
Genetic Diversity and Fine-Scale Spatial Genetic Structure of the Endangered Shrub Birch (Betula humilis Schrk.) Populations in Protected and Unprotected Areas


Abstract
:1. Introduction
2. Materials and Methods
2.1. Study Area
2.2. Sample Collection
2.3. Molecular Laboratory Analyses
2.4. Genetic Diversity Analyses
2.5. Genetic and Spatial Genetic Structure Analyses
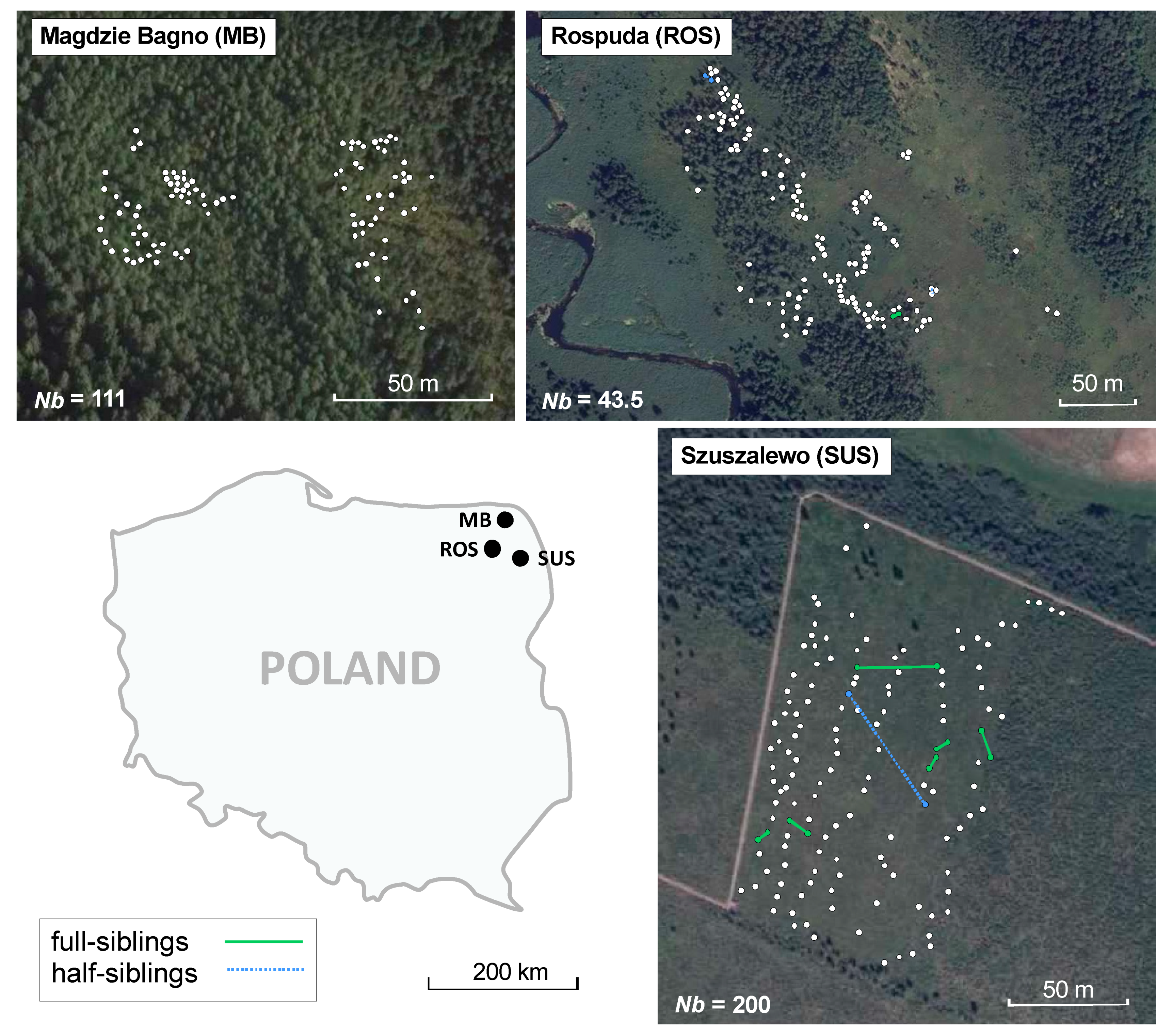
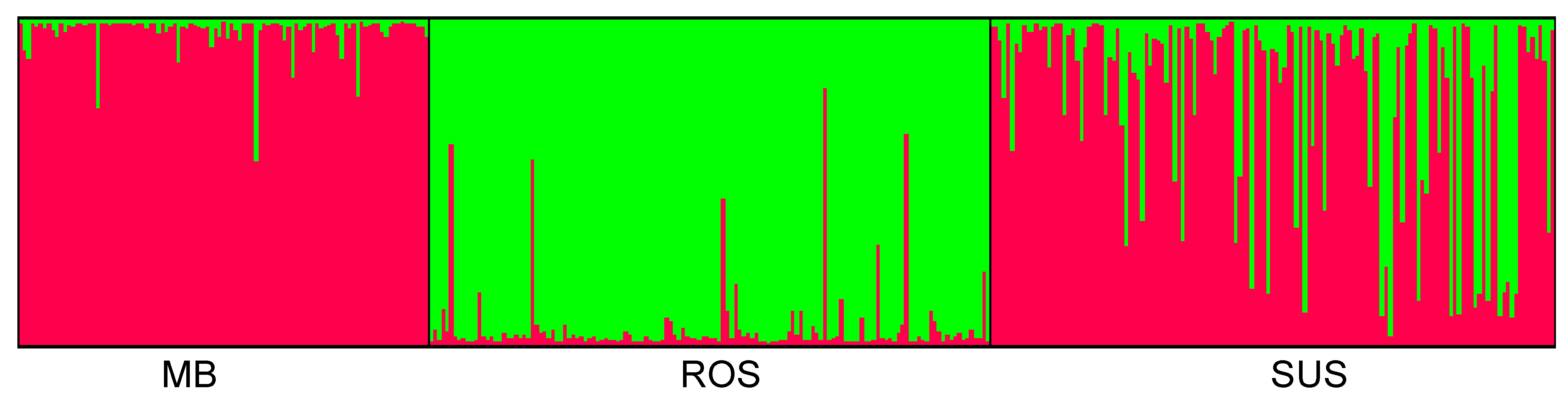
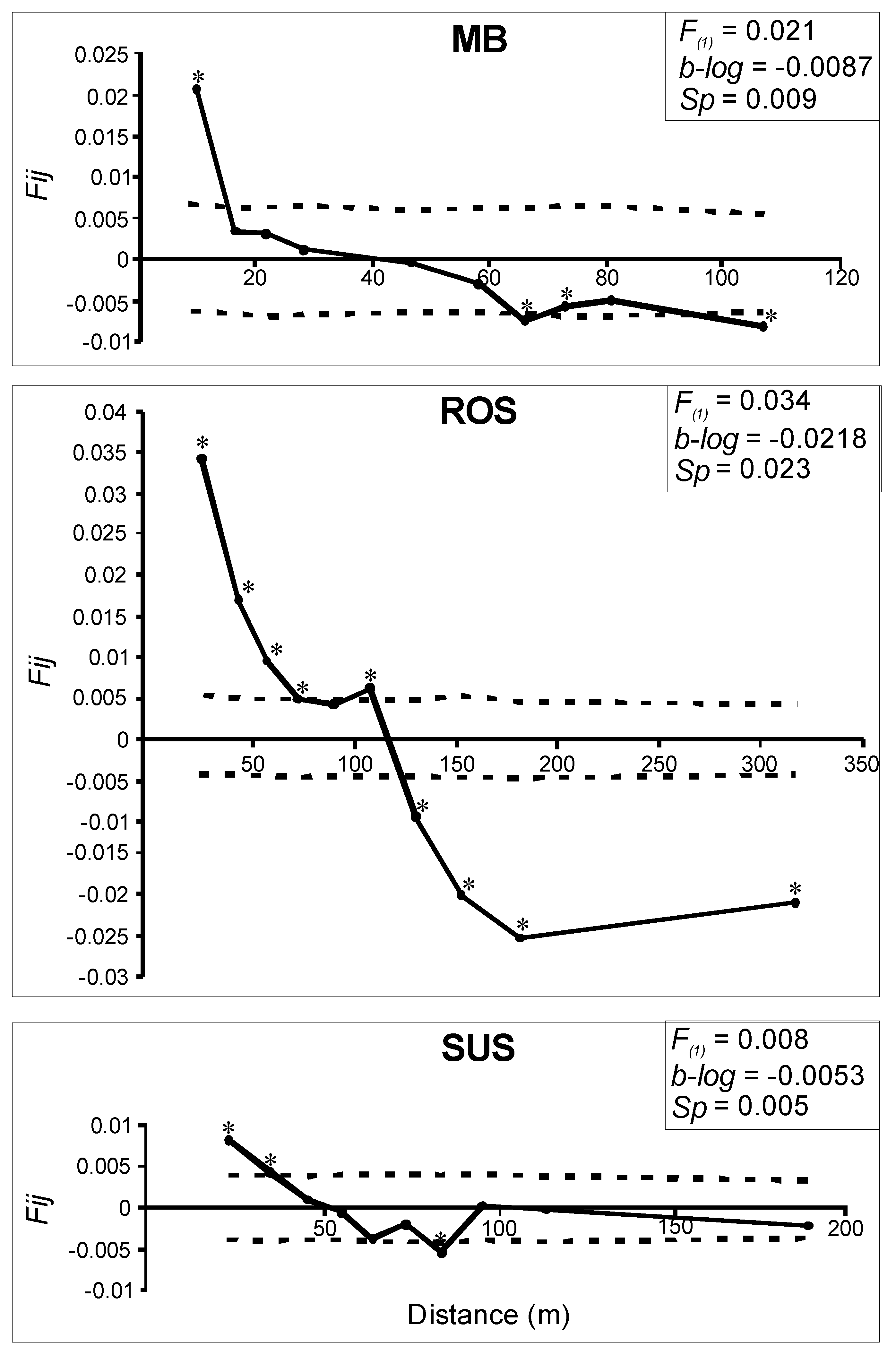
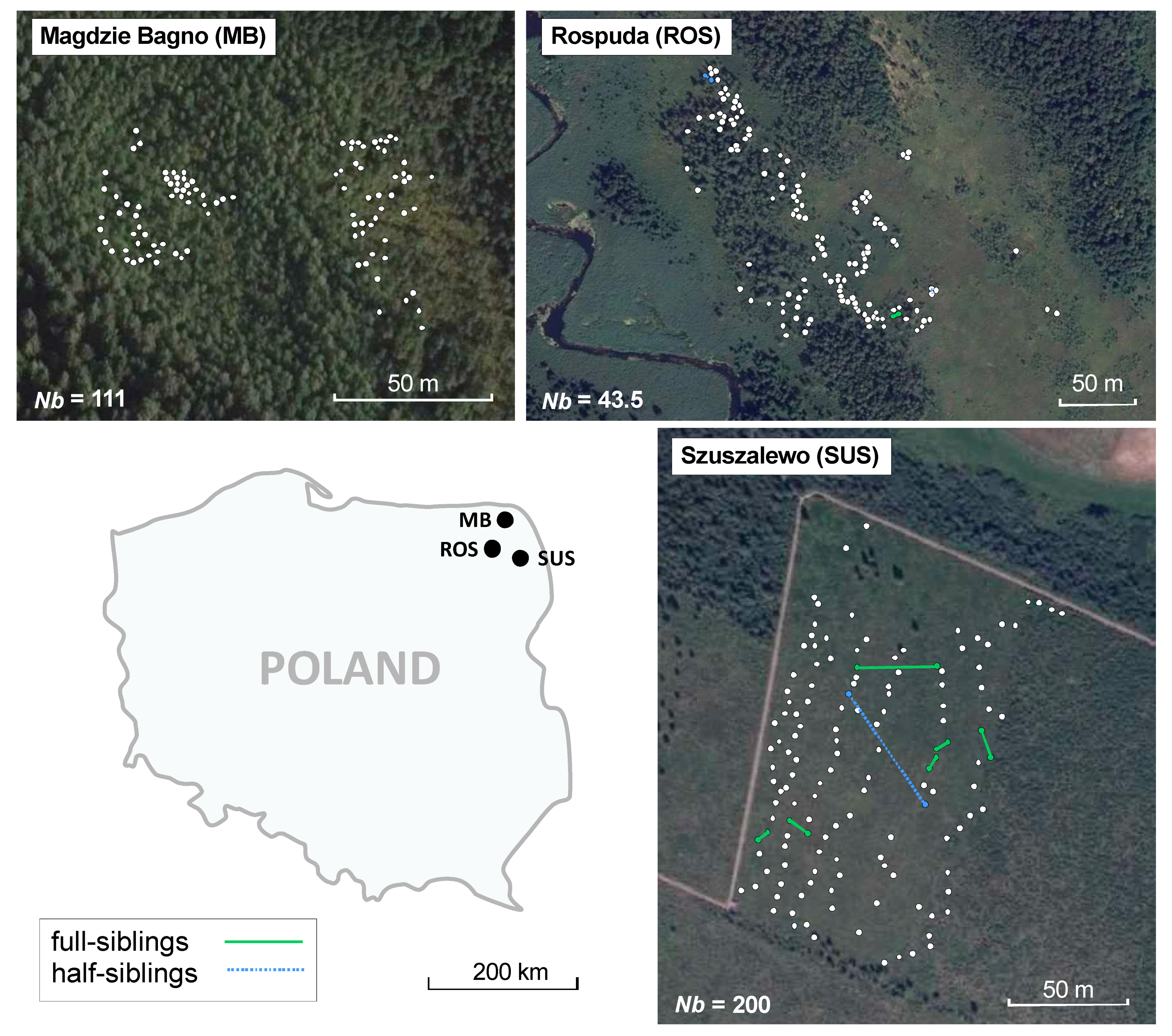
3. Results
3.1. Genetic Diversity
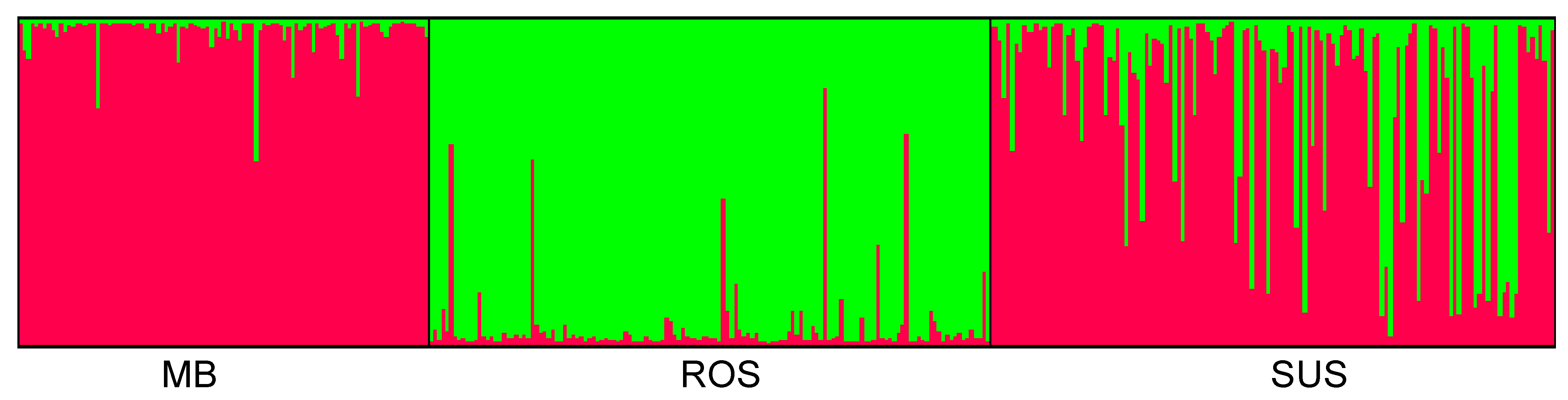
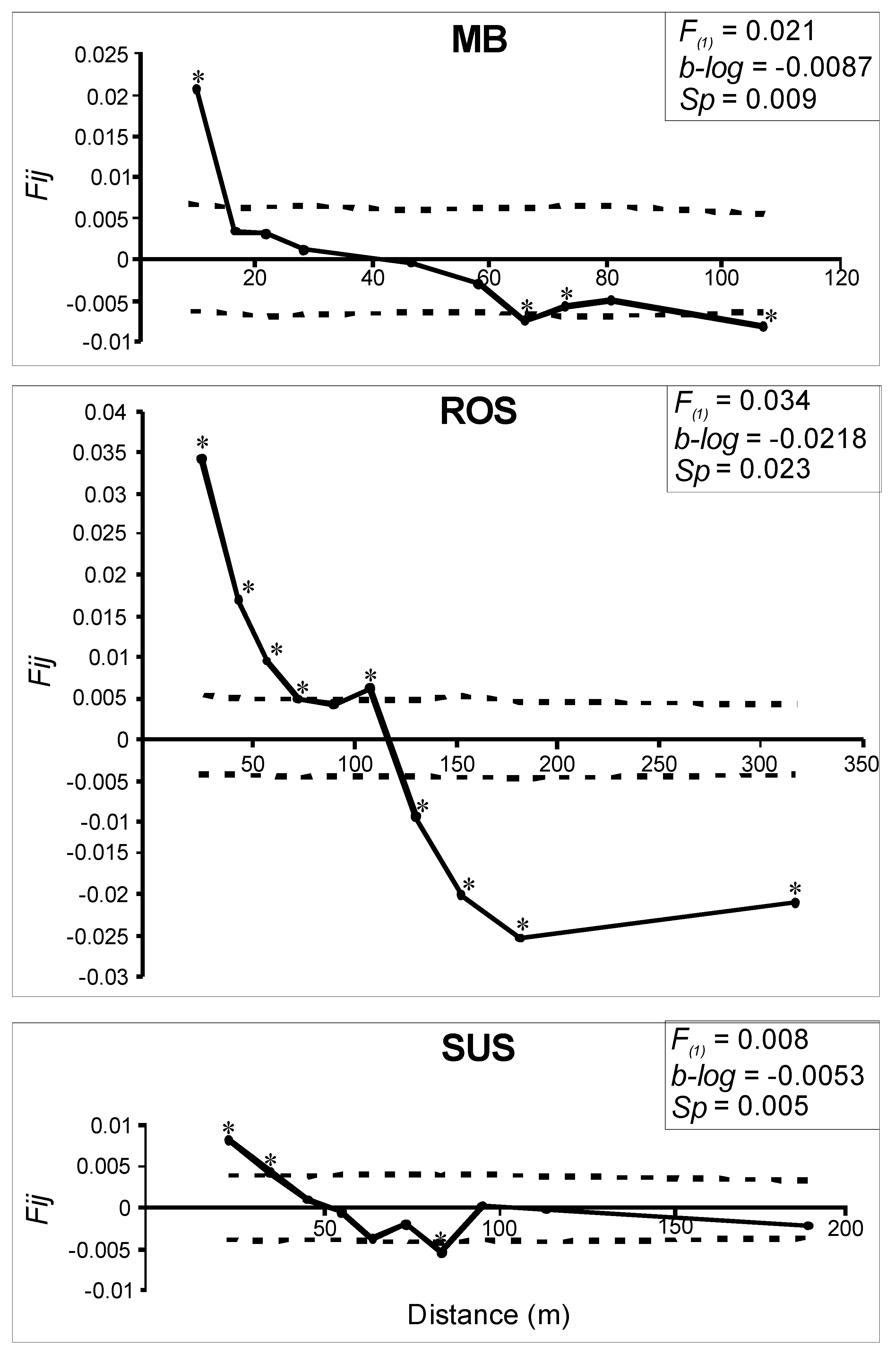
3.2. Genetic and Spatial Genetic Structure
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gardner, R.C.; Finlayson, C. Global Wetland Outlook: State of the World’s Wetlands and Their Services to People; Ramsar Convention Secretariat: Gland, Switzerland, 2018; pp. 1–89. Available online: https://ssrn.com/abstract=3261606 (accessed on 1 December 2021).
- Silva, J.; Jones, W.; Phillips, L. LIFE and Europe’s Wetlands: Restoring a Vital Ecosystem; Office for Official Publications of the European Communities: Luxembourg, 2008; pp. 1–68. [Google Scholar] [CrossRef]
- Verhoeven, J.T. Wetlands in Europe: Perspectives for restoration of a lost paradise. Ecol. Eng. 2014, 66, 6–9. [Google Scholar] [CrossRef]
- Salimi, S.; Almuktar, S.A.A.A.N.; Scholz, M. Impact of climate change on wetland ecosystems: A critical review of experimental wetlands. J. Environ. Manag. 2021, 286, 112160. [Google Scholar] [CrossRef]
- Mantyka-Pringle, C.S.; Martin, T.G.; Rhodes, J.R. Interactions between climate and habitat loss effects on biodiversity: A systematic review and meta-analysis. Glob. Chang. Biol. 2012, 18, 1239–1252. [Google Scholar] [CrossRef] [Green Version]
- Pimm, S.L.; Russell, G.J.; Gittleman, J.L.; Brooks, T.M. The future of biodiversity. Science 1995, 269, 347–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanski, I. Habitat loss, the dynamics of biodiversity, and a perspective on conservation. Ambio 2011, 40, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Vekemans, X.; Hardy, O.J. New insights from fine-scale spatial genetic structure analyses in plant populations. Mol. Ecol. 2004, 13, 921–935. [Google Scholar] [CrossRef]
- Aguilar, R.; Quesada, M.; Ashworth, L.; Herrerias-Diego, Y.; Lobo, J. Genetic consequences of habitat fragmentation in plant populations: Susceptible signals in plant traits and methodological approaches. Mol. Ecol. 2008, 17, 5177–5188. [Google Scholar] [CrossRef]
- Pometti, C.; Bessega, C.; Cialdella, A.; Ewens, M.; Saidman, B.; Vilardi, J. Spatial genetic structure within populations and management implications of the South American species Acacia aroma (Fabaceae). PLoS ONE 2018, 13, e0192107. [Google Scholar] [CrossRef] [Green Version]
- Lynch, M.; Conery, J.; Burger, R. Mutation accumulation and the extinction of small populations. Am. Nat. 1995, 146, 489–518. [Google Scholar] [CrossRef] [Green Version]
- Barrett, S.C.H. The evolution, maintenance, and loss of self-incompatibility systems. In Plant Reproductive Ecology: Patterns and Strategies; Lovett Doust, J., Lovett-Doust, L., Eds.; Oxford University Press: New York, NY, USA, 1988; pp. 98–124. [Google Scholar]
- Załuski, T.; Jabłońska, E.; Pawlikowski, P.; Pisarek, W.; Kucharczyk, M. Betula humilis Schrank. In Polska Czerwona Księga Roślin (Polish Plant Red Book); Kaźmierczakowa, R., Zarzycki, K., Mirek, Z., Eds.; W. Szafer Institute of Botany, Polish Academy of Sciences: Kraków, Poland, 2014; pp. 92–95. [Google Scholar]
- Jadwiszczak, K.A.; Banaszek, A.; Jabłońska, E.; Sozinov, O.V. Chloroplast DNA variation of Betula humilis Schrk. in Poland and Belarus. Tree Genet. Genomes 2012, 8, 1017–1030. [Google Scholar] [CrossRef] [Green Version]
- Jadwiszczak, K.A.; Banaszek, A.; Jabłońska, E.; Sozinov, O.V. Could Betula humilis Schrk. have survived the last glaciation at a current margin of its distribution? Testing the hypothesis of glacial refugium using nuclear microsatellites. Plant Syst. Evol. 2011, 297, 147–156. [Google Scholar] [CrossRef]
- Jadwiszczak, K.A.; Banaszek, A.; Chrzanowska, A.; Kłosowski, S.; Sozinov, O.V. The admixture zone of Betula humilis Schrk. phylogenetic lineages follows the eastern central European suture zone. Plant Ecol. Divers. 2015, 8, 323–329. [Google Scholar] [CrossRef]
- Petit, R.J.; Aguinagalde, I.; de Beaulieu, J.; Bittkau, C.; Brewer, S.; Cheddadi, R.; Ennos, R.; Fineschi, S.; Grivet, D.; Lascoux, M.; et al. Glacial refugia: Hotspots but not melting pots of genetic diversity. Science 2003, 300, 1563–1565. [Google Scholar] [CrossRef] [Green Version]
- Jabłońska, E. Vegetation with Betula humilis in central Europe. Phytocoenologia 2012, 42, 259–277. [Google Scholar] [CrossRef]
- Chrzanowska, A.; Jadwiszczak, K.A.; Kłosowski, S.; Banaszek, A.; Sozinov, O.V. Sexual reproduction efficiency and genetic diversity of endangered Betula humilis Schrk. populations from edge and sub-central parts of its range. Folia Geobot. 2016, 51, 161–173. [Google Scholar] [CrossRef] [Green Version]
- Bona, A.; Kłosowski, S.; Jadwiszczak, K.A.; Petrova, G. Flowering and quality of seeds and pollen in endangered populations of Betula humilis. Trees 2022, 36, 313–324. [Google Scholar] [CrossRef]
- Smouse, P.E.; Dyer, R.J.; Westfall, R.D.; Sork, V.L. Two-generation analysis of pollen flow across a landscape. I. Male gamete heterogeneity among females. Evolution 2001, 55, 260–271. [Google Scholar] [CrossRef]
- Chybicki, I.J.; Oleksa, A. Seed and pollen gene dispersal in Taxus baccata, a dioecious conifer in the face of strong population fragmentation. Ann. Bot. 2018, 122, 409–421. [Google Scholar] [CrossRef] [Green Version]
- Șofletea, N.; Mihai, G.; Ciocîrlan, E.; Curtu, A.L. Genetic Diversity and Spatial Genetic Structure in Isolated Scots Pine (Pinus sylvestris L.) Populations Native to Eastern and Southern Carpathians. Forests 2020, 11, 1047. [Google Scholar] [CrossRef]
- Truong, C.; Palmé, A.E.; Felber, F. Recent invasion of the mountain birch Betula pubescens ssp. tortuosa above the treeline due to climate change: Genetic and ecological study in northern Sweden. J. Evol. Biol. 2007, 20, 369–380. [Google Scholar] [CrossRef]
- Jabłońska, E.; Pawlikowski, P.; Jarzombkowski, F.; Chormański, J.; Okruszko, T.; Kłosowski, S. Importance of water level dynamics for vegetation patterns in a natural percolation mire (Rospuda fen, NE Poland). Hydrobiologia 2011, 674, 105–117. [Google Scholar] [CrossRef] [Green Version]
- Bojsza, A.; Lićwinko, M.; Pińkowska, J. LIFE11/NAT/PL/422. Final Report—Results of Hydrological Monitoring Conducted in the Upper Basin of the Biebrza River Valley in 2013–2018. 2019. Available online: https://www.gorna.biebrza.org.pl/plik,4784,raport-koncowy-z-analizy-warunkow-meteorologicznych-i-stanow-wod-w-latach-hydrologicznych-2013-2018.pdf (accessed on 27 December 2021).
- Jarzombkowski, F. Torfowiska w basenie górnym Doliny Biebrzy (Peat bogs in the upper basin of the Biebrza river valley). In Z Mazowsza na Polesie i Wileńszczyznę. Zróżnicowanie i Ochrona Szaty Roślinnej Pogranicza Europy Środkowej i Północno-Wschodniej (From Mazovia to Polesie and the Vilnus Region. Diversification and Protection of Vegetation on the Border of Central and North-Eastern Europe; Obidziński, A., Ed.; Polish Botanical Society: Warsaw, Poland, 2010; pp. 331–340. [Google Scholar]
- Bojsza, A. LIFE11/NAT/PL/422 (Górna Biebrza–Upper Biebrza River). Final Report Covering the Project Activities from 01/09/2012 to 30/03/2019. 2019. Available online: https://www.gorna.biebrza.org.pl/plik,4836,raport-koncowy-life11-nat-pl-422.pdf (accessed on 27 December 2021).
- Kulju, K.K.M.; Pekkinen, M.; Varvio, S. Twenty-three microsatellite primer pairs for Betula pendula (Betulaceae). Mol. Ecol. Notes 2004, 4, 471–473. [Google Scholar] [CrossRef]
- Truong, C.; Palmé, A.E.; Felber, F.; Naciri-Graven, Y. Isolation and characterization of microsatellite markers in the tetraploid birch, Betula pubescens ssp. tortuosa. Mol. Ecol. Notes 2005, 5, 96–98. [Google Scholar] [CrossRef]
- Bona, A.; Kulesza, U.; Jadwiszczak, K.A. Clonal diversity, gene flow and seed production in endangered populations of Betula humilis Schrk. Tree Genet. Genomes 2019, 15, 50. [Google Scholar] [CrossRef] [Green Version]
- Arnaud-Haond, S.; Belkhir, K. GenClone: A computer program to analyse genotypic data, test for clonality and describe spatial clonal organization. Mol. Ecol. Notes 2007, 7, 15–17. [Google Scholar] [CrossRef]
- van Oosterhout, C.; Hutchinson, W.F.; Wills, P.M.; Shipley, P. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Kalinowski, S.T.; Taper, M.L. Maximum likelihood estimation of the frequency of null alleles at microsatellite loci. Conserv. Genet. 2006, 7, 991–995. [Google Scholar] [CrossRef]
- Dąbrowski, M.J.; Bornelöv, S.; Kruczyk, M.; Baltzer, N.; Komorowski, J. ‘True’ null allele detection in microsatellite loci: A comparison of methods, assessment of difficulties and survey of possible improvements. Mol. Ecol. Resour. 2015, 15, 477–488. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. GENEPOP (version 1.2): Population genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Rousset, F. Genepop’007: A complete reimplementation of the Genepop software for Windows and Linux. Mol. Ecol. Resour 2008, 8, 103–106. [Google Scholar] [CrossRef]
- Rice, W.R. Analyzing tables of statistical tests. Evolution 1989, 43, 223–225. [Google Scholar] [CrossRef] [PubMed]
- Peakall, R.; Smouse, P.E. GENALEX 6: Genetic analysis in Excel. Population genetic software for teaching and research. Mol. Ecol. Notes 2006, 6, 288–295. [Google Scholar] [CrossRef]
- Chybicki, I.J.; Burczyk, J. Simultaneous estimation of null alleles and inbreeding coefficients. J. Hered. 2009, 100, 106–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Rienzo, A.; Peterson, A.C.; Garza, J.C.; Valdes, A.M.; Slatkin, M.; Freimer, N.B. Mutational processes of simple sequence repeat loci in human populations. Proc. Natl. Acad. Sci. USA 1994, 91, 3166–3170. [Google Scholar] [CrossRef] [Green Version]
- Garza, J.C.; Williamson, E.G. Detection of reduction in population size using data from microsatellite loci. Mol. Ecol. 2001, 10, 305–318. [Google Scholar] [CrossRef]
- Cornuet, J.-M.; Luikart, G. Description and power analysis of two tests for detecting recent population bottlenecks from allele frequency data. Genetics 1996, 144, 2001–2014. [Google Scholar] [CrossRef]
- Piry, S.; Luikart, G.; Cornuet, J.M. BOTTLENECK: A computer program for detecting recent reductions in the effective population size using allele frequency data. J. Hered. 1999, 90, 502–503. [Google Scholar] [CrossRef]
- Jones, O.R.; Wang, J. COLONY: A program for parentage and sibship inference from multilocus genotype data. Mol. Ecol. Resour. 2010, 10, 551–555. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef] [Green Version]
- Earl, D.A.; vonHoldt, B.M. STRUCTURE HARVESTER: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Jakobsson, M.; Rosenberg, N.A. CLUMPP: A cluster matching and permutation programfor dealing with label switching and multimodality in analysis of population structure. Bioinformatics 2007, 23, 1801–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenberg, N.A. DISTRUCT: A program for the graphical display of population structure. Mol. Ecol. Notes 2004, 4, 137–138. [Google Scholar] [CrossRef]
- Chapuis, M.-P.; Estoup, A. Microsatellite null alleles and estimation of population differentiation. Mol. Biol. Evol. 2007, 24, 621–631. [Google Scholar] [CrossRef] [Green Version]
- Hardy, O.J.; Vekemans, X. SPAGeDi: A versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2002, 2, 618. [Google Scholar] [CrossRef] [Green Version]
- Loiselle, B.A.; Sork, V.L.; Nason, J.; Graham, C. Spatial genetic structure of a tropical understory shrub, Psychotria officinalis (Rubiaceae). Am. J. Bot. 1995, 82, 1420–1425. [Google Scholar] [CrossRef]
- Fenster, C.B.; Vekemans, X.; Hardy, O.J. Quantifying gene flow from spatial genetic structure data in a metapopulation of Chamaecrista fasciculata (Leguminosae). Evolution 2003, 57, 995–1007. [Google Scholar] [CrossRef]
- Dick, C.W.; Hardy, O.; Jones, F.; Petit, R. Spatial scales of pollen and seed-mediated gene flow in tropical rain forest trees. Trop. Plant Biol. 2008, 1, 20–33. [Google Scholar] [CrossRef]
- Aavik, T.; Helm, A. Restoration of plant species and genetic diversity depends on land scape-scale dispersal. Restor. Ecol. 2018, 26, 92–102. [Google Scholar] [CrossRef]
- Lovett-Doust, L. Population dynamics and local specialization in a clonal perennial (Ranunculus repens). I. The dynamics of ramets in contrasting habitats. J. Ecol. 1981, 69, 743–755. [Google Scholar] [CrossRef]
- Barrett, S.C.H. Influences of clonality on plant sexual reproduction. Proc. Natl. Acad. Sci. USA 2015, 112, 8859–8866. [Google Scholar] [CrossRef] [Green Version]
- Kettenring, K.M.; Mercer, K.L.; Adams, C.R.; Hines, J. Application of genetic diversity—Ecosystem function research to ecological restoration. J. Appl. Ecol. 2014, 51, 339–348. [Google Scholar] [CrossRef]
- Morente-López, J.; Lara-Romero, C.; García-Fernández, A.; Teso, M.L.R.; Prieto-Benítez, S.; Iriondo, J.M. Gene flow effects on populations inhabiting marginal areas: Origin matters. J. Ecol. 2021, 109, 139–153. [Google Scholar] [CrossRef]
- Jabłońska, E. Aktualny wykaz stanowisk Betula humilis (Betulaceae) w Polsce (Current list of localities of Betula humilis (Betulaceae) in Poland). Fragm. Flor. Geobot. Pol. 2014, 21, 77–90. [Google Scholar]
- Slatkin, M. Rare alleles as indicators of gene flow. Evolution 1985, 39, 53–65. [Google Scholar] [CrossRef]
- Jadwiszczak, K.A.; Drzymulska, D.; Banaszek, A.; Jadwiszczak, P. Population history, genetic variation and conservation status of the endangered birch species Betula nana L. in Poland. Silva Fenn. 2012, 4, 465–477. [Google Scholar] [CrossRef] [Green Version]
- Jarni, K.; Jakše, J.; Brus, R. Vegetative propagation: Linear barriers and somatic mutation affect the genetic structure of a Prunus avium L. stand. Forestry 2015, 88, 612–621. [Google Scholar] [CrossRef] [Green Version]
- Yu, L.; Boström, C.; Franzenburg, S.; Bayer, T.; Dagan, T.; Reusch, T.B.H. Somatic genetic drift and multilevel selection in a clonal seagrass. Nat. Ecol. Evol. 2020, 4, 952–962. [Google Scholar] [CrossRef]
- Paluch, J.; Zarek, M. Seed dispersal of anemochoric Abies alba Mill.: Lessons from seed tracking, seed trap experiments and the genetic parentage assignment of seedlings. Ann. For. Res. 2020, 63, 19–38. [Google Scholar] [CrossRef]
- Cavers, S.; Degen, B.; Caron, H.; Lemers, M.R.; Margis, R.; Salgueiro, F.; Lowe, A.J. Optimal sampling strategy for estimation of spatial genetic structure in tree populations. Heredity 2005, 95, 281–289. [Google Scholar] [CrossRef]
- Curtu, A.L.; Craciunesc, I.; Enescu, C.M.; Vidalis, A.; Sofletea, N. Fine-scale spatial genetic structure in a multi-oak-species (Quercus spp.) forest. iForest-Biogeosci. For. 2015, 8, 324–332. [Google Scholar] [CrossRef] [Green Version]
- Nei, M.; Maruyama, T.; Chakraborty, R. The bottleneck effect and genetic variability in populations. Evolution 1975, 29, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Serafin, A.; Urban, D.; Bronowicka-Mielniczuk, U.; Szczurowska, A. To what degree can the specifics of occurrence of glacial relic Betula humilis Schrank be an indicator of habitat conditions of moderate climate peatlands? Water 2018, 10, 1062. [Google Scholar] [CrossRef] [Green Version]
- Kapowicz, E. Nowe stanowiska brzozy niskiej Betula humilis na terenie Niecki Gródecko-Michałowskiej (The new stands of shrubby birch Betula humilis in the area of Gródecko-Michałowska Basin). Przegląd Przyr. 2013, 24, 61–67. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population Name | Code | Coordinates | Altitude | NR | NG | Habitat Protection Type | |
|---|---|---|---|---|---|---|---|
| Latitude | Longitude | ||||||
| Magdzie Bagno | MB | 54°08′41″ N | 23°16′05″ E | 138 m | 109 | 102 | No protection |
| Rospuda river valley | ROS | 53°54′23″ N | 22°56′38″ E | 123 m | 138 | 138 | Natura 2000 network |
| Szuszalewo | SUS | 53°43′07″ N | 23°21′23″ E | 119 m | 139 | 139 | Biebrza National Park |
| Pop | Locus | A | AP | AN | HO | HE | P(HWE) |
|---|---|---|---|---|---|---|---|
| MB | L1.10 | 14 | 0 | 0.0001 | 0.873 | 0.825 | 0.2617 |
| L021 | 12 | 0 | 0.0526 * | 0.716 | 0.830 | 0.0000 * | |
| L022 | 15 | 1 | 0.0001 | 0.833 | 0.851 | 0.1812 | |
| L5.4 | 16 | 0 | 0.0000 | 0.892 | 0.900 | 0.1085 | |
| L5.1 | 16 | 1 | 0.0013 | 0.912 | 0.847 | 0.0535 | |
| Bo.F394 | 18 | 1 | 0.0042 | 0.745 | 0.745 | 0.6725 | |
| Bo.G182 | 9 | 3 | 0.0058 | 0.706 | 0.716 | 0.7330 | |
| 14.3 ± 1.13 SE | 0.86 | 0.0019 | 0.811 ± 0.033 SE | 0.816 ± 0.024 SE | <0.0001 | ||
| ROS | L1.10 | 15 | 0 | 0.0000 | 0.884 | 0.878 | 0.1355 |
| L021 | 11 | 0 | 0.0000 | 0.761 | 0.828 | 0.0228 | |
| L022 | 13 | 0 | 0.0018 | 0.833 | 0.862 | 0.6196 | |
| L5.4 | 18 | 3 | 0.0035 | 0.928 | 0.917 | 0.5972 | |
| L5.1 | 13 | 1 | 0.0012 | 0.862 | 0.810 | 0.0347 | |
| Bo.F394 | 23 | 2 | 0.0216 | 0.891 | 0.929 | 0.5092 | |
| Bo.G182 | 8 | 0 | 0.0580 | 0.587 | 0.661 | 0.1553 | |
| 14.4 ± 1.95 SE | 0.86 | 0.0123 | 0.821 ± 0.044 SE | 0.841 ± 0.034 SE | 0.0313 | ||
| SUS | L1.10 | 20 | 4 | 0.0087 | 0.906 | 0.894 | 0.1613 |
| L021 | 23 | 8 | 0.0660 * | 0.784 | 0.878 | 0.0000 * | |
| L022 | 17 | 2 | 0.0239 * | 0.827 | 0.918 | 0.2888 | |
| L5.4 | 22 | 3 | 0.0225 | 0.935 | 0.908 | 0.6367 | |
| L5.1 | 24 | 7 | 0.0532 * | 0.842 | 0.905 | 0.0000 * | |
| Bo.F394 | 28 | 5 | 0.0218 | 0.871 | 0.911 | 0.2300 | |
| Bo.G182 | 11 | 2 | 0.0346 | 0.669 | 0.705 | 0.0000 * | |
| 20.7 ± 2.07 SE | 4.42 | 0.0219 | 0.834 ± 0.033 SE | 0.874 ± 0.029 SE | <0.0001 |
| Pop | p Value for TPM | M-Ratio | ||
|---|---|---|---|---|
| MR | ∆MR | p Value | ||
| MB | 0.8515 | 0.6954 | 0.0984 | 0.0543 |
| ROS | 0.0195 | 0.6678 | 0.1365 | 0.0387 |
| SUS | 0.7656 | 0.7469 | 0.0500 | 0.4066 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bona, A.; Brzeziński, D.; Jadwiszczak, K.A. Genetic Diversity and Fine-Scale Spatial Genetic Structure of the Endangered Shrub Birch (Betula humilis Schrk.) Populations in Protected and Unprotected Areas. Diversity 2022, 14, 684. https://doi.org/10.3390/d14080684
Bona A, Brzeziński D, Jadwiszczak KA. Genetic Diversity and Fine-Scale Spatial Genetic Structure of the Endangered Shrub Birch (Betula humilis Schrk.) Populations in Protected and Unprotected Areas. Diversity. 2022; 14(8):684. https://doi.org/10.3390/d14080684
Chicago/Turabian StyleBona, Agnieszka, Damian Brzeziński, and Katarzyna A. Jadwiszczak. 2022. "Genetic Diversity and Fine-Scale Spatial Genetic Structure of the Endangered Shrub Birch (Betula humilis Schrk.) Populations in Protected and Unprotected Areas" Diversity 14, no. 8: 684. https://doi.org/10.3390/d14080684
APA StyleBona, A., Brzeziński, D., & Jadwiszczak, K. A. (2022). Genetic Diversity and Fine-Scale Spatial Genetic Structure of the Endangered Shrub Birch (Betula humilis Schrk.) Populations in Protected and Unprotected Areas. Diversity, 14(8), 684. https://doi.org/10.3390/d14080684