

Cross Cultivation on Homologous/Heterologous Plant-Based Culture Media Empowers Host-Specific and Real Time In Vitro Signature of Plant Microbiota
 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Hypothesis and Experimental Design
2.2. Tested Plant Materials
2.3. Plant Broth
2.4. Culture Media
2.4.1. Plant Broth-Based Culture Media (Elsawey et al. [12])
2.4.2. Chemically-Synthetic Standard Culture Medium
2.5. In Situ Recovery and Cultivability of Endophytes of Maize Endo-Rhizosphere and Endo-Phyllosphere
2.6. In Situ Diversity of Culturable Endophytes of Maize Developed on Homologous (Maize) and Heterologous (Sunflower) Plant Broth
2.7. DNA Extraction
2.8. Amplification of the 16S rRNA Gene and DGGE Fingerprinting
2.9. Illumina MiSeq Sequencing and Analysis of 16S rRNA Gene Amplicons from TC-DNA
2.10. Chemical Analysis of Dehydrated Plant Powders
2.11. Statistical Analysis
3. Results
3.1. In Situ Diversity of Culturable Endophytes of Maize Developed on Culture Media Based on Homologous Broth of Maize and Heterologous Broth of Sunflower
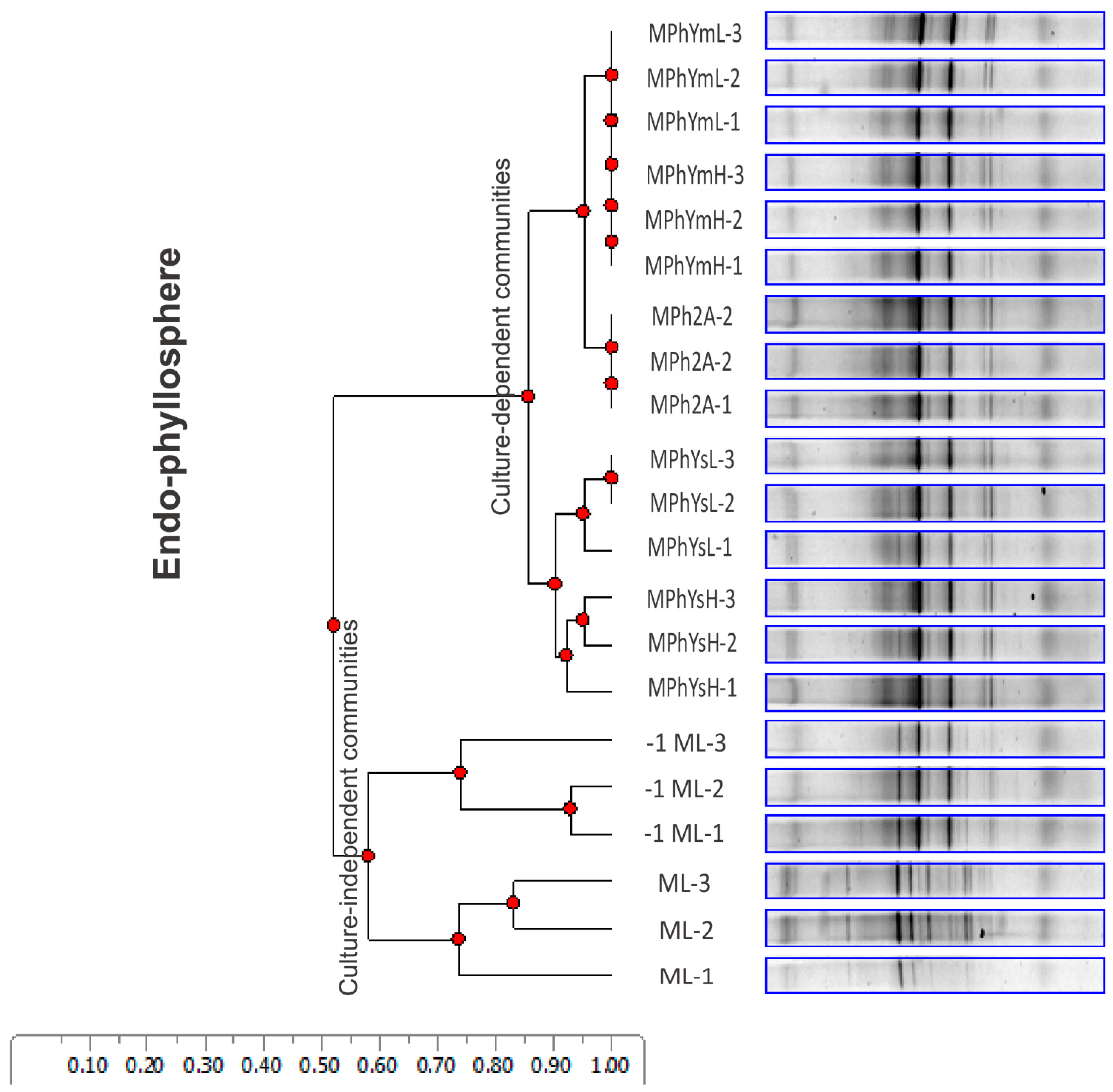
3.2. Divergence in Culturable Community Composition of Maize Bacterial Endophytes as Indicated by DGGE Analysis
3.3. Amplicon Sequence Data Analysis of Maize Endo-Phyllosphere Bacteria
3.3.1. Total OTUs Obtained
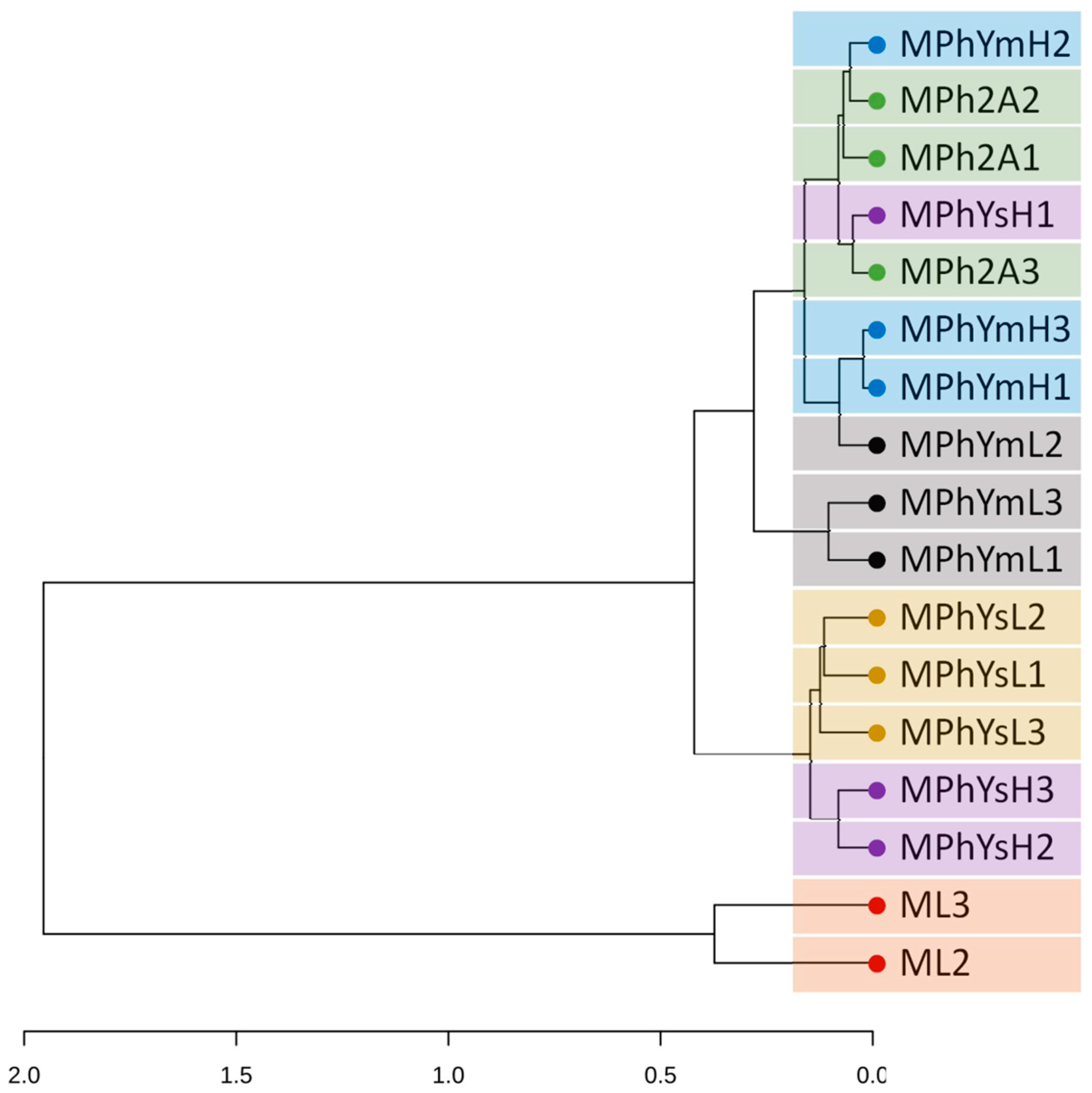
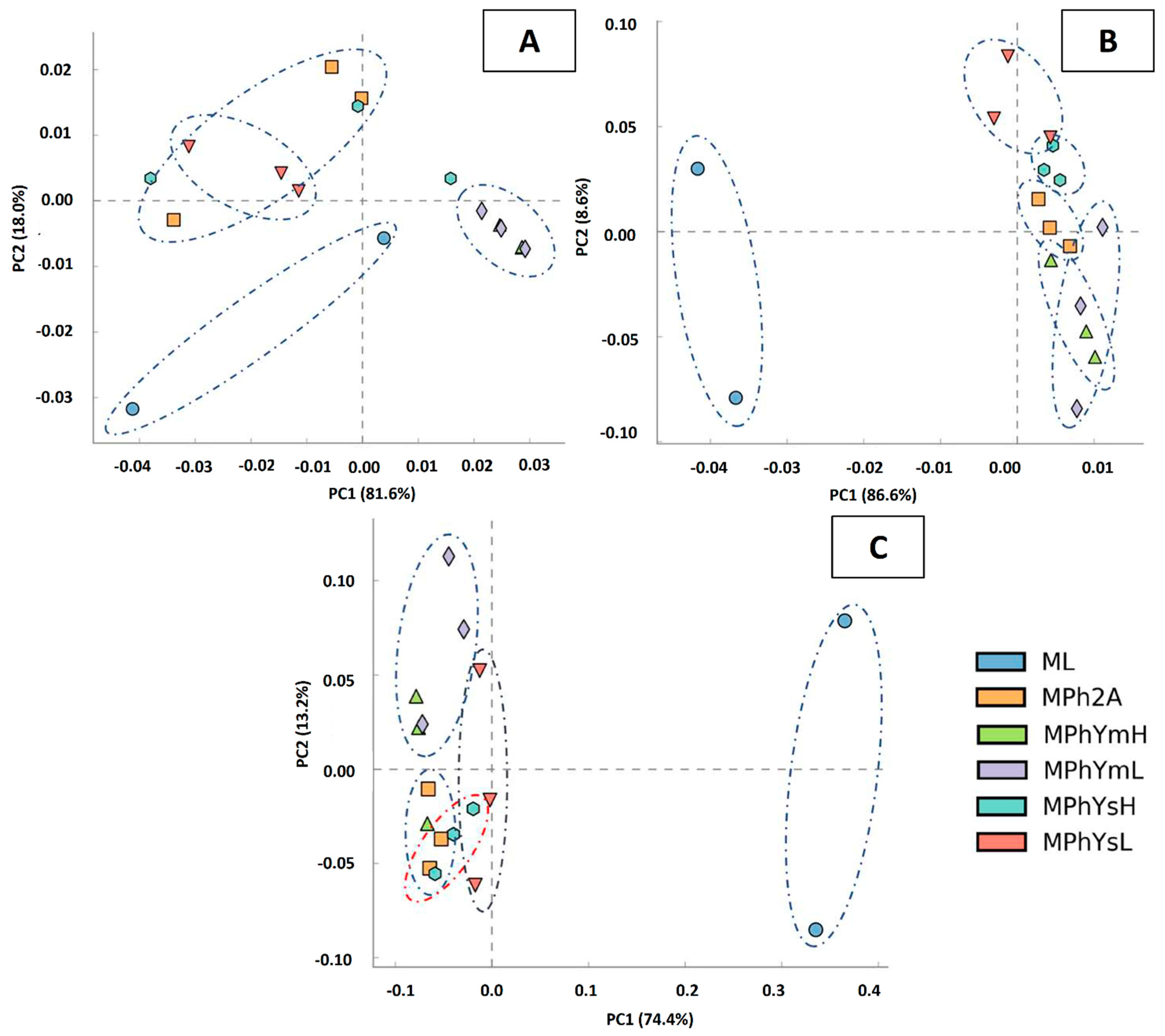
3.3.2. UPGMA and PCA Analyses
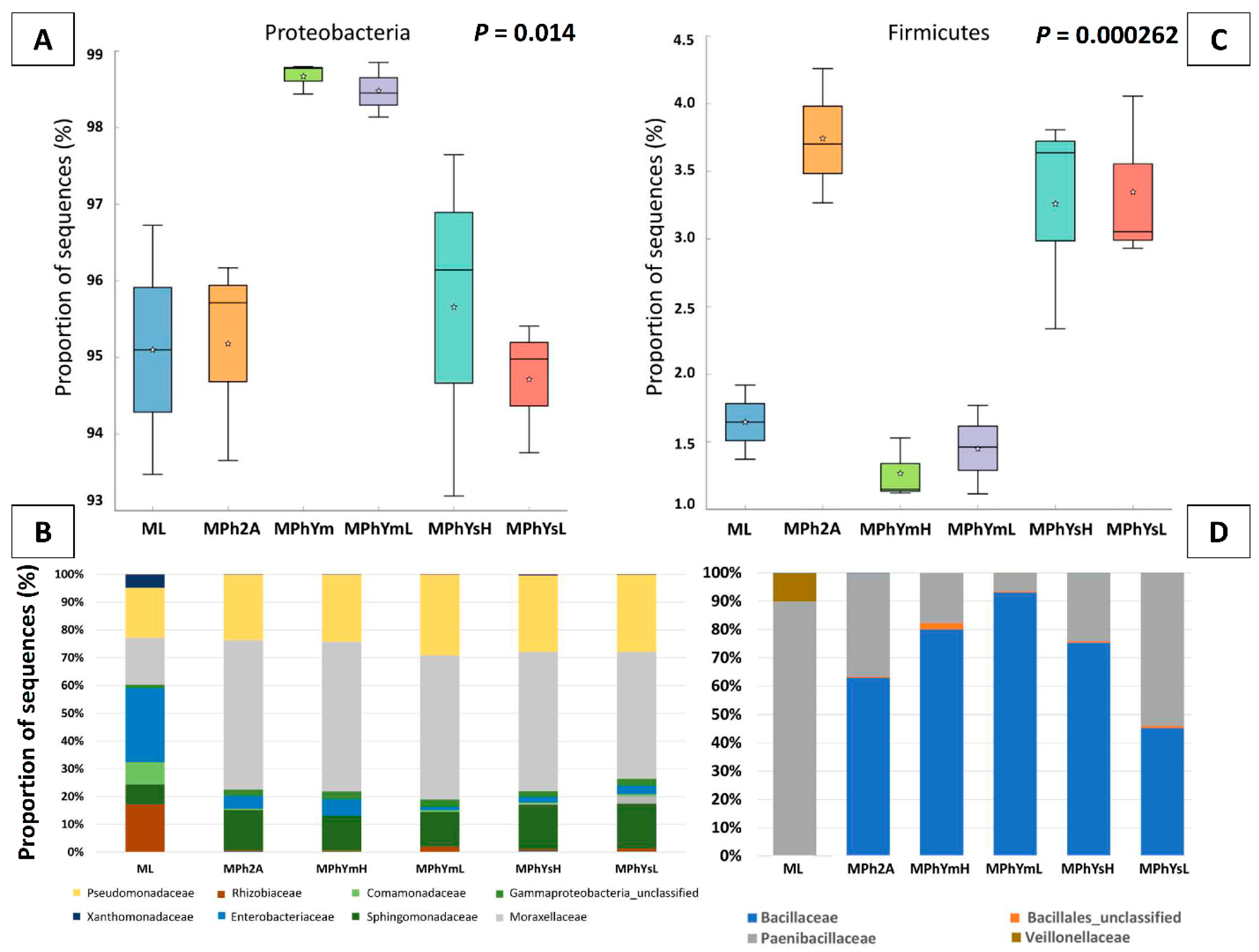
3.3.3. Maize Endo-Phyllosphere Bacterial Community Composition: Culture-Independent Community
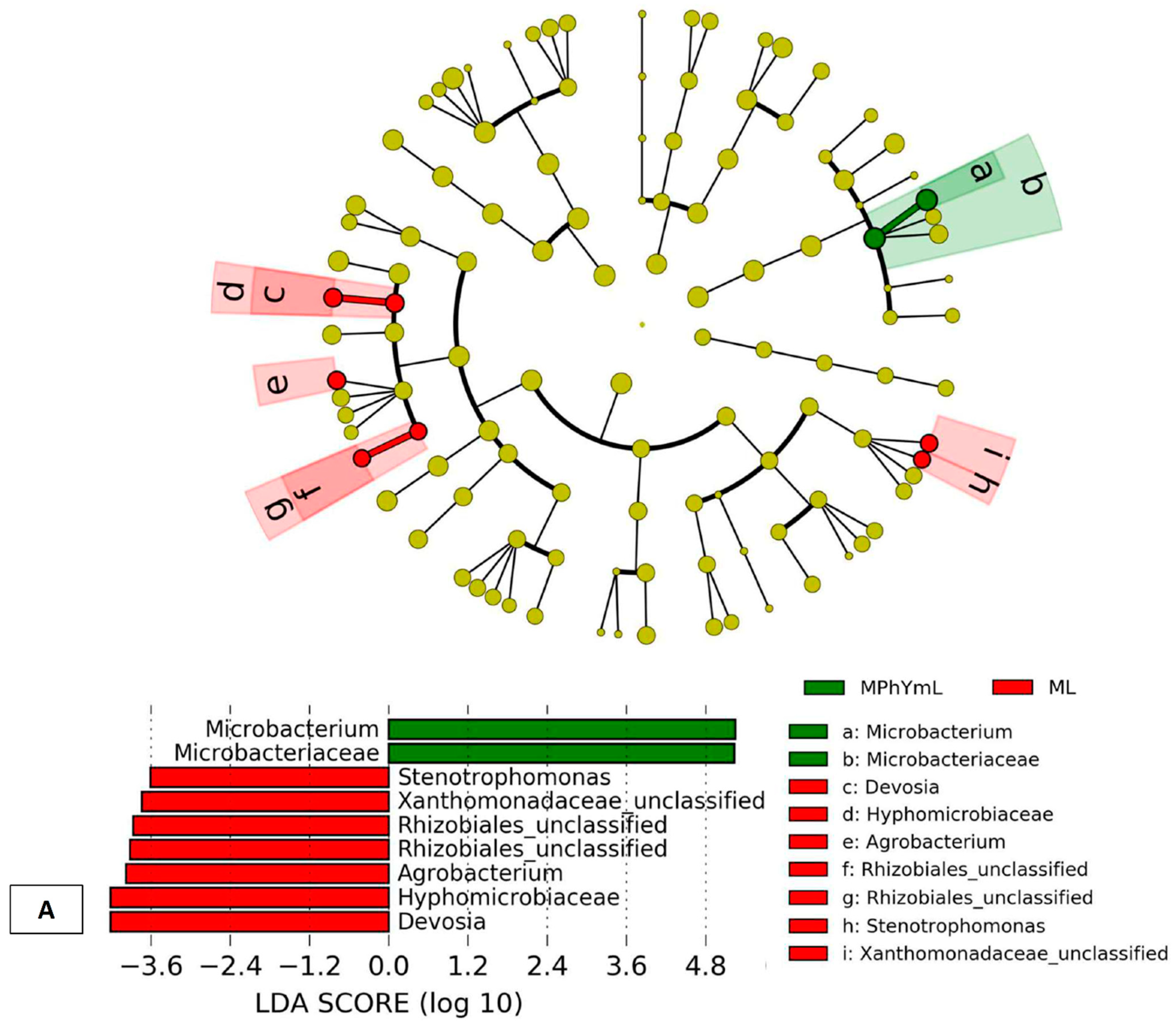
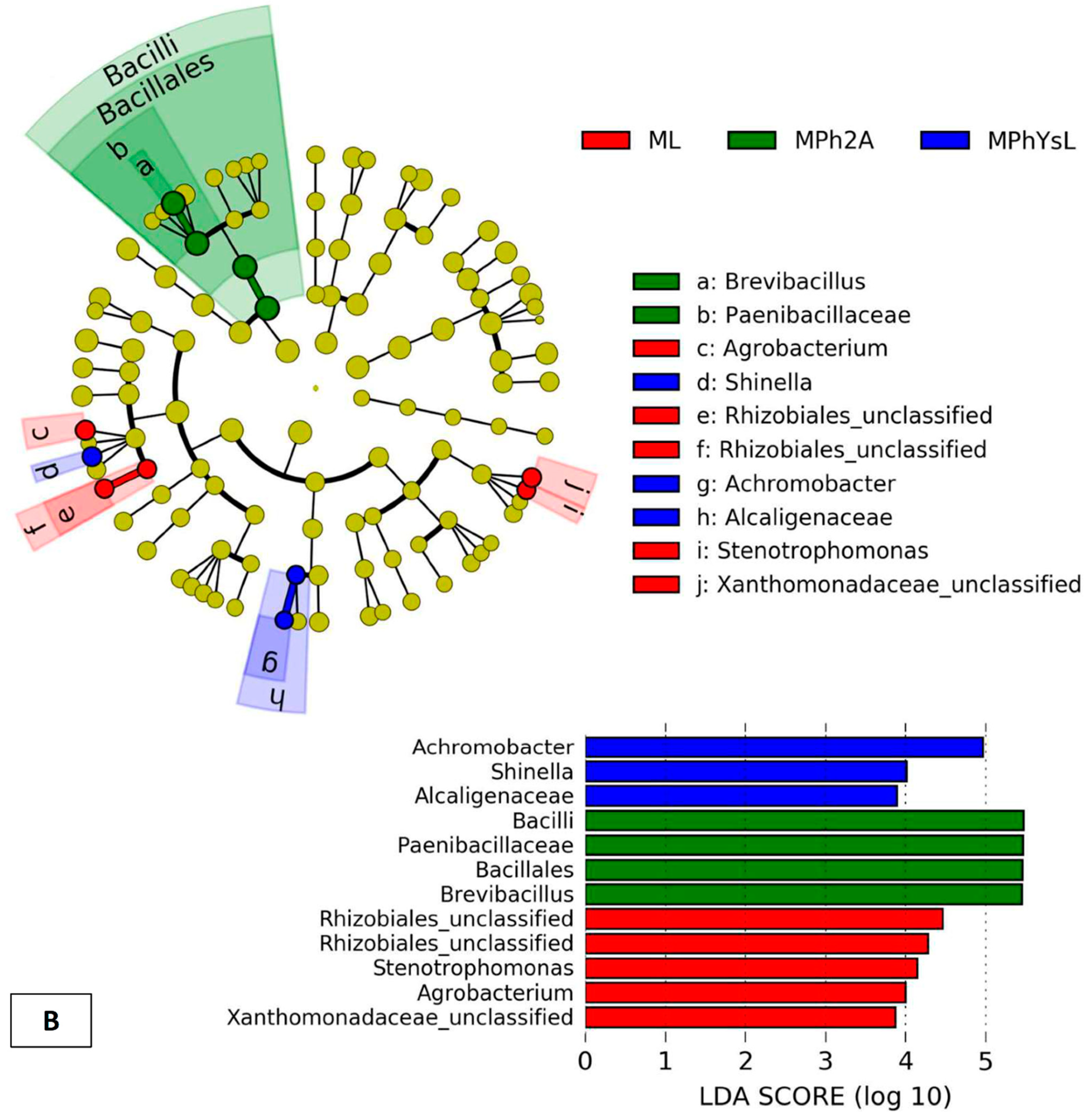
3.3.4. Differential In Vitro Culturability of Bacterial Communities of Maize Endo-Phyllosphere in Response to Cross Cultivation on Homologous/Heterologous Culture Media
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Solden, L.; Lloyd, K.; Wrighton, K. The bright side of microbial dark matter: Lessons learned from the uncultivated majority. Curr. Opin. Microbiol. 2016, 31, 217–226. [Google Scholar] [CrossRef] [PubMed]
- Abt, B.; Sikorski, J. Present and Future of Culturing Bacteria. Annu. Rev. Microbiol. 2017, 71, 711–730. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Y.-X.; Guo, X.; Qin, Y. High-throughput cultivation and identification of bacteria from the plant root microbiota. Nat. Protoc. 2021, 16, 988–1012. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, M.S.; Hamza, M.A.; Youssef, H.H.; Patz, S.; Becker, M.; ElSawey, H.; Nemr, R.; Daanaa, H.-S.A.; Mourad, E.F.; Morsi, A.T.; et al. Culturomics of the plant prokaryotic microbiome and the dawn of plant-based culture media—A review. J. Adv. Res. 2019, 19, 15–27. [Google Scholar] [CrossRef] [PubMed]
- Van Der Gast, C.J.; Walker, A.; A Stressmann, F.; Rogers, G.; Scott, P.; Daniels, T.W.; Carroll, M.P.; Parkhill, J.; Bruce, K.D. Partitioning core and satellite taxa from within cystic fibrosis lung bacterial communities. ISME J. 2010, 5, 780–791. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Neu, A.T.; Allen, E.E.; Roy, K. Defining and quantifying the core microbiome: Challenges and prospects. Proc. Natl. Acad. Sci. USA 2021, 118, e2104429118. [Google Scholar] [CrossRef]
- Zhang, L.; Zhang, M.; Huang, S.; Li, L.; Gao, Q.; Wang, Y.; Zhang, S.; Huang, S.; Yuan, L.; Wen, Y.; et al. A highly conserved core bacterial microbiota with nitrogen-fixation capacity inhabits the xylem sap in maize plants. Nat. Commun. 2022, 13, 3361. [Google Scholar] [CrossRef]
- Nichols, D.; Lewis, K.; Orjala, J.; Mo, S.; Ortenberg, R.; O’Connor, P.; Zhao, C.; Vouros, P.; Kaeberlein, T.; Epstein, S.S. Short peptide induces an “uncultivable” microorganism to grow in vitro. Appl. Environ. Microbiol. 2008, 74, 4889–4897. [Google Scholar] [CrossRef]
- Aoi, Y.; Kinoshita, T.; Hata, T.; Ohta, H.; Obokata, H.; Tsuneda, S. Hollow-fiber membrane chamber as a device for in situ environmental cultivation. Appl. Environ. Microbiol. 2009, 75, 3826–3833. [Google Scholar] [CrossRef]
- Stewart, E.J. Growing Unculturable Bacteria. J. Bacteriol. 2012, 194, 4151–4160. [Google Scholar] [CrossRef] [PubMed]
- Elsawey, H.; Patz, S.; Nemr, R.; Sarhan, M.; Hamza, M.; Youssef, H.; Abdelfadeel, M.; Daanaa, H.-S.; El-Tahan, M.; Abbas, M.; et al. Plant broth- (Not bovine-) based culture media provide the most compatible vegan nutrition for in vitro culturing and in situ probing of plant microbiota. Diversity 2020, 12, 418. [Google Scholar] [CrossRef]
- Nemr, R.A.; Khalil, M.; Sarhan, M.S.; Abbas, M.; Elsawey, H.; Youssef, H.H.; Hamza, M.A.; Morsi, A.T.; El-Tahan, M.; Fayez, M.; et al. “In situ similis” Culturing of Plant Microbiota: A Novel Simulated Environmental Method Based on Plant Leaf Blades as Nutritional Pads. Front. Microbiol. 2020, 11, 454. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Hugon, P.; Khelaifia, S.; Fournier, P.-E.; La Scola, B.; Raoult, D. The rebirth of culture in microbiology through the example of culturomics to study human gut microbiota. Clin. Microbiol. Rev. 2015, 28, 237–264. [Google Scholar] [CrossRef] [PubMed]
- Lagier, J.-C.; Khelaifia, S.; Alou, M.T.; Ndongo, S.; Dione, N.; Hugon, P.; Caputo, A.; Cadoret, F.; Traore, S.I.; Seck, E.H.; et al. Culture of previously uncultured members of the human gut microbiota by culturomics. Nat. Microbiol. 2016, 1, 16203. [Google Scholar] [CrossRef]
- Lagier, J.-C.; Dubourg, G.; Million, M.; Cadoret, F.; Bilen, M.; Fenollar, F.; Levasseur, A.; Rolain, J.-M.; Fournier, P.-E.; Raoult, D. Culturing the human microbiota and culturomics. Nat. Rev. Microbiol. 2018, 16, 540–550. [Google Scholar] [CrossRef]
- Khelaifia, S.; Lagier, J.-C.; Nkamga, V.D.; Guilhot, E.; Drancourt, M.; Raoult, D. Aerobic culture of methanogenic archaea without an external source of hydrogen. Eur. J. Clin. Microbiol. 2016, 35, 985–991. [Google Scholar] [CrossRef]
- Bai, Y.; Müller, D.B.; Srinivas, G.; Garrido-Oter, R.; Potthoff, E.; Rott, M.; Dombrowski, N.; Münch, P.C.; Spaepen, S.; Remus-Emsermann, M.; et al. Functional overlap of the Arabidopsis leaf and root microbiota. Nature 2015, 528, 364–369. [Google Scholar] [CrossRef]
- Mauchline, T.H.; Chedom-Fotso, D.; Chandra, G.; Samuels, T.; Greenaway, N.; Backhaus, A.; McMillan, V.; Canning, G.; Powers, S.J.; Hammond-Kosack, K.E.; et al. An analysis of Pseudomonas genomic diversity in take-all infected wheat fields reveals the lasting impact of wheat cultivars on the soil microbiota. Environ. Microbiol. 2015, 17, 4764–4778. [Google Scholar] [CrossRef]
- Levy, A.; Conway, J.M.; Dangl, J.L.; Woyke, T. Elucidating Bacterial Gene Functions in the Plant Microbiome. Cell Host Microbe 2018, 24, 475–485. [Google Scholar] [CrossRef]
- Cross, K.L.; Campbell, J.H.; Balachandran, M.; Campbell, A.G.; Cooper, C.J.; Griffen, A.; Heaton, M.; Joshi, S.; Klingeman, D.; Leys, E.; et al. Targeted isolation and cultivation of uncultivated bacteria by reverse genomics. Nat. Biotechnol. 2019, 37, 1314–1321. [Google Scholar] [CrossRef] [PubMed]
- Jiang, C.-Y.; Dong, L.; Zhao, J.-K.; Hu, X.; Shen, C.; Qiao, Y.; Zhang, X.; Wang, Y.; Ismagilov, R.F.; Liu, S.-J.; et al. High-Throughput Single-Cell Cultivation on Microfluidic Streak Plates. Appl. Environ. Microbiol. 2016, 82, 2210–2218. [Google Scholar] [CrossRef] [PubMed]
- Zengler, K.; Toledo, G.; Rappé, M.; Elkins, J.; Mathur, E.J.; Short, J.M.; Keller, M. Cultivating the uncultured. Proc. Natl. Acad. Sci. USA 2002, 99, 15681–15686. [Google Scholar] [CrossRef] [PubMed]
- Ferrari, B.C.; Binnerup, S.J.; Gillings, M. Microcolony cultivation on a soil substrate membrane system selects for previously uncultured soil bacteria. Appl. Environ. Microbiol. 2005, 71, 8714–8720. [Google Scholar] [CrossRef]
- Nichols, D.; Cahoon, N.; Trakhtenberg, E.M.; Pham, L.; Mehta, A.; Belanger, A.; Kanigan, T.; Lewis, K.; Epstein, S.S. Use of Ichip for High-Throughput In Situ Cultivation of “Uncultivable” Microbial Species. Appl. Environ. Microbiol. 2010, 76, 2445–2450. [Google Scholar] [CrossRef]
- Polrot, A.; Kirby, J.R.; Olorunniji, F.J.; Birkett, J.W.; Sharples, G.P. iChip increases the success of cultivation of TBT-resistant and TBT-degrading bacteria from estuarine sediment. World J. Microbiol. Biotechnol. 2022, 38, 1–9. [Google Scholar] [CrossRef]
- Eevers, N.; Gielen, M.; Sánchez-López, A.; Jaspers, S.; White, J.C.; Vangronsveld, J.; Weyens, N. Optimization of isolation and cultivation of bacterial endophytes through addition of plant extract to nutrient media. Microb. Biotechnol. 2015, 8, 707–715. [Google Scholar] [CrossRef]
- Murphy, B.R.; Batke, S.P.; Doohan, F.M.; Hodkinson, T.R. Media Manipulations and the Culture of Beneficial Fungal Root Endophytes. Int. J. Biol. 2015, 7, 94–102. [Google Scholar] [CrossRef]
- Koch, A.L. Oligotrophs versus copiotrophs. Bioessays 2001, 23, 657–661. [Google Scholar] [CrossRef]
- Connon, S.A.; Giovannoni, S.J. High-Throughput Methods for Culturing Microorganisms in Very-Low-Nutrient Media Yield Diverse New Marine Isolates. Appl. Environ. Microbiol. 2002, 68, 3878–3885. [Google Scholar] [CrossRef]
- Gerna, D.; Clara, D.; Allwardt, D.; Mitter, B.; Roach, T. Tailored Media Are Key to Unlocking the Diversity of Endophytic Bacteria in Distinct Compartments of Germinating Seeds. Microbiol. Spectr. 2022, 10, e00172-22. [Google Scholar] [CrossRef]
- Nour, E.H.; Hamza, M.A.; Fayez, M.; Monib, M.; Ruppel, S.; Hegazi, N.A. The crude plant juices of desert plants as appropriate culture media for the cultivation of rhizospheric microorganisms. J. Adv. Res. 2012, 3, 35–43. [Google Scholar] [CrossRef]
- Youssef, H.H.; Hamza, M.A.; Fayez, M.; Mourad, E.F.; Saleh, M.Y.; Sarhan, M.S.; Suker, R.M.; Eltahlawy, A.A.; Nemr, R.A.; El-Tahan, M.; et al. Plant-based culture media: Efficiently support culturing rhizobacteria and correctly mirror their in-situ diversity. J. Adv. Res. 2016, 7, 305–316. [Google Scholar] [CrossRef] [PubMed]
- Mourad, E.F.; Sarhan, M.S.; A Daanaa, H.-S.; Abdou, M.; Morsi, A.T.; Abdelfadeel, M.R.; Elsawey, H.; Nemr, R.; El-Tahan, M.; A Hamza, M.; et al. Plant Materials are Sustainable Substrates Supporting New Technologies of Plant-Only-Based Culture Media for in vitro Culturing of the Plant Microbiota. Microbes Environ. 2018, 33, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Sarhan, M.S.; Mourad, E.F.; Nemr, R.A.; Abdelfadeel, M.R.; Daanaa, H.-S.A.; Youssef, H.H.; Goda, H.A.; Hamza, M.A.; Fayez, M.; Eichler-Löbermann, B.; et al. An inoculum-dependent culturing strategy (IDC) for the cultivation of environmental microbiomes and the isolation of novel endophytic Actinobacteria. J. Antibiot. 2019, 73, 66–71. [Google Scholar] [CrossRef]
- Sarhan, M.S.; Mourad, E.F.; Hamza, M.A.; Youssef, H.H.; Scherwinski, A.-C.; El-Tahan, M.; Fayez, M.; Ruppel, S.; Hegazi, N.A. Plant powder teabags: A novel and practical approach to resolve culturability and diversity of rhizobacteria. Physiol. Plant. 2016, 157, 403–413. [Google Scholar] [CrossRef]
- Daanaa, H.-S.A.; Abdou, M.; Goda, H.A.; Abbas, M.T.; Hamza, M.A.; Sarhan, M.S.; Youssef, H.H.; Hamed, R.; El-Tahan, M.; Fayez, M.; et al. Plant pellets: A compatible vegan feedstock for preparation of plant-based culture media and production of value-added biomass of rhizobia. Sustainability 2020, 12, 8389. [Google Scholar] [CrossRef]
- Cleary, J.L.; Condren, A.R.; Zink, K.E.; Sanchez, L.M. Calling All Hosts: Bacterial Communication In Situ. Chem 2017, 2, 334–358. [Google Scholar] [CrossRef]
- Nemr, R.A.; Patz, S.; Abdelwakeel, S.M.; Khalil, M.; Ben Djadid, A.; Abdelfadeel, M.R.; Morsi, A.T.; Goda, H.A.; Youssef, H.H.; Hamza, M.; et al. Culture Media Based on Leaf Strips/Root Segments Create Compatible Host/Organ Setup for in vitro Cultivation of Plant Microbiota. Front. Sustain. Food Syst. 2021, 5, 660790. [Google Scholar] [CrossRef]
- Zhalnina, K.; Louie, K.B.; Hao, Z.; Mansoori, N.; da Rocha, U.N.; Shi, S.; Cho, H.; Karaoz, U.; Loqué, D.; Bowen, B.P.; et al. Dynamic root exudate chemistry and microbial substrate preferences drive patterns in rhizosphere microbial community assembly. Nat. Microbiol. 2018, 3, 470–480. [Google Scholar] [CrossRef]
- Neumann, G.; Römheld, V. The release of root exudates as affected by the plant physiological status. In The Rhizosphere: Biochemic and Organic Substances at the Soil-Plant Interface; Pinton, R., Varani, Z., Nannipieri, P., Eds.; Marrcel Dekker, Inc.: New York, NY, USA; Basel, Switzerland, 2000; pp. 41–98. ISBN 0849338557. [Google Scholar]
- Tesfaye, M.; Dufault, N.S.; Dornbusch, M.R.; Allan, D.L.; Vance, C.P.; A Samac, D. Influence of enhanced malate dehydrogenase expression by alfalfa on diversity of rhizobacteria and soil nutrient availability. Soil Biol. Biochem. 2003, 35, 1103–1113. [Google Scholar] [CrossRef]
- Watve, M.; Shejval, V.; Sonawane, C.; Rahalkar, M.; Matapurkar, A.; Shouche, Y.; Patole, M.; Phadnis, N.; Champhenkar, A.; Damle, K.; et al. The “K” selected oligophilic bacteria: A key to uncultured diversity? Curr. Sci. 2000, 78, 1535–1542. [Google Scholar] [CrossRef]
- Magarelli, R.A.; Trupo, M.; Ambrico, A.; Larocca, V.; Martino, M.; Palazzo, S.; Balducchi, R.; Joutsjoki, V.; Pihlanto, A.; Bevivino, A. Designing a Waste-Based Culture Medium for the Production of Plant Growth Promoting Microorganisms Based on Cladodes Juice from Opuntia ficus-indica Pruning. Fermentation 2022, 8, 225. [Google Scholar] [CrossRef]
- Fang, C.; Fernie, A.R.; Luo, J. Exploring the Diversity of Plant Metabolism. Trends Plant Sci. 2018, 24, 83–98. [Google Scholar] [CrossRef] [PubMed]
- Maeda, H.A. Evolutionary diversification of primary metabolism and its contribution to plant chemical diversity. Front. Plant Sci. 2019, 10, 881. [Google Scholar] [CrossRef]
- Fang, C.; Luo, J. Metabolic GWAS-based dissection of genetic bases underlying the diversity of plant metabolism. Plant J. 2018, 97, 91–100. [Google Scholar] [CrossRef] [PubMed]
- Zhou, X.; Liu, Z. Unlocking plant metabolic diversity: A (pan)-genomic view. Plant Commun. 2022, 3, 100300. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Alseekh, S.; Fernie, A.R.; Luo, J. The Structure and Function of Major Plant Metabolite Modifications. Mol. Plant 2019, 12, 899–919. [Google Scholar] [CrossRef]
- van der Hooft, J.J.J.; Mohimani, H.; Bauermeister, A.; Dorrestein, P.C.; Duncan, K.R.; Medema, M.H. Linking genomics and metabolomics to chart specialized metabolic diversity. Chem. Soc. Rev. 2020, 49, 3297–3314. [Google Scholar] [CrossRef]
- Reasoner, D.J.; Geldreich, E.E. A new medium for the enumeration and subculture of bacteria from potable water. Appl. Environ Microbiol. 1985, 49, 1–7. [Google Scholar] [CrossRef]
- Youssef, H.H.; Fayez, M.; Monib, M.; Hegazi, N. Gluconacetobacter diazotrophicus: A natural endophytic diazotroph of Nile Delta sugarcane capable of establishing an endophytic association with wheat. Biol. Fertil. Soils 2004, 39, 391–397. [Google Scholar] [CrossRef]
- de Oliveira Costa, L.E.; de Queiroz, M.V.; Borges, A.C.; de Moraes, C.A.; de Araújo, E.F. Isolation and characterization of endophytic bacteria isolated from the leaves of the common bean (Phaseolus vulgaris). Braz. J. Microbiol. 2012, 43, 1562–1575. [Google Scholar] [CrossRef] [PubMed]
- Jackson, C.R.; Randolph, K.C.; Osborn, S.L.; Tyler, H.L. Culture dependent and independent analysis of bacterial communities associated with commercial salad leaf vegetables. BMC Microbiol. 2013, 13, 274. [Google Scholar] [CrossRef] [PubMed]
- Hegazi, N.A.; Hamza, M.A.; Osman, A.; Ali, S.; Sedik, M.Z. Modified combined carbon N-deficient medium for isolation, enumeration and biomass production of diazotrophs. In Nitrogen Fixation with Non-Legumes; Springer: Dordrecht, The Netherlands, 1998; Volume 1, pp. 247–253. [Google Scholar]
- Mühling, M.; Woolven-Allen, J.; Murrell, J.C.; Joint, I. Improved group-specific PCR primers for denaturing gradient gel electrophoresis analysis of the genetic diversity of complex microbial communities. ISME J. 2008, 2, 379–392. [Google Scholar] [CrossRef]
- Muyzer, G.; de Waal, E.C.; Uitterlinden, A.G. Profiling of Complex Microbial Populations by Denaturing Gradient Gel Electrophoresis Analysis of Polymerase Chain Reaction-Amplified Genes Coding for 16S rRNA. Appl. Environ. Microbiol. 1993, 59, 695–700. [Google Scholar] [CrossRef]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Čech, M.; Chilton, J.; Clements, D.; Coraor, N.; Grüning, B.A.; et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W537–W544. [Google Scholar] [CrossRef]
- Schloss, P.D.; Westcott, S.L.; Ryabin, T.; Hall, J.R.; Hartmann, M.; Hollister, E.B.; Lesniewski, R.A.; Oakley, B.B.; Parks, D.H.; Robinson, C.J.; et al. Introducing mothur: Open-Source, Platform-Independent, Community-Supported Software for Describing and Comparing Microbial Communities. Appl. Environ. Microbiol. 2009, 75, 7537–7541. [Google Scholar] [CrossRef]
- DeSantis, T.Z.; Hugenholtz, P.; Larsen, N.; Rojas, M.; Brodie, E.L.; Keller, K.; Huber, T.; Dalevi, D.; Hu, P.; Andersen, G.L. Greengenes, a Chimera-Checked 16S rRNA Gene Database and Workbench Compatible with ARB. Appl. Environ. Microbiol. 2006, 72, 5069–5072. [Google Scholar] [CrossRef]
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef]
- Elsayed, T.R.; Jacquiod, S.; Nour, E.H.; Sørensen, S.J.; Smalla, K. Biocontrol of Bacterial Wilt Disease Through Complex Interaction Between Tomato Plant, Antagonists, the Indigenous Rhizosphere Microbiota, and Ralstonia solanacearum. Front. Microbiol. 2020, 10, 2835. [Google Scholar] [CrossRef]
- Segata, N.; Izard, J.; Waldron, L.; Gevers, D.; Miropolsky, L.; Garrett, W.S.; Huttenhower, C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011, 12, R60. [Google Scholar] [CrossRef] [PubMed]
- Hassani, M.A.; Durán, P.; Hacquard, S. Microbial interactions within the plant holobiont. Microbiome 2018, 6, 58. [Google Scholar] [CrossRef] [PubMed]
- Xiong, C.; Singh, B.K.; He, J.-Z.; Han, Y.-L.; Li, P.-P.; Wan, L.-H.; Meng, G.-Z.; Liu, S.-Y.; Wang, J.-T.; Wu, C.-F.; et al. Plant developmental stage drives the differentiation in ecological role of the maize microbiome. Microbiome 2021, 9, 171. [Google Scholar] [CrossRef] [PubMed]
- Thrall, P.H.; Hochberg, M.E.; Burdon, J.J.; Bever, J.D. Coevolution of symbiotic mutualists and parasites in a community context. Trends Ecol. Evol. 2007, 22, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Reinhold-Hurek, B.; Bünger, W.; Burbano, C.S.; Sabale, M.; Hurek, T. Roots Shaping Their Microbiome: Global Hotspots for Microbial Activity. Annu. Rev. Phytopathol. 2015, 53, 403–424. [Google Scholar] [CrossRef]
- Fang, C.; Luo, J.; Wang, S. The Diversity of Nutritional Metabolites: Origin, Dissection, and Application in Crop Breeding. Front. Plant Sci. 2019, 10, 1028. [Google Scholar] [CrossRef]
- He, M.; Zhang, K.; Tan, H.; Hu, R.; Su, J.; Wang, J.; Huang, L.; Zhang, Y.; Li, X. Nutrient levels within leaves, stems, and roots of the xeric species Reaumuria soongorica in relation to geographical, climatic, and soil conditions. Ecol. Evol. 2015, 5, 1494–1503. [Google Scholar] [CrossRef]
- Orlewska, K.; Piotrowska-Seget, Z.; Cycoń, M. Use of the PCR-DGGE Method for the Analysis of the Bacterial Community Structure in Soil Treated With the Cephalosporin Antibiotic Cefuroxime and/or Inoculated With a Multidrug-Resistant Pseudomonas putida Strain MC1. Front. Microbiol. 2018, 9, 1387. [Google Scholar] [CrossRef]
- Vischetti, C.; Casucci, C.; De Bernardi, A.; Monaci, E.; Tiano, L.; Marcheggiani, F.; Ciani, M.; Comitini, F.; Marini, E.; Taskin, E.; et al. Sub-Lethal Effects of Pesticides on the DNA of Soil Organisms as Early Ecotoxicological Biomarkers. Front. Microbiol. 2020, 11, 1892. [Google Scholar] [CrossRef]
- Valaskova, V.; Baldrian, P. Denaturing gradient gel electrophoresis as a fingerprinting method for the analysis of soil microbial communities. Plant Soil Environ. 2009, 55, 413–423. [Google Scholar] [CrossRef]
- Pereira, S.I.A.; Castro, P.M.L. Diversity and characterization of culturable bacterial endophytes from Zea mays and their potential as plant growth-promoting agents in metal-degraded soils. Environ. Sci. Pollut. Res. 2014, 21, 14110–14123. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Treatments | Log No. CFUs g−1DW. | |
|---|---|---|
| Total Colonies | Micro-Colonies | |
| Incubation time | ||
| 1 day | 6.96 ± 0.090 c | 6.01 ± 0.300 a |
| 4 days | 7.28 ± 0.104 b | 4.36 ± 0.304 b |
| 7 days | 7.30 ± 0.105 ab | 3.67 ± 0.300 b |
| 14 day | 7.33 ± 0.107 a | 3.63 ± 0.300 b |
| LSD (p value ≤ 0.05) | 0.041 | 0.84 |
| Plant sphere | ||
| Endo-rhizosphere | 7.77 ± 033 a | 6.20 ± 0.214 a |
| Endo-phyllosphere | 6.67 ± 0.059 b | 2.69 ± 0.212 b |
| LSD (p value ≤ 0.05) | 0.029 | 0.60 |
| Culture medium | ||
| R2A | 7.47 ± 0.020 b | 2.85 ± 0.336 b |
| MPhYsH | 7.60 ± 0.089 a | 3.13 ± 0.341 b |
| MPhYsL | 7.36 ± 0.089 c | 5.18 ± 0.336 a |
| MPhYmH | 6.79 ± 0.127 e | 5.10 ± 0.336 a |
| MPhYmL | 6.87 ± 0.148 d | 5.82 ± 0.336 a |
| LSD (p value ≤ 0.05) | 0.06 | 0.94 |
| Phylum | ML | MPh2A | MPhYmH | MphYmL | MphYsH | MphYsL |
|---|---|---|---|---|---|---|
| Proteobacteria | 94.81 ± 2.16 ab | 94.76 ± 1.32 ab | 98.26 ± 0.25 a | 98 ± 0.41 ab | 95.27 ± 2.26 ab | 94.32 ± 0.82 b |
| Firmicutes | 1.63 ± 0.38 bc | 3.73 ± 0.5 a | 1.25 ± 0.22 c | 1.43 ± 0.32 c | 3.25 ± 0.8 ab | 3.34 ± 0.6 a |
| Bacteroidetes | 3.09 ± 2.82 | 0.96 ± 1.66 | 0 ± 0 | 0.01 ± 0.01 | 0.92 ± 1.49 | 1.58 ± 0.4 |
| Actinobacteria | 0.11 ± 0.15 | 0.12 ± 0.09 | 0.06 ± 0.04 | 0.07 ± 0.03 | 0.17 ± 0.19 | 0.34 ± 0.29 |
| Class | ML | MPh2A | MphYmH | MphYmL | MphYsH | MphYsL |
| Gammaproteobacteria | 66.06 ± 12.71 b | 79.61 ± 1.55 ab | 85.8 ± 2.73 a | 84.78 ± 3.85 a | 77.02 ± 1.39 ab | 72.45 ± 3.06 b |
| Alphaproteobacteria | 21.34 ± 4.12 a | 14.75 ± 1.1 ab | 12.35 ± 2.82 b | 12.49 ± 4.33 b | 16.98 ± 0.83 ab | 18.43 ± 2.16 ab |
| Betaproteobacteria | 7.3 ± 6.42 a | 0.3 ± 0.51 b | 0 ± 0 b | 0.6 ± 0.54 b | 1.16 ± 0.92 ab | 3.33 ± 1.58 ab |
| Proteobacteria_unclassified | 0.11 ± 0.02 | 0.1 ± 0.01 | 0.1 ± 0.01 | 0.12 ± 0.03 | 0.1 ± 0 | 0.11 ± 0.02 |
| Bacilli | 1.47 ± 0.15 b | 3.73 ± 0.5 a | 1.25 ± 0.22 b | 1.43 ± 0.32 b | 3.25 ± 0.8 a | 3.33 ± 0.62 a |
| Clostridia | 0.16 ± 0.23 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| Flavobacteria | 3.09 ± 2.82 | 0.95 ± 1.65 | 0 ± 0 | 0 ± 0 | 0.87 ± 1.4 | 1.53 ± 0.43 |
| Saprospirae | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.05 ± 0.08 | 0 ± 0 |
| Sphingobacteria | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0.05 ± 0.09 |
| Actinobacteria | 0.11 ± 0.15 | 0.12 ± 0.09 | 0.06 ± 0.04 | 0.07 ± 0.03 | 0.17 ± 0.19 | 0.34 ± 0.29 |
| Order | ML | MPh2A | MphYmH | MphYmL | MphYsH | MphYsL |
| Pseudomonadales | 38.24 ± 4.92 c | 72.78 ± 2.2 ab | 77.24 ± 3.61 a | 78.37 ± 0.59 a | 71.98 ± 1.11 ab | 66.99 ± 3.51 b |
| Enterobacteriales | 22.68 ± 7.92 a | 4.6 ± 0.84 b | 5.84 ± 0.72 b | 3.37 ± 3.69 b | 2.81 ± 1.74 b | 2.94 ± 1.2 b |
| Gammaproteobacteria_unclassified | 1.05 ± 0.07 b | 2.21 ± 0.18 ab | 2.72 ± 0.19 a | 3.03 ± 0.62 a | 2.05 ± 0.25 ab | 2.47 ± 0.56 a |
| Xanthomonadales | 4.09 ± 0.2 a | 0.01 ± 0.01 b | 0.01 ± 0 b | 0.01 ± 0 b | 0.17 ± 0.27 b | 0.05 ± 0.02 b |
| Rhizobiales | 15.21 ± 4.58 a | 0.45 ± 0.35 b | 0.14 ± 0.16 b | 1.91 ± 3.11 b | 0.76 ± 0.51 b | 1.3 ± 0.73 b |
| Sphingomonadales | 6.11 ± 0.46 bc | 14.12 ± 1.2 ab | 11.96 ± 2.3 ab | 10.55 ± 3.78 bc | 16.17 ± 0.88 a | 17.09 ± 2.14 a |
| Alphaproteobacteria_unclassified | 0.02 ± 0 | 0.03 ± 0 | 0.03 ± 0.01 | 0.02 ± 0.01 | 0.04 ± 0 | 0.04 ± 0.01 |
| Caulobacterales | 0 ± 0 | 0.13 ± 0.23 | 0 ± 0 | 0 ± 0 | 0.01 ± 0.01 | 0 ± 0 |
| Rhodobacterales | 0 ± 0 | 0.01 ± 0.01 | 0.22 ± 0.37 | 0.01 ± 0 | 0.01 ± 0.01 | 0 ± 0 |
| Burkholderiales | 7.24 ± 6.35 a | 0.29 ± 0.5 b | 0 ± 0 b | 0.59 ± 0.53 b | 1.15 ± 0.91 ab | 3.3 ± 1.58 ab |
| Betaproteobacteria_unclassified | 0.06 ± 0.07 | 0 ± 0.01 | 0 ± 0 | 0.01 ± 0.01 | 0.01 ± 0.01 | 0.03 ± 0.01 |
| Proteobacteria_unclassified | 0.11 ± 0.02 | 0.1 ± 0.01 | 0.1 ± 0.01 | 0.12 ± 0.03 | 0.1 ± 0 | 0.11 ± 0.02 |
| Bacillales | 1.47 ± 0.15 | 3.73 ± 0.5 | 1.25 ± 0.22 | 1.43 ± 0.32 | 3.25 ± 0.8 | 3.33 ± 0.62 |
| Clostridiales | 0.16 ± 0.23 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 | 0 ± 0 |
| Actinomycetales | 0.11 ± 0.15 | 0.12 ± 0.09 | 0.06 ± 0.04 | 0.07 ± 0.03 | 0.17 ± 0.19 | 0.34 ± 0.29 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Elsawey, H.; Nour, E.H.; Elsayed, T.R.; Nemr, R.A.; Youssef, H.H.; Hamza, M.A.; Abbas, M.; El-Tahan, M.; Fayez, M.; Ruppel, S.; et al. Cross Cultivation on Homologous/Heterologous Plant-Based Culture Media Empowers Host-Specific and Real Time In Vitro Signature of Plant Microbiota. Diversity 2023, 15, 46. https://doi.org/10.3390/d15010046
Elsawey H, Nour EH, Elsayed TR, Nemr RA, Youssef HH, Hamza MA, Abbas M, El-Tahan M, Fayez M, Ruppel S, et al. Cross Cultivation on Homologous/Heterologous Plant-Based Culture Media Empowers Host-Specific and Real Time In Vitro Signature of Plant Microbiota. Diversity. 2023; 15(1):46. https://doi.org/10.3390/d15010046
Chicago/Turabian StyleElsawey, Hend, Eman H. Nour, Tarek R. Elsayed, Rahma A. Nemr, Hanan H. Youssef, Mervat A. Hamza, Mohamed Abbas, Mahmoud El-Tahan, Mohamed Fayez, Silke Ruppel, and et al. 2023. "Cross Cultivation on Homologous/Heterologous Plant-Based Culture Media Empowers Host-Specific and Real Time In Vitro Signature of Plant Microbiota" Diversity 15, no. 1: 46. https://doi.org/10.3390/d15010046
APA StyleElsawey, H., Nour, E. H., Elsayed, T. R., Nemr, R. A., Youssef, H. H., Hamza, M. A., Abbas, M., El-Tahan, M., Fayez, M., Ruppel, S., & Hegazi, N. A. (2023). Cross Cultivation on Homologous/Heterologous Plant-Based Culture Media Empowers Host-Specific and Real Time In Vitro Signature of Plant Microbiota. Diversity, 15(1), 46. https://doi.org/10.3390/d15010046