
Characterization of Captive Breeders to Preserve the Residual Genetic Diversity of Adriatic Sturgeon (Acipenser naccarii)

 ,
,  ,
,  and
and 
Abstract
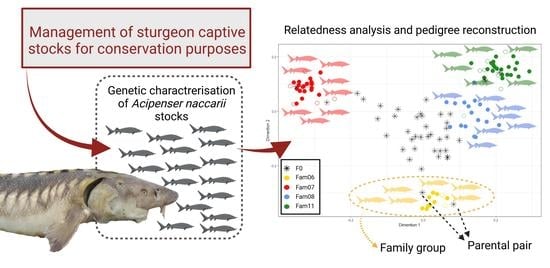
1. Introduction
2. Materials and Methods
2.1. Sampling and Molecular Markers Used
2.2. Parental Allocation Analysis
2.3. Analyses and Visualization of Pairwise Genetic Distances
3. Results
3.1. Pedigree Reconstruction
3.2. Multi-Dimensional Scaling and Relatedness Inference
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Arlati, G.; Bronzi, P.; Colombo, L.; Giovannini, G. Induzione della riproduzione nello storione italiano (Acipenser naccarii) allevato in cattività. Riv. Ital. Acquacol. 1988, 23, 94–96. [Google Scholar]
- Giovannini, G.; Colombo, L.; Bronzi, P.; Arlati, G. Growth of hatchery-produced juveniles of Italian sturgeon, Acipenser naccarii Bonaparte, reared intensively in fresh water. In Acipenser; Cemagref Publication: Bordeaux, France, 1991; pp. 401–404. [Google Scholar]
- Boscari, E.; Pujolar, J.M.; Dupanloup, I.; Corradin, R.; Congiu, L. Captive breeding programs based on family groups in polyploid sturgeons. PLoS ONE 2014, 9, e110951. [Google Scholar] [CrossRef]
- Arlati, G.; Poliakova, L. Restoration of Adriatic Sturgeon (Acipenser naccarii) in Italy: Situation and Perspectives. In Biology, Conservation and Sustainable Development of Sturgeons; Carmona, R., García-Gallego, M., Ruiz-Rejón, M., Eds.; Springer: Dordrecht, The Netherlands, 2009; Volume 14, pp. 237–238. [Google Scholar]
- Congiu, L.; Boscari, E.; Pagani, S.; Gazzola, M.; Bronzi, P. Resumption of natural reproduction of the Adriatic sturgeon in the River Po. Oryx 2021, 55, 816. [Google Scholar] [CrossRef]
- Congiu, L.; Pujolar, J.M.; Forlani, A.; Cenadelli, S.; Dupanloup, I.; Barbisan, F.; Galli, A.; Fontana, F. Managing polyploidy in ex situ conservation genetics: The case of the critically endangered Adriatic sturgeon (Acipenser naccarii). PLoS ONE 2011, 6, e18249. [Google Scholar] [CrossRef]
- Boscari, E.; Congiu, L. The need for genetic support in restocking activities and ex situ conservation programmes: The case of the Adriatic sturgeon (Acipenser naccarii Bonaparte, 1836) in the Ticino River Park. J. Appl. Ichthyol. 2014, 30, 1416–1422. [Google Scholar] [CrossRef]
- World Sturgeon Conservation Society; WWF. Pan-European Action Plan for Sturgeons. In Proceedings of the Convention on the Conservation of European Wildlife and Natural Habitats Standing Committee: 38th Meeting, Strasbourg, France, 27–30 November 2018. [Google Scholar]
- Cabrera-Castro, R.; Zabal, C.; Soriguer, M.C.; Domezain, A.; Hernando, J.A. Morphological development in the first phase of Adriatic sturgeon Acipenser naccarii under controlled conditions. J. Fish Biol. 2018, 92, 1956–1974. [Google Scholar] [CrossRef]
- Forlani, A.; Fontana, F.; Congiu, L. Isolation of microsatellite loci from the endemic and endangered Adriatic sturgeon (Acipenser naccarii). Conserv. Genet. 2007, 9, 461–463. [Google Scholar] [CrossRef]
- McQuown, E.C.; Sloss, B.L.; Sheehan, R.J.; Rodzen, J.; Tranah, G.J.; May, B. Microsatellite analysis of genetic variation in sturgeon: New primer sequences for Scaphirhynchus and Acipenser. Trans. Am. Fish. Soc. 2000, 129, 1380–1388. [Google Scholar] [CrossRef]
- Welsh, A.B.; Blumberg, M.; May, B. Identification of microsatellite loci in lake sturgeon, Acipenser fulvescens, and their variability in green sturgeon, A. medirostris. Mol. Ecol. Notes 2003, 3, 47–55. [Google Scholar] [CrossRef]
- Boscari, E.; Vidotto, M.; Martini, D.; Papetti, C.; Ogden, R.; Congiu, L. Microsatellites from the genome and the transcriptome of the tetraploid Adriatic sturgeon, Acipenser naccarii (Bonaparte, 1836) and cross-species applicability to the diploid beluga sturgeon, Huso huso (Linnaeus, 1758). J. Appl. Ichthyol. 2015, 31, 977–983. [Google Scholar] [CrossRef]
- Henderson-Arzapalo, A.; King, T.L. Novel microsatellite markers for Atlantic sturgeon (Acipenser oxyrinchus) population delineation and broodstock management. Mol. Ecol. Notes 2022, 2, 437–439. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547. [Google Scholar] [CrossRef]
- Sørensen, T. A Method of Establishing Groups of Equal Amplitude in Plant Sociology Based on Similarity of Species Content and Its Application to Analyses of the Vegetation on Danish Commons. Biol. Skr. K. Dan. Vidensk. Selsk. 1948, 5, 1–34. [Google Scholar]
- Thioulouse, J.; Dray, S.; Dufour, A.-B.; Siberchicot, A.; Jombart, T.; Pavoine, S. Multivariate Analysis of Ecological Data with ade4; Springer: New York, NY, USA, 2018. [Google Scholar] [CrossRef]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016; Available online: https://ggplot2.tidyverse.org (accessed on 5 September 2020)ISBN 978-3-319-24277-4.
- Tortonese, E. Acipenser naccarii (Bonaparte, 1836). In The Freshwater Fishes of Europe; Holcik, J., Ed.; AULA_Verlag: Wiesbaden, Germany, 1989; pp. 286–293. [Google Scholar]
- Boscari, E.; Wu, J.; Jiang, T.; Zhang, S.; Cattelan, S.; Wang, C.; Du, H.; Li, C.; Li, J.; Ruan, R.; et al. The last giants of the Yangtze River: A multidisciplinary picture of what remains of the endemic Chinese sturgeon. Sci. Total Environ. 2022, 843, 157011. [Google Scholar] [CrossRef]
- Fontana, F.; Congiu, L.; Mudrak, V.A.; Quattro, J.M.; Smith, T.I.; Ware, K.; Doroshov, S.I. Evidence of hexaploid karyotype in shortnose sturgeon. Genome 2008, 51, 113–119. [Google Scholar] [CrossRef]
- Palle, S.D.; Boscari, E.; Bordignon, S.G.; Muñoz-Mora, V.H.; Bertorelle, G.; Congiu, L. Different chromosome segregation patterns coexist in the tetraploid Adriatic sturgeon Acipenser naccarii. Diversity 2022, 14, 745. [Google Scholar] [CrossRef]
- Stift, M.; Berenos, C.; Kuperus, P.; Van Tienderen, P.H. Segregation models for disomic, tetrasomic and intermediate inheritance in tetraploids: A general procedure applied to Rorippa (yellow cress) microsatellite data. Genetics 2008, 179, 2113–2123. [Google Scholar] [CrossRef] [PubMed]
- May, B.; Krueger, C.C.; Kincaid, H.L. Genetic variation at microsatellite loci in sturgeon: Primer sequence homology in Acipenser and Scaphirhynchus. Can. J. Fish. Aquat. Sci. 1997, 54, 1542–1547. [Google Scholar] [CrossRef]
- Zane, L.; Patarnello, T.; Ludwig, A.; Fontana, F.; Congiu, L. Isolation and characterization of microsatellites in the Adri-atic sturgeon (Acipenser naccarii). Mol. Ecol. Notes 2022, 2, 586–588. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Family Group | Parental Pair | N F1 Tot (Alive) | mtDNA Haplotype | Family Group | Parental Pair | N F1 Tot (Alive) | mtDNA Haplotype |
|---|---|---|---|---|---|---|---|
| 01 | Pelvienne (F) × NaccS18 (M) | 14 (5) | 2 | 17 | NaccS8 (F) × 740 (M) | 21 (14) | 2 |
| 02 | NaccS8 (F) × Matto (M) | 9 (6) | 2 | 18 | NaccS3 (F) × Matto (M) | 5 (1) | 3 |
| 03 | NaccS7 (F) × NaccS6 (M) | 12 (12) | 3 | 19 | NaccS33 (F) × 740 (M) | 4 (2) | 3 |
| 04 | NaccS8 (F) × NaccS31 (M) | 17 (15) | 2 | 20 | NaccS7 (F) × VerdeBis (M) | 2 (1) | 3 |
| 05 | NaccS33 (F) × NaccS11 (M) | 10 (4) | 3 | 21 | NaccS12 (F) × NaccS17 (M) | 1 (1) | 2 |
| 06 | O2 (F) × Matto (M) | 12 (9) | 5 | 22 | Indecisa (U) × Matto (M) | 2 (2) | 2 |
| 07 | NaccS26 (F) × NaccS29 (M) | 27 (18) | 5 | 23 | Indecisa (U) × AnS11 (M) | 1 (1) | 2 |
| 08 | NaccS19 (F) × NaccS17 (M) | 18 (17) | 5/7 | 24 | NaccS12 (F) × Matto (M) | 1 (0) | 2 |
| 09 | NaccS28 (F) × NaccS17 (M) | 51 (30) | 3 | 25 | Naccs3 (F)–Naccs29 (M) | 1 (0) | 3 |
| 10 | NaccS16 (F) × NaccS23 (M) | 39 (15) | 3 | 26 | NaccS16 (F) × 740 (M) | 1 (1) | 3 |
| 11 | NaccS19 (F) × NaccS31 (M) | 30 (21) | 5/7 | 27 | NaccS16 (F) × NaccS29 (M) | 1 (1) | 3 |
| 12 | NaccS16 (F) × NaccS30 (M) | 13 (2) | 3 | 28 | Naccs8 (F)–Naccs11 (M) | 6 (6) | 2 |
| 13 | NaccS33 (F) × NaccS9 (M) | 17 (12) | 3 | 29 | Pelvienne (F)–Naccs23 (M) | 3 (3) | 2 |
| 14 | NaccS8 (F) × NaccS17 (M) | 8 (8) | 2 | 30 | Non-allocated | 141 (108) | - |
| 15 | NaccS12 (F) × NaccS27 (M) | 10 (3) | 2 | 31 | Multi-allocated | 7 (7) | - |
| 16 | NaccS3 (F) × 740 (M) | 21 (6) | 3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barca, F.; Dalle Palle, S.; Schiavon, L.; Samassa, C.; Castaldelli, G.; Boscari, E.; Congiu, L. Characterization of Captive Breeders to Preserve the Residual Genetic Diversity of Adriatic Sturgeon (Acipenser naccarii). Diversity 2022, 14, 829. https://doi.org/10.3390/d14100829
Barca F, Dalle Palle S, Schiavon L, Samassa C, Castaldelli G, Boscari E, Congiu L. Characterization of Captive Breeders to Preserve the Residual Genetic Diversity of Adriatic Sturgeon (Acipenser naccarii). Diversity. 2022; 14(10):829. https://doi.org/10.3390/d14100829
Chicago/Turabian StyleBarca, Federica, Stefano Dalle Palle, Luca Schiavon, Chiara Samassa, Giuseppe Castaldelli, Elisa Boscari, and Leonardo Congiu. 2022. "Characterization of Captive Breeders to Preserve the Residual Genetic Diversity of Adriatic Sturgeon (Acipenser naccarii)" Diversity 14, no. 10: 829. https://doi.org/10.3390/d14100829
APA StyleBarca, F., Dalle Palle, S., Schiavon, L., Samassa, C., Castaldelli, G., Boscari, E., & Congiu, L. (2022). Characterization of Captive Breeders to Preserve the Residual Genetic Diversity of Adriatic Sturgeon (Acipenser naccarii). Diversity, 14(10), 829. https://doi.org/10.3390/d14100829