergence between Wild and Domestic Bactrian Camels. Sci. Rep. 2017, 7, 9970. [Google Scholar] [CrossRef]
- Legesse, Y.W.; Dunn, C.D.; Mauldin, M.R.; Ordoñez-Garza, N.; Rowden, G.R.; Mekasha Gebre, Y.; Kurtu, M.Y.; Ali, S.M.; Whibesilassie, W.D.; Ballou, M.; et al. Morphometric and genetic variation in 8 breeds of Ethiopian camels (Camelus dromedarius). J. Anim. Sci. 2018, 96, 4925–4934. [Google Scholar] [CrossRef]
- Burger, P.A.; Ciani, E.; Faye, B. Old World camels in a modern world—A balancing act between conservation and genetic improvement. Anim. Genet. 2019, 50, 598–612. [Google Scholar] [CrossRef]
- Zeller, U.; Göttert, T. The relations between evolution and domestication reconsidered—Implications for systematics, ecology, and nature conservation. Glob. Ecol. Cons. 2019, 20, e00756. [Google Scholar] [CrossRef]
- Groves, C.P. Systematic relationships in the Bovini (Artiodactyla, Bovidae). J. Zool. Syst. Evol. Res. 1981, 19, 264–278. [Google Scholar] [CrossRef]
- Zhang, Y.; Hu, Y.; Wang, X.; Jiang, Q.; Zhao, H.; Wang, J.; Ju, Z.; Yang, L.; Gao, Y.; Wei, X.; et al. Population Structure and Selection Signatures Underlying High-Altitude Adaptation Inferred From Genome-Wide Copy Number Variations in Chinese Indigenous Cattle. Front. Genet. 2019, 10, 1404. [Google Scholar] [CrossRef] [PubMed]
- D’Elia, G.; Fabre, P.; Lessa, E.P. Rodent systematics in an age of discovery: Recent advances and prospects. J. Mamm. 2019, 100, 852–871. [Google Scholar] [CrossRef]
- Shen, Y.; Liang, L.; Zhu, Z.; Zhou, E.; Irwin, D.M.; Zhang, Y. Adaptive evolution of energy metabolism genes and the origin of flight in bats. Proc. Natl. Acad. Sci. USA 2010, 11, 8666–8671. [Google Scholar] [CrossRef] [PubMed]
- Hecker, N.; Sharma, V.; Hiller, M. Convergent gene losses illuminate metabolic and physiological changes in herbivores and carnivores. Proc. Natl. Acad. Sci. USA 2019, 116, 3036–3041. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y. Diet evolution of carnivorous and herbivorous mammals in Laurasiatheria. BMC Ecol. Evol. 2022, 22, 82. [Google Scholar] [CrossRef]
- Fu, Y. Estimating Mutation Rate and Generation Time from Longitudinal Samples of DNA Sequences. Mol. Biol. Evol. 2001, 18, 620–626. [Google Scholar] [CrossRef][Green Version]
- Okazaki, A.; Yamazaki, S.; Inoue, I.; Ott, J. Population genetics: Past, present, and future. Hum. Genet. 2021, 140, 231–240. [Google Scholar] [CrossRef]
- Springer, M.S.; Burk, A.; Kavanagh, J.R.; Waddell, V.G.; Stanhope, M.J. The interphotoreceptor retinoid binding protein gene in therian mammals: Implications for higher level relationships and evidence for loss of function in the marsupial mole. Proc. Natl. Acad. Sci. USA 1997, 9, 13754–13759. [Google Scholar] [CrossRef]
- Bradley, R.D.; Baker, R.J. A Test of the Genetic Species Concept: Cytochrome-b Sequences and Mammals. J. Mamm. 2001, 82, 960–973. [Google Scholar] [CrossRef]
- Chan, A.H.; Chaisiri, K.; Saralamba, S.; Morand, S.; Thaenkham, U. Assessing the suitability of mitochondrial and nuclear DNA genetic markers for molecular systematics and species identification. Parasites Vectors 2021, 14, 233. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Species | Families | Length (nt) | Gene | Species | Families | Length (nt) |
|---|---|---|---|---|---|---|---|
| Acr | 145 | 60 | 2133 | Spaca6 | 133 | 51 | 2787 |
| Adam2 | 105 | 51 | 2413 | Spam1 | 120 | 50 | 2288 |
| Adam18 | 82 | 41 | 2939 | Spink2 | 147 | 55 | 646 |
| Adam32 | 106 | 47 | 3378 | Sprr4 | 119 | 50 | 414 |
| Adgre1 | 147 | 57 | 6871 | Tas1r2 | 85 | 42 | 2370 |
| C5ar1 | 146 | 59 | 1812 | Tchhl1 | 137 | 54 | 3294 |
| Ccdc54 | 155 | 62 | 1749 | Tcn1 | 132 | 60 | 1833 |
| Ccl1 | 140 | 55 | 465 | Tcte1 | 162 | 65 | 1972 |
| Cfh | 95 | 45 | 4884 | Tecta | 110 | 61 | 4317 |
| Cr2 | 118 | 46 | 8016 | Tectb | 166 | 63 | 1102 |
| Cytb | 114 | 62 | 1095 | Tpx1 | 87 | 38 | 846 |
| Gpr50 | 155 | 62 | 2798 | Tex11 | 130 | 53 | 4760 |
| Izumo1 | 144 | 60 | 2150 | Tex14 | 166 | 67 | 4518 |
| Izumo1R | 159 | 59 | 1605 | Tnp2 | 124 | 49 | 1213 |
| Lelp1 | 131 | 55 | 549 | Usp26 | 139 | 58 | 3389 |
| Man2b1 | 103 | 48 | 4368 | Wbp2nl | 155 | 60 | 1916 |
| Prdm9 | 57 | 27 | 2872 | Zan | 170 | 66 | 6873 |
| Prm1 | 66 | 34 | 890 | Zp2 | 125 | 59 | 3639 |
| Prm2 | 77 | 28 | 609 | Zp3 | 159 | 64 | 1667 |
| S100a2 | 137 | 59 | 394 | ||||
| Slc6a5 | 103 | 57 | 3128 | Total: | 104,962 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Roberts, E.K.; Wright, E.A.; Worsham, A.E.; Hardy, D.M.; Bradley, R.D. Gamete Recognition Gene Divergence Yields a Robust Eutherian Phylogeny across Taxonomic Levels. Diversity 2023, 15, 1145. https://doi.org/10.3390/d15111145
Roberts EK, Wright EA, Worsham AE, Hardy DM, Bradley RD. Gamete Recognition Gene Divergence Yields a Robust Eutherian Phylogeny across Taxonomic Levels. Diversity. 2023; 15(11):1145. https://doi.org/10.3390/d15111145
Chicago/Turabian StyleRoberts, Emma K., Emily A. Wright, Asha E. Worsham, Daniel M. Hardy, and Robert D. Bradley. 2023. "Gamete Recognition Gene Divergence Yields a Robust Eutherian Phylogeny across Taxonomic Levels" Diversity 15, no. 11: 1145. https://doi.org/10.3390/d15111145
APA StyleRoberts, E. K., Wright, E. A., Worsham, A. E., Hardy, D. M., & Bradley, R. D. (2023). Gamete Recognition Gene Divergence Yields a Robust Eutherian Phylogeny across Taxonomic Levels. Diversity, 15(11), 1145. https://doi.org/10.3390/d15111145